
GBE1 Promotes Glioma Progression by Enhancing Aerobic Glycolysis through Inhibition of FBP1


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid Construction and Lentiviral Transfection
2.3. RNA Extraction and Real−Time Quantitative PCR (qRT−PCR)
2.4. Protein Extraction and Western Blot (WB)
2.5. Cell Proliferation Assay and Colony Formation Assay
2.6. Cell Cycle Analysis and Apoptosis Detection
2.7. Wound Healing Assay and Transwell Assay
2.8. Oxygen Deprivation Assay and Tubule Formation Assay
2.9. Immunofluorescence Staining and Immunohistochemistry Staining
2.10. Glycolytic Stress Test and Mitochondrial Stress Test
2.11. Intracranial Tumor Formation
2.12. Bioinformatics Analysis Based on Public Databases
2.13. Statistical Analysis
3. Results
3.1. GBE1 Expression Was Associated with Glioma Malignancy
3.2. GBE1 Knockdown Inhibited Glioma Cell Proliferation and Induced Cell Cycle Arrest and Apoptosis
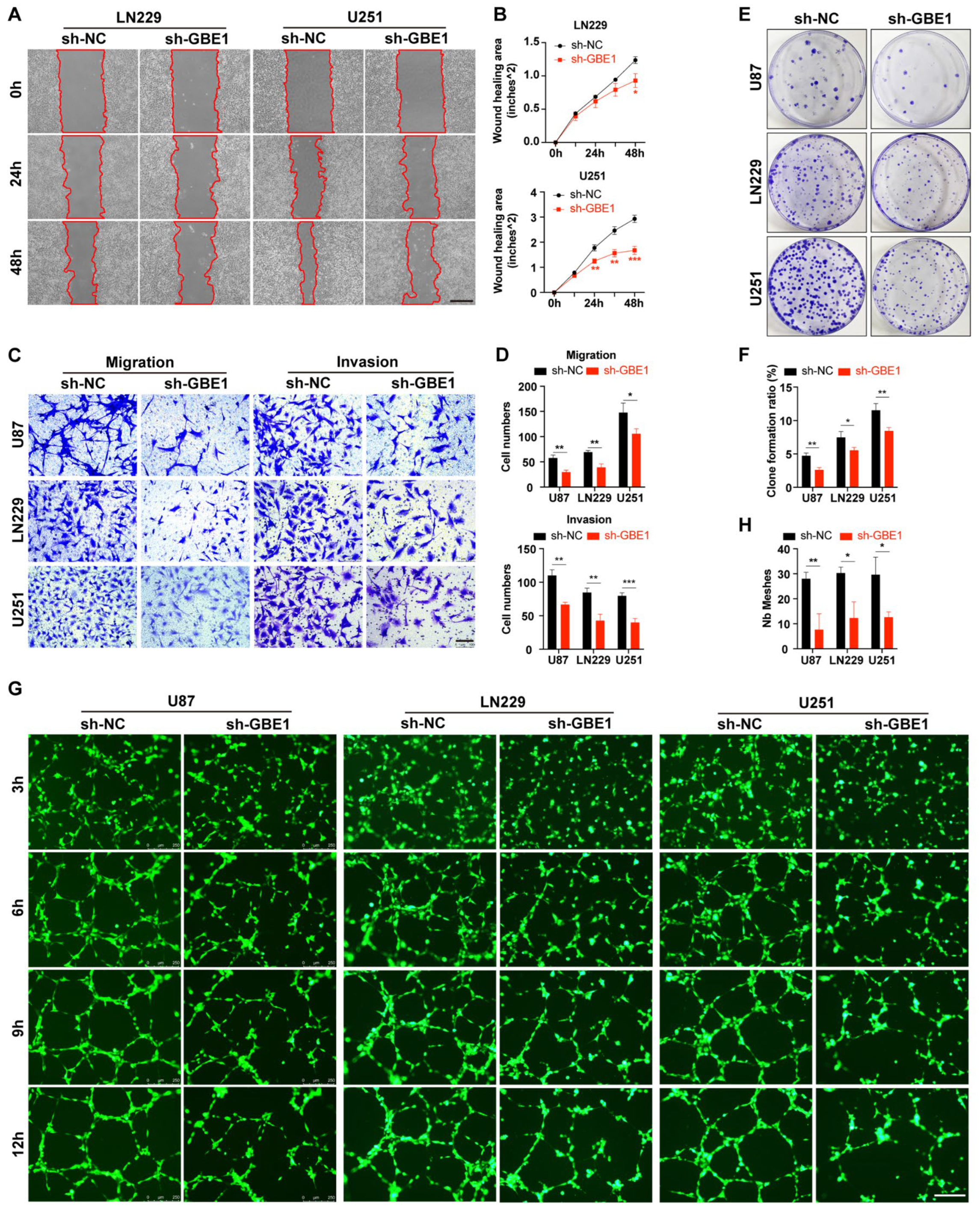
3.3. GBE1 Knockdown Affected Various Biological Behaviors of Glioma Cells
3.4. GBE1 Knockdown Affected the Biological Behavior of Glioma by Regulating Various Proteins and Affected the Expression of FBP1 through the NF−κB Pathway
3.5. FBP1 Suppressed Malignant Phenotypes of Glioma Cells
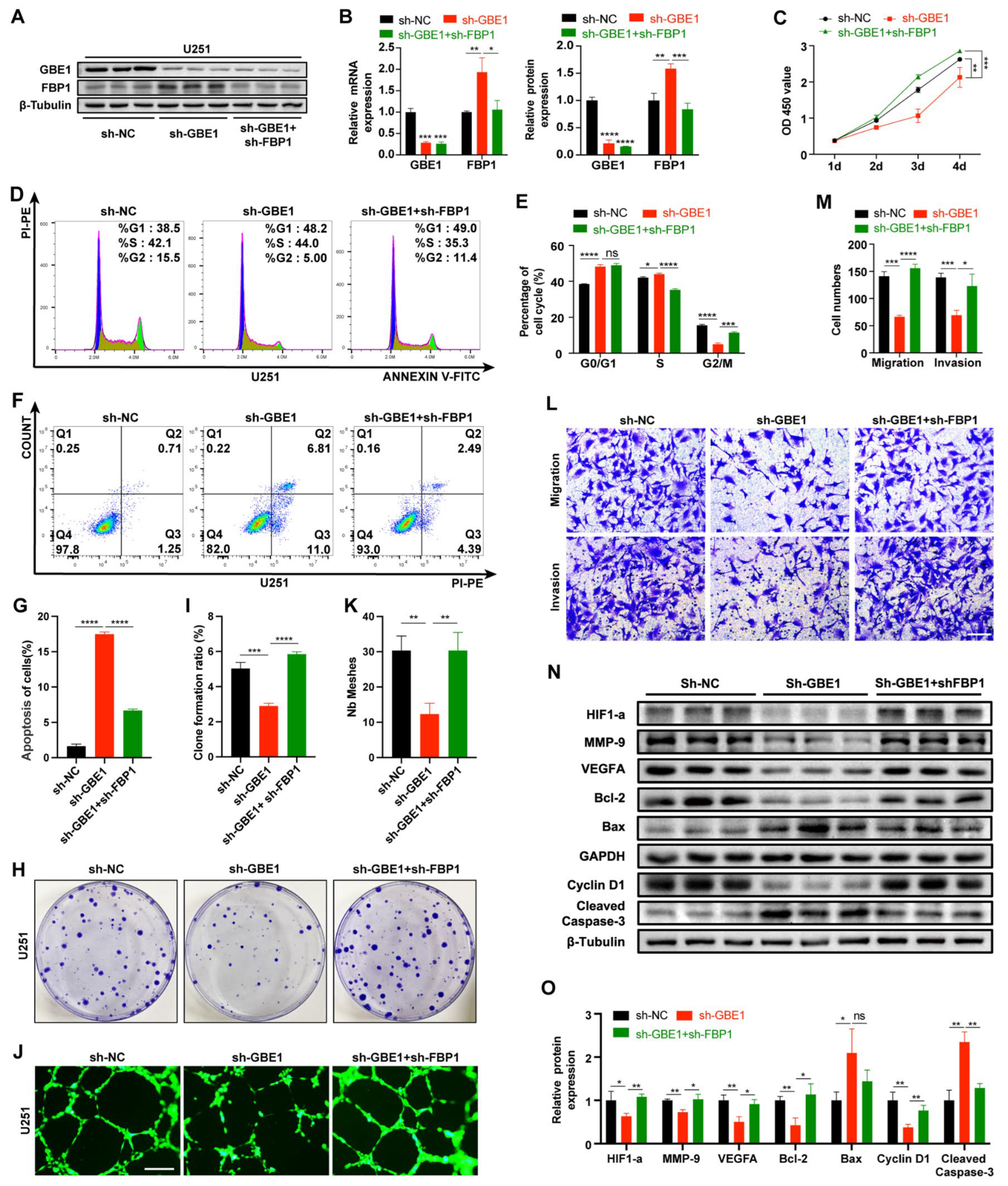
3.6. FBP1 Knockdown Reversed the Glioma Inhibition Caused by GBE1 Knockdown
3.7. Elevated FBP1 Expression Caused by GBE1 Knockdown Induced Metabolic Reprogramming of Glioma Cells
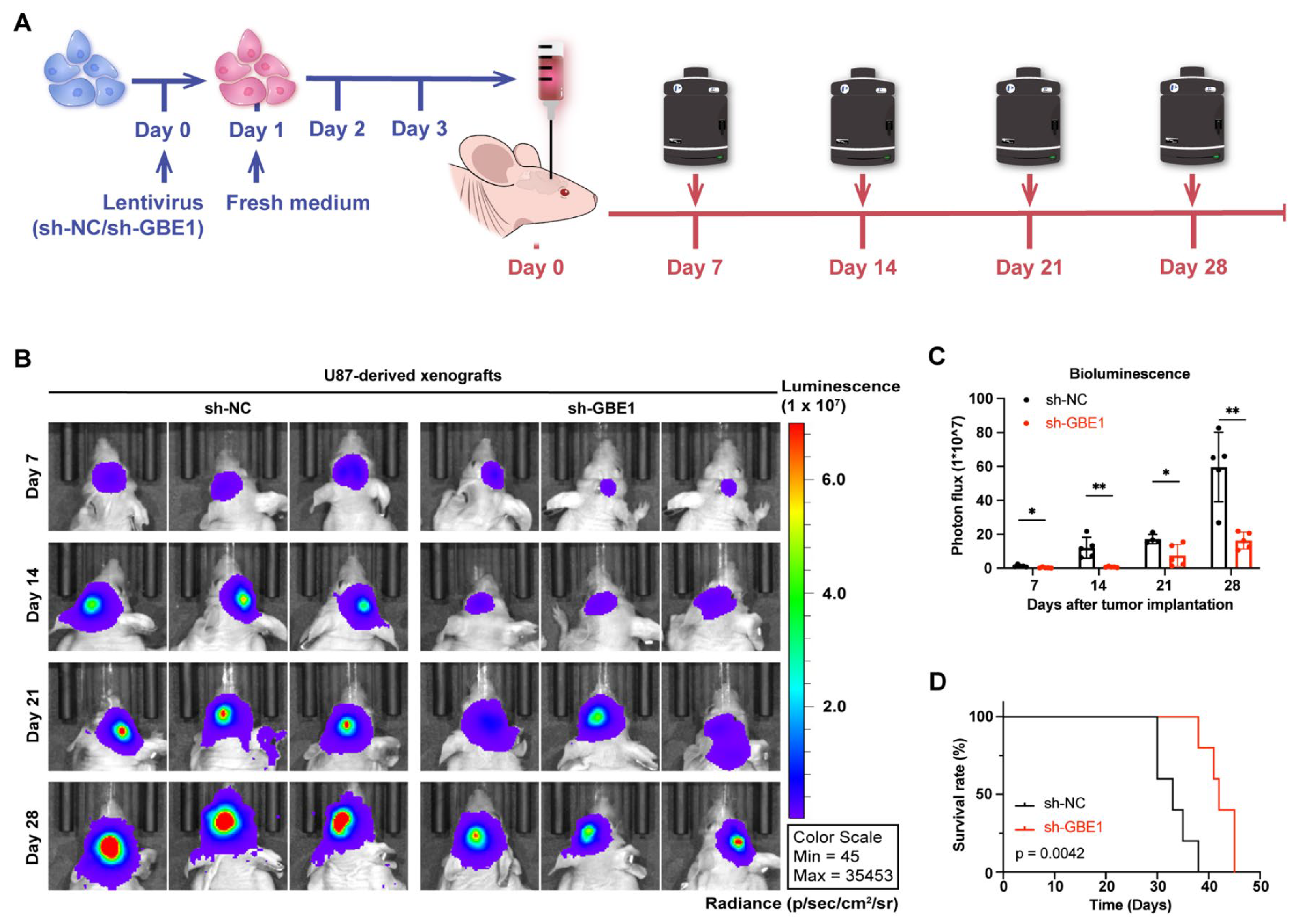
3.8. GBE1 Knockdown Significantly Inhibited the Growth of Glioma Xenograft In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—Implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022, 24, v1–v95. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Smith, Z.D.; Shi, J.; Gu, H.; Donaghey, J.; Clement, K.; Cacchiarelli, D.; Gnirke, A.; Michor, F.; Meissner, A. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 2017, 549, 543–547. [Google Scholar] [CrossRef]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Li, Z.; Ge, Y.; Dong, J.; Wang, H.; Zhao, T.; Wang, X.; Liu, J.; Gao, S.; Shi, L.; Yang, S.; et al. BZW1 Facilitates Glycolysis and Promotes Tumor Growth in Pancreatic Ductal Adenocarcinoma Through Potentiating eIF2α Phosphorylation. Gastroenterology 2022, 162, 1256–1271.e1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Luo, F.; Luo, S.; Li, L.; Ren, X.; Lin, J.; Liang, Y.; Ma, C.; Ding, L.; Zhang, D.; et al. Transcriptional Repression of Aerobic Glycolysis by OVOL2 in Breast Cancer. Adv. Sci. 2022, 9, e2200705. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, W.; Zuo, L.; Xu, M.; Wu, Y.; Huang, J.; Zhang, X.; Li, Y.; Wang, J.; Chen, J.; et al. The Fibrillin-1/VEGFR2/STAT2 signaling axis promotes chemoresistance via modulating glycolysis and angiogenesis in ovarian cancer organoids and cells. Cancer Commun. 2022, 42, 245–265. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, J.; Cui, X.; Zhan, Q.; Yi, K.; Wang, Q.; Xiao, M.; Tan, Y.; Hong, B.; Fang, C.; et al. TCA-phospholipid-glycolysis targeted triple therapy effectively suppresses ATP production and tumor growth in glioblastoma. Theranostics 2022, 12, 7032–7050. [Google Scholar] [CrossRef]
- Akman, H.O.; Kakhlon, O.; Coku, J.; Peverelli, L.; Rosenmann, H.; Rozenstein-Tsalkovich, L.; Turnbull, J.; Meiner, V.; Chama, L.; Lerer, I.; et al. Deep intronic GBE1 mutation in manifesting heterozygous patients with adult polyglucosan body disease. JAMA Neurol. 2015, 72, 441–445. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Oldfors, A. Polyglucosan storage myopathies. Mol. Aspects Med. 2015, 46, 85–100. [Google Scholar] [CrossRef]
- Bhanot, H.; Reddy, M.M.; Nonami, A.; Weisberg, E.L.; Bonal, D.; Kirschmeier, P.T.; Salgia, S.; Podar, K.; Galinsky, I.; Chowdary, T.K.; et al. Pathological glycogenesis through glycogen synthase 1 and suppression of excessive AMP kinase activity in myeloid leukemia cells. Leukemia 2015, 29, 1555–1563. [Google Scholar] [CrossRef]
- Liang, Y.; Lei, Y.; Liang, M.; Du, M.; Liu, Z.; Li, X.; Meng, X.; Zhou, B.; Gao, Y. GBE1 Is an Independent Prognostic Marker and Associated with CD163(+) Tumor-Associated Macrophage Infiltration in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 781344. [Google Scholar] [CrossRef]
- Lando, M.; Holden, M.; Bergersen, L.C.; Svendsrud, D.H.; Stokke, T.; Sundfør, K.; Glad, I.K.; Kristensen, G.B.; Lyng, H. Gene dosage, expression, and ontology analysis identifies driver genes in the carcinogenesis and chemoradioresistance of cervical cancer. PLoS Genet. 2009, 5, e1000719. [Google Scholar] [CrossRef]
- Yang, J.; Smith, D.K.; Ni, H.; Wu, K.; Huang, D.; Pan, S.; Sathe, A.A.; Tang, Y.; Liu, M.L.; Xing, C.; et al. SOX4-mediated repression of specific tRNAs inhibits proliferation of human glioblastoma cells. Proc. Natl. Acad. Sci. USA 2020, 117, 5782–5790. [Google Scholar] [CrossRef]
- Wang, K.; Pan, S.; Zhao, P.; Liu, L.; Chen, Z.; Bao, H.; Wang, H.; Zhang, Y.; Zhuge, Q.; Yang, J. PTBP1 knockdown promotes neural differentiation of glioblastoma cells through UNC5B receptor. Theranostics 2022, 12, 3847–3861. [Google Scholar] [CrossRef]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Ebus, M.E.; Koster, J.; van Sluis, P.; van Noesel, C.J.; Versteeg, R.; Caron, H.N. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008, 68, 2599–2609. [Google Scholar] [CrossRef]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011, 474, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, L.; Fan, Z.; Xue, W.; Shen, Z.; Yuan, Y.; Sun, X.; Wang, D.; Lian, J.; Wang, L.; et al. Hypoxia-induced GBE1 expression promotes tumor progression through metabolic reprogramming in lung adenocarcinoma. Signal Transduct. Target. Ther. 2020, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.T.; Chiang, I.T.; Kuo, Y.C.; Hsia, T.C.; Lin, C.C.; Liu, Y.C.; Chung, J.G. Amentoflavone Effectively Blocked the Tumor Progression of Glioblastoma via Suppression of ERK/NF-κ B Signaling Pathway. Am. J. Chin. Med. 2019, 47, 913–931. [Google Scholar] [CrossRef]
- Chen, R.; Li, J.; Zhou, X.; Liu, J.; Huang, G. Fructose-1,6-Bisphosphatase 1 Reduces (18)F FDG Uptake in Hepatocellular Carcinoma. Radiology 2017, 284, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Zhang, J.; Lam, E.K.; Shin, V.Y.; Cheng, A.S.; Yu, J.; Chan, F.K.; Sung, J.J.; Jin, H.C. Warburg effect revisited: An epigenetic link between glycolysis and gastric carcinogenesis. Oncogene 2010, 29, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, X.; Zheng, J.; Song, J.; Liu, Y.; Ruan, X.; Shen, S.; Shao, L.; Yang, C.; Wang, D.; et al. Lin28A promotes IRF6-regulated aerobic glycolysis in glioma cells by stabilizing SNHG14. Cell Death Dis. 2020, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, J.; Liu, X.; Li, M.; Wang, D.; Kang, Z.; Yu, J.; Chen, J.; Pan, H.; Wang, H.; et al. PTEN loss promotes Warburg effect and prostate cancer cell growth by inducing FBP1 degradation. Front. Oncol. 2022, 12, 911466. [Google Scholar] [CrossRef]
- Hirata, H.; Sugimachi, K.; Komatsu, H.; Ueda, M.; Masuda, T.; Uchi, R.; Sakimura, S.; Nambara, S.; Saito, T.; Shinden, Y.; et al. Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res. 2016, 76, 3265–3276. [Google Scholar] [CrossRef]
- Alderton, G.K. Tumorigenesis: FBP1 is suppressed in kidney tumours. Nat. Rev. Cancer 2014, 14, 575. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Lai, X.; Zhang, J.; Meng, Q.; Wang, P.; Qin, S.; Liu, W.; Wang, Y.; Yao, Z.; Wang, D.; et al. FBP1 knockdown decreases ovarian cancer formation and cisplatin resistance through EZH2-mediated H3K27me3. Biosci. Rep. 2022, 42, BSR20221002. [Google Scholar] [CrossRef]
- Damanakis, A.; Plum, P.S.; Gebauer, F.; Schröder, W.; Büttner, R.; Zander, T.; Bruns, C.J.; Quaas, A. Fructose-1,6-bisphosphatase 1 (FBP1) is an independent biomarker associated with a favorable prognosis in esophageal adenocarcinoma. J. Cancer Res. Clin. Oncol. 2022, 148, 2287–2293. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Kim, H.; Kang, H.; Jeon, J.; Jo, S.; Seong, K.M.; Lee, S.J.; Youn, H.; Youn, B. Decreased FBP1 expression rewires metabolic processes affecting aggressiveness of glioblastoma. Oncogene 2020, 39, 36–49. [Google Scholar] [CrossRef]
- Zhong, X.; He, X.; Wang, Y.; Hu, Z.; Huang, H.; Zhao, S.; Wei, P.; Li, D. Warburg effect in colorectal cancer: The emerging roles in tumor microenvironment and therapeutic implications. J. Hematol. Oncol. 2022, 15, 160. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- de Padua, M.C.; Delodi, G.; Vučetić, M.; Durivault, J.; Vial, V.; Bayer, P.; Noleto, G.R.; Mazure, N.M.; Ždralević, M.; Pouysségur, J. Disrupting glucose-6-phosphate isomerase fully suppresses the “Warburg effect” and activates OXPHOS with minimal impact on tumor growth except in hypoxia. Oncotarget 2017, 8, 87623–87637. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Ruiz, R.; Avéret, N.; Araiza, D.; Pinson, B.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in Crabtree effect induction? J. Biol. Chem. 2008, 283, 26948–26955. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Bao, H.; Long, J.; Zhao, P.; Hu, X.; Wang, H.; Zhang, Y.; Yang, J.; Zhuge, Q.; Xia, L. GBE1 Promotes Glioma Progression by Enhancing Aerobic Glycolysis through Inhibition of FBP1. Cancers 2023, 15, 1594. https://doi.org/10.3390/cancers15051594
Chen Z, Bao H, Long J, Zhao P, Hu X, Wang H, Zhang Y, Yang J, Zhuge Q, Xia L. GBE1 Promotes Glioma Progression by Enhancing Aerobic Glycolysis through Inhibition of FBP1. Cancers. 2023; 15(5):1594. https://doi.org/10.3390/cancers15051594
Chicago/Turabian StyleChen, Zhen, Han Bao, Jingfang Long, Peiqi Zhao, Xiaowei Hu, Hao Wang, Ying Zhang, Jianjing Yang, Qichuan Zhuge, and Lei Xia. 2023. "GBE1 Promotes Glioma Progression by Enhancing Aerobic Glycolysis through Inhibition of FBP1" Cancers 15, no. 5: 1594. https://doi.org/10.3390/cancers15051594
APA StyleChen, Z., Bao, H., Long, J., Zhao, P., Hu, X., Wang, H., Zhang, Y., Yang, J., Zhuge, Q., & Xia, L. (2023). GBE1 Promotes Glioma Progression by Enhancing Aerobic Glycolysis through Inhibition of FBP1. Cancers, 15(5), 1594. https://doi.org/10.3390/cancers15051594