1. Introduction
Colorectal cancer (CRC) is one of the most common types of cancer and causes of cancer deaths worldwide, with nearly 1,250,000 new CRC cases every year and a mortality rate as high as 50% of all cases [
1]. The molecular basis of CRC involves in most cases the activation of the Wnt–β-catenin signaling pathway by the mutation of intracellular components such as APC, AXIN, and CTNNB1/β-catenin genes or the epigenetic alteration of Wnt inhibitors such as DKK-1, SFRPs, and WIF, considered the initial steps in colorectal tumorigenesis [
2]. These changes result in the activation of c-myc and K-ras, which lead to adenoma, adenocarcinoma, and colon carcinoma [
2]. Myc activation induces the overexpression of multiple genes, including ornithine decarboyxlase (ODC), the limiting step in the synthesis of the polyamines putrescine, spermine, and spermidine. ODC activation can be efficiently prevented by the suicide inhibitor Difluoromethylornithine (DFMO, also named eflornithine) [
3]. Substantial evidence links ODC overexpression, excess polyamines synthesis, and CRC. For instance, ODC is overexpressed in most CRCs, and different tumor promoters induce ODC1 and tumor formation [
4,
5]. ODC polymorphisms have also been reported in CRC. In addition, targeting ODC and polyamines using cell lines, animal models, and even clinical trials may efficiently prevent CRC [
6,
7,
8]. For instance, DFMO inhibits colon carcinogenesis in ApcMin/+ mice with increased levels of ODC and polyamines in intestinal tissues and suppresses carcinogenesis in the small intestines of these mice [
3,
4]. Interestingly, the grade of intestinal polyps is polyamine-dependent, and the anti-carcinogenic effects can be rescued by putrescine. DFMO may work in humans as well. An ongoing trial is presently evaluating the effectiveness of the combination of DFMO and sulindac in preventing colon adenomas [
9]. However, the mechanisms by which polyamines promote carcinogenesis remain to be fully established.
Polyamines are physiological molecules that are produced transiently during epithelial restitution for epithelial tissue repair. This process involves the transient activation of cell migration and/or proliferation after wounding. Evidence suggests that this process could be mediated by Ca
2+ signaling induced by changes in the expression of TRPC1 channels as well as an increased ratio of STIM1 to STIM2 [
10]; molecular players involved in SOCE in epithelial cells, mediated by caveolin [
11]; and the small guanosine-5′-triphosphate-binding protein RhoA [
12]. Interestingly, SOCE and its molecular players have recently been found to be involved in carcinogenesis in CRC and other forms of cancer [
13,
14,
15]. For instance, we recently reported that intracellular Ca
2+ homeostasis is remodeled in CRC cells [
14,
16,
17]. Specifically, CRC cells display enhanced SOCE and decreased Ca
2+ store content relative to normal colonic cells, and these changes contribute to cancer hallmarks, such as increased cell proliferation, cell invasion, and resistance to apoptosis [
16,
17]. At the molecular level, enhanced SOCE was linked to the increased expression of TRPC1 and an increased ratio of STIM1 to STIM2 in CRC cells [
16,
17], thus mimicking changes previously reported to be induced by transient polyamine exposure during epithelial restitution [
10]. In addition, we also reported that store-operated currents (SOCs) are quite different in normal and colon cancer cells. Specifically, normal colonic cells display typical CRAC-like currents driven by Orai1 channels, which are very small, Ca
2+-selective, inward-rectifying currents. In contrast, CRC cells display larger, non-selective currents with both inward and outward components that are mediated by both Orai1 and TRPC1 channels [
16]. We recently conducted a transcriptomic analysis of 77 selected gene transcripts involved in intracellular Ca
2+ transport that provided the first insights into the transcriptional basis of this remodeling [
18]. In short, we found the differential expression of selected voltage-operated Ca
2+ channels and SOCE players, transient receptor potential (TRP) channels, Ca
2+ release channels, Ca
2+ pumps, Na
+/Ca
2+ exchanger isoforms, and genes involved in mitochondrial Ca
2+ transport [
18]. Therefore, the evidence suggests that intracellular Ca
2+ homeostasis is largely remodeled in CRC, and these changes could be mediated by excess polyamine synthesis linked to CRC.
To address this issue, we recently tested the effects of polyamine synthesis inhibition on Ca
2+ remodeling in CRC cells [
19]. In accordance with this hypothesis, we reported that CRC cells overexpressed ODC1 relative to normal cells. In addition, polyamine synthesis inhibition in CRC cells that were resistant to cell death reversed this phenotype and sensitized CRC cells to apoptosis. Importantly, polyamine synthesis inhibition promoted changes in intracellular Ca
2+ homeostasis consistent with phenotype reversal, including changes in store-operated currents and SOCE, Ca
2+ store content, and the expression of a few proteins involved in SOCE [
19]. However, whether polyamine synthesis inhibition reverses the whole calcium signature linked to carcinogenesis remains to be addressed.
Here, we combined calcium imaging and transcriptomic analysis using next-generation sequencing and microarray technology to determine the molecular basis of Ca2+ remodeling in CRC and the effects of polyamine synthesis inhibition on transcriptomic remodeling and changes in intracellular Ca2+ homeostasis. Our results indicated that polyamine synthesis inhibition partially reversed the remodeling of intracellular Ca2+ in CRC cells. In addition, polyamine synthesis inhibition induced expression changes in 25% of the whole transcriptome of CRC cells but had nearly negligible effects on normal cells. Finally, we found that the reversal of Ca2+ remodeling depended on the changes in a dozen genes, including SOCE modulators, several TRP channels, two Ca2+ pumps, and two channels involved in mitochondrial Ca2+ transport.
2. Materials and Methods
2.1. Materials
The HT29 cell line was obtained from LONZA (Basel, Switzerland). NCM460 cells were from INCELL Corporation (San Antonio, TX, USA). SW480 cells were a kind gift from Prof. Alberto Muñoz (CSIC, Madrid, Spain). Dulbecco’s modified Eagle’s medium (DMEM), penicillin, streptomycin, and fetal bovine serum (FBS) were sourced from Lonza (Basel, Switzerland). L-glutamine was from Gibco (Barcelona, Spain). Trypsin-EDTA was from LONZA (Verbiers, Belgium). Poly-L-Lysin was from Marlenfeld GmbH (Lauda-Könlgshofen, Germany). Six-well plates were from NUNC (Thermo Scientific, Waltham, MA, USA). Dishes 10 cm2 in diameter were from Corning (NY, USA). DFMO was from TOCRIS (Bristol, UK). Fura2/AM and qPCR primers are from Invitrogen (Eugene, OR, USA). Cyclopiazonic acid (CPA) was from Sigma-Aldrich (Steinheim, Alemania). Antibodies against MCU and β actin were from Sigma (Madrid, Spain). The RNA extraction kit was a GeneMATRIX Universal RNA Purification Kit from EURx (Gdansk, Poland). Clariom D human microarrays (Affymetrix) were supplied by CABIMER (Andalucía, Spain). RNA-seq (Illumina) was provided by Sistemas Genómicos S.L (Valencia, Spain). PolyamineRED was from Funakoshi Co., Ltd., Tokyo, Japan). All other reagents were obtained from Sigma and Merck.
2.2. Cell Models and Sample Preparation
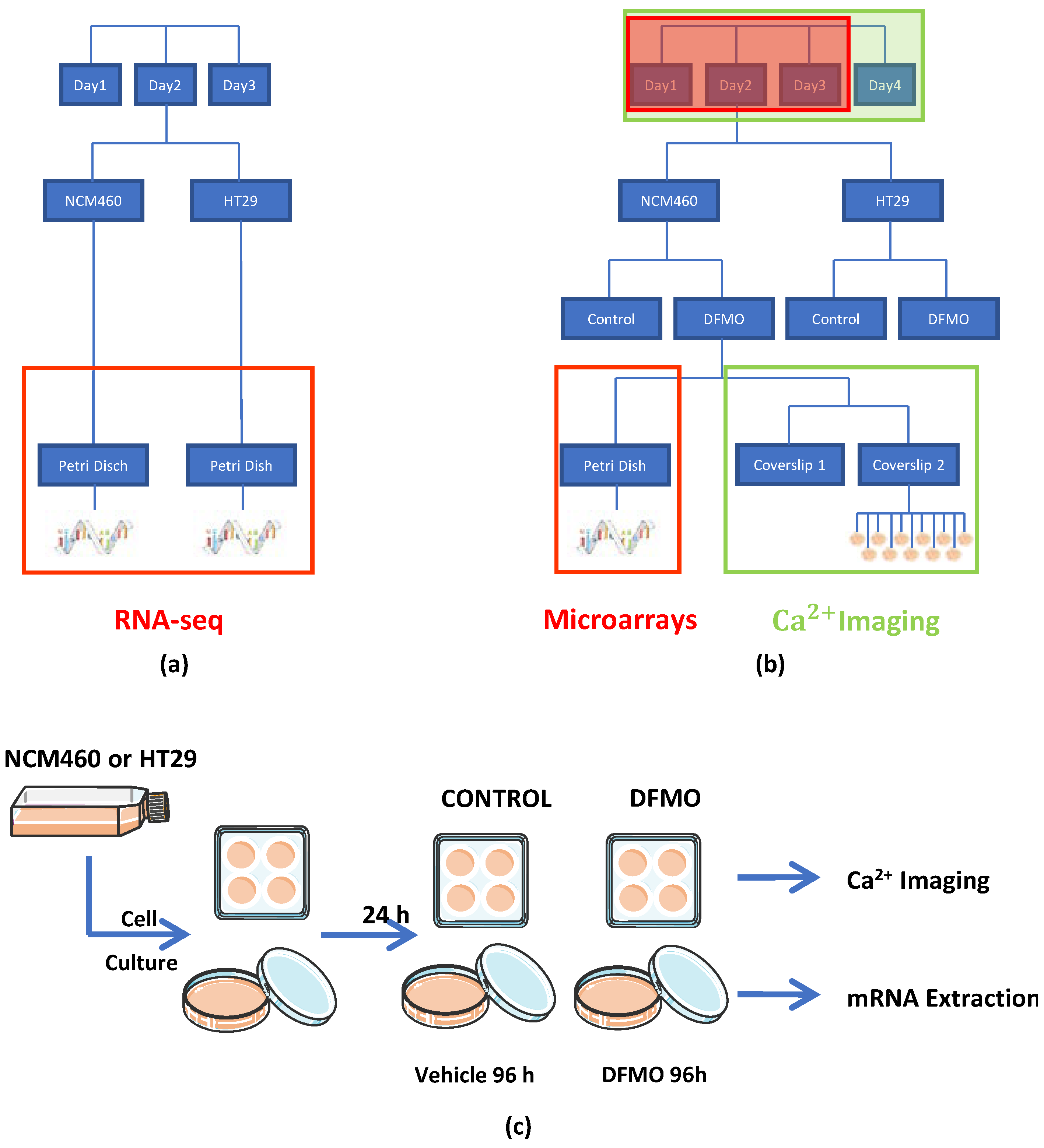
As human colon cancer models, we used the HT29 and SW480 colon cancer cell lines, and the NCM460 cell line was employed as the normal control; all of these have been widely used and validated in the colon cancer research field. Cells were cultured in a 25 cm2 flask with DMEM plus 1 g/L of glucose, 10% FBS, 1% penicillin/streptomycin, and 1% L-glutamine, which were placed into an incubator at 37 °C under a 10% CO2 atmosphere. All cells were used in passage 2. From each cell culture seeded in one flask, cells were detached with trypsin-EDTA and broken into two parts. One of these parts, which was used for the calcium imaging experiment, was seeded on four glass coverslips pretreated with poly-L-Lysin; each of them was then placed into a well in a 6-well plate with a cell density of 3000 cells per coverslip. The other part, which was used for the transcriptomic experiment (microarrays), was seeded on two dishes 10 cm2 in diameter with a cell density of 10.5 × 105 cells per dish. Notably, two flasks, one flask of HT29 and the other of NCM460 cells, were processed on the same day. Then, two flasks (one from HT29 cell cultures and the other from NCM460) were processed every day on four different days.
After an entire day at 37 °C under a 10% CO
2 atmosphere, of the eight coverslips and four dishes obtained from the two flasks (with one flask per cell line), two coverslips and one dish from each flask were treated with DFMO (DFMO 5 mM in DMEM 1 g/L glucose plus 10% dialyzed FBS), whereas the other two coverslips and dish from each flask were used to represent the non-treated conditions, i.e., treated without DFMO (DMEM 1 g/L glucose plus 10% dialyzed FBS). Then, the eight coverslips and four dishes were kept in an incubator at 37 °C under a 10% CO
2 atmosphere for 96 h. Next, the coverslips were used for calcium imaging experiments, and the dishes were used to extract and isolate their mRNA for transcriptomic analysis (see procedure schematic in
Figure 1a–c). Notice that a transcriptome analysis was previously carried out for both the NCM460 and HT29 cell lines without DFMO treatment using Illumina RNA-seq for comparison.
2.3. Intracellular Polyamine Levels
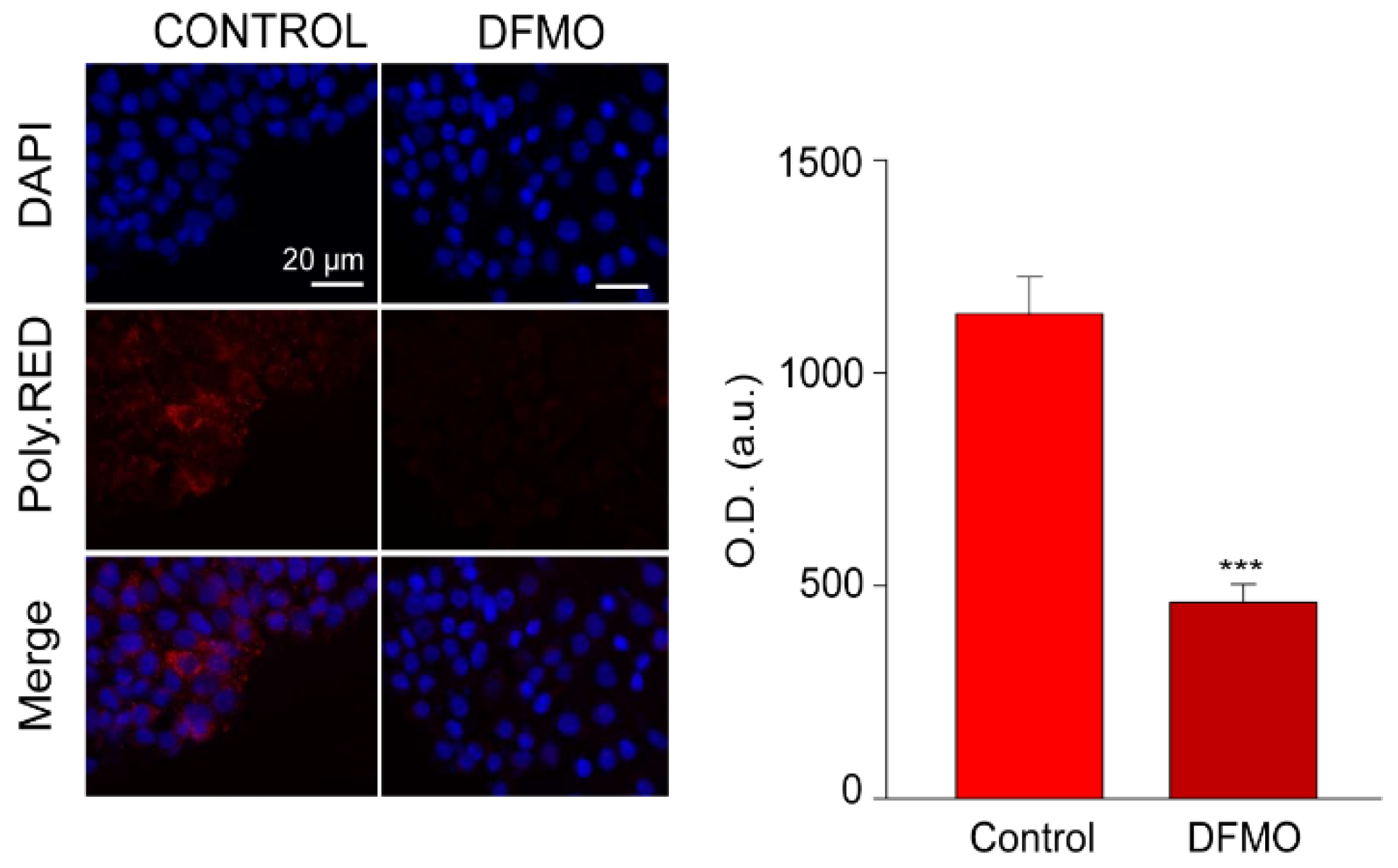
Intracellular polyamines were estimated using fluorescence imaging and PolyamineRED (Funakoshi Co., Ltd., Japan), an intracellular polyamine detection reagent. Treated and control cells were cultured with 30 µM of the reagent in free-serum media for 30 min according to manufacturer’s procedure. Then, cells were fixed, and nuclei were stained with DAPI. Fluorescence images were obtained using a Nikon Eclipse 90i fluorescence microscope and analyzed with ImageJ software.
Figure 2 shows that PolyamineRED fluorescence in cells treated with DFMO was largely reduced relative to untreated cells, in accordance with polyamine depletion. Similar results were obtained in SW480 and NCM460 cells.
2.4. Experimental Design
The effect of the inhibition of the polyamine synthesis pathway through DFMO on both HT29 and NCM460 cells was assessed at both the transcriptomic and functional levels (
Figure 1). On the one hand, the transcriptomic experiment was conducted using Clarion D human microarrays from Affymetrix and, on the other hand, the functional test was carried out using calcium imaging. Then, four experimental conditions were evaluated: NCM460 cells, both treated and non-treated with DFMO, and HT29 cells, again both treated and non-treated with DFMO. Furthermore, since we were interested in obtaining the transcriptomic expression profile through calcium imaging assays, we acquired from the same flask, at the same time, a replicate for every experimental condition (for the same cell line) and for both transcriptomic and calcium imaging assays, as shown in
Figure 1c. Specifically, on the same day, a replicate for both treated and non-treated experimental conditions was extracted from the same flask, and two replicates for calcium imaging for the experimental condition “g” were obtained and processed from one replicate for the same experimental conditions in the transcriptomic experiment. Furthermore, prior to the experiments described above, both non-treated HT29 and NCM460 cells were assessed at the transcriptomic level through Illumina RNA-seq technology. Regarding the factors that needed to be considered for later data analysis, on the one hand, in the calcium imaging experiment, there were two random factors (day and coverslip) and another two fixed factors (cell line and treatment). Then, we had to use linear mixed models to control the effects of random factors. On the other hand, concerning the transcriptomic experiment, there were two fixed factors (cell line and treatment) and just one random factor. We took into account this random factor, since it was necessary to control it. The data analysis for each type of experiment is explained more in detail in the corresponding paragraph.
2.5. Single-Cell Calcium Measurements
Fluorescence calcium measurements were conducted in colon cancer HT29 and SW480 cells as well as normal NCM460 cells, all treated and non-treated with DFMO. First of all, cells were washed with a standard external medium (SEM) containing (in mM) NaCl 145, KCl 5, CaCl
2 1, MgCl
2 1, glucose 10, and Hepes/NaOH 10 (pH 7.4). Then, cells were loaded with 4 μM of Fura2/AM, the acetoxymethyl ester form of the calcium-sensitive dye fura2, in SEM for 45 min at room temperature and in the dark. After loading, coverslips with attached cells were mounted in the perfusion chamber of a Zeiss Axiovert 100 TV inverted microscope and perfused with prewarmed (37 °C) SEM. Then, the cells in the chamber were excited alternately by light at 340 and 380 nm using a xenon lamp and a filter wheel, and the light emitted by the cells at 520 nm was filtered through a dichroic mirror and recorded every 5 s by a Hamamatsu ER camera (Hamamatsu Photonics France). Finally, the 340 nm/380 nm ratios between each pair of pixels were calculated, and all those belonging to the same region of interest (ROI), corresponding to a single cell, were averaged and interpreted as an intracellular [Ca
2+] measurement from that ROI, as detailed before [
20,
21]. For resting [Ca
2+] measurements, we used the median 340 nm/380 nm ratio of the first 30 s from each cell. After that, to obtain a measure of the Ca
2+ store content from each cell, cells were treated with the reversible sarcoplasmic and endoplasmic reticulum Ca
2+ ATPase (SERCA) inhibitor cyclopiazonic acid (CPA) at 10 μM in a calcium-free SEM. Accordingly, the Ca
2+ store content from each cell was measured as both the increment in and area under the curve (AUC) of the 340 nm/380 nm ratio for the signal corresponding to the rise in intracellular [Ca
2+] induced by CPA in Ca
2+-free medium. Finally, after store depletion, cells were perfused with SEM containing CPA and 1 mM Ca
2+ to induce SOCE. Then, the SOCE corresponding to every single cell was measured as both the rise in and the area under the curve (AUC) of the F340 nm/F380 nm ratio for the signal corresponding to the rise in intracellular [Ca
2+].
2.6. Transcriptomic Experiments
After RNA isolation (using a GeneMATRIX Universal RNA Purification Kit from EURx), microarrays were carried out in Centro Andaluz de Medicina Regenerativa (CABIMER, Seville, Spain), and RNA-seq assays were conducted by Sistemas Genómicos S.L (Valencia, Spain). Then, data analysis based on the microarrays was performed by our group using .CEL files, whereas data analysis for RNA-seq was performed using count-matrix, provided by Sistemas Genómicos S.L. (Valencia, Spain).
2.7. Western Blotting
HT29 cells were treated with vehicle or DFMO (5 mM, 96 h) and used for western blotting. Total protein was extracted from cells and used to quantify the expression of MCU. Whole-cell lysate was obtained using RIPA buffer (20 mM Tris–HCl, pH 7.8, 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, 1 mM EDTA, 0.05% SDS) supplemented with the Halt™ Protease and Phosphatase Inhibitor Cocktail (100×) from ThermoFisher Scientific (ref #1861281) (Waltham, MC, USA). Protein concentrations were determined by a Bradford protein assay. Proteins were fractionated by SDS-PAGE; electroblotted onto PVDF membranes; and probed with the antibodies at a dilution of 1/200, except for the anti-β-actin, which was used at a dilution of 1/5000. The antibody against MCU (SC-246071) had been previously characterized and was visualized by the addition of goat anti-rabbit IgG or rabbit anti-mouse IgG. Detection was performed using Pierce ECL Western Blotting substrate (Thermo Scientific) and a VersaDoc Imaging System (BioRad, Munich, Germany). The quantification of protein expression was carried out using Quantity One software (BioRad, Munich, Germany). The datasets were analyzed by adjusting a linear model to fit the data, the model assumptions were evaluated, the response variables were transformed with Box–Cox family transformations because the normality assumption was violated, and outlier detection was carried out by an analysis of the Cook’s distance and the studentized residual. After transformation, the model assumptions were fulfilled as follows: residual normality with the Shapiro–Wilks test (p value = 0.1961) and homoscedasticity with the Bartlett test (p value = 0.8771). The sample size was equal to 6:3, corresponding to HT29 cells without treatment and HT29 cells treated with DFMO, respectively; p < 0.01.
2.8. RNA Isolation, RT, and Real-Time PCR
Total RNA from HT29 and NCM460 cells was isolated with a GeneMatrix Universal RNA purification kit (Eurx®, Molecular biology products) following the manufacturer’s instructions. A confluent 60 mm Petri dish per condition was employed for each assay. The quality of the RNA was determined by optical density measurements at 260 and 280 nm and by electrophoresis on agarose gels. After DNAse I treatment with a RapidOut DNA removal kit (Thermo Scientific), 1000 ng of RNA was reverse-transcribed using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) at 37 °C for 120 min to obtain cDNA.
Quantitative PCR was carried out on equal amounts of cDNA in triplicate for each sample using a Kapa Sybr
® Fast qPCR kit (Kapa Biosystems, Wilmington, MA, USA) in a LightCyler
® 480 (Roche, Basel, Switzerland) thermocycler. The primers used are shown in
Table 1 and were designed using OligoPerfect Primer Designer (ThermoFisher Scientific). The cycling conditions were 5 min at 95 °C, 40 cycles of 95 °C for 15 s, and 60 °C for 20 s. These amplifications were used to compare the different samples and melting curves to determine the specificity of the PCR products.
The qPCR data were analyzed using the threshold cycle (Ct) relative quantification method (ΔΔCt). The gene expression levels were normalized by the housekeeping gene RP18S. The relative abundance of the genes was calculated as 2ΔCt, where ΔCt = Ctgene–Ct18S. Differences between cancer (HT29) and control (NCM460) cells were calculated from 2ΔΔCt, where ΔΔCt = ΔCt(HT29)–ΔCt(NCM460), using ΔCt(NCM460) as the calibrator. In this analysis, a value of 0 indicated no change, negative values indicated decreased expression, and positive values indicated increased expression relative to the calibrator. The statistical analysis method used was a Student’s t test with Bonferroni’s correction for two independent samples (ΔCt(HT29) and ΔCt(NCM460)) obtained with RP18S as endogenous control. * p < 0.05, ** p < 0.01, *** p < 0.001
2.9. Data Analysis: Calcium Imaging
For the calcium imaging experiments, we were interested in testing which experimental conditions were different from each other in terms of resting intracellular [Ca2+], Ca2+ store content, and SOCE. Thus, as response variables, at the single-cell level, we considered the median from the absolute value of the signal across the first 30 s for resting intracellular [Ca2+], and the corresponding increase in [Ca2+] levels and the area under the curve (AUC) for both Ca2+ store content and SOCE. We evaluated the effect of two factors, treatment and cellular line, on the response variable, each of them with just two levels (control and DFMO for the treatment factor, and NCM460 and HT29 for the cellular line factor), so there was a total of 4 experimental conditions: control-NCM460, DFMO-NCM460, control-HT29, and DFMO-HT29. Due to the experimental design, the dataset showed a hierarchical structure, since observations were clustered into, or organized at, different levels: the experiment was carried out on 4 different days; each day we performed 2 replicates corresponding to each of the 4 experimental conditions; and within each replicate, we measured several cells from the same experimental conditions. Furthermore, both replicates and day were random factors.
Since there were random factors and there could be a correlation between observations belonging to the same cluster (i.e., they were non-independent), it became necessary to use linear mixed models, also known as multilevel models. Regarding the most adequate random structure, we followed the top-down strategy suggested by Brady and West [
22]: several models with the same full structure (i.e., with all possible variables and interactions) but with a different random structure were estimated using restricted maximum likelihood estimation (REML) and compared using the conditional Akaike criterion information (cAIC), such that the model whose cAIC was lower was selected.
We fitted the linear mixed models for each variable response: both Ca
2+ rise and the AUC of the calcium signal for SOCE and Ca
2+ store content or the median of the first 30 s of the recording. Specifically, we evaluated the assumptions of the models, such as the normality and homoscedasticity, through both graphical and test methods, since the non-independence of the observations from the same cluster was controlled, precisely, by the linear mixed models. On the one hand, for the normality assumption of both the Pearson residuals and random effects, we used QQ plots, histograms, Pearson residuals against fitted values plots, Shapiro–Wilks tests, and Jarke Bera tests. On the other hand, we employed Pearson residuals versus fitted values plots and Breush–Pagan and Barlett tests for the homoscedasticity assumption. If any model assumption was violated, different approaches were followed depending on the assumption violated: normality violations were addressed by removing outliers and transforming the response with Box–Cox transformation; heteroscedasticity violations were solved using generalized linear mixed models and different variance–covariance matrix structures and weighting observations. Furthermore, re-sampling methods, such as parametric bootstrapping for mixed models, were performed to evaluate the models and to estimate both the coefficients and their confidence intervals. Notably, the results from the bootstrapping procedures agreed with those from the classic linear mixed models that slightly violated the assumptions. That is why, in the real world, some deviations from assumptions are expected, since, if these deviations are not too large, the inferences extracted from the analysis will be acceptable, which is known as the robustness of validity [
23]. For example, the non-normality of the residual distribution results in neither bias nor inefficiency from models; indeed, several authors claim that the violation of normality is not a serious problem as a consequence of the central limit theorem and can even be disregarded when the sample size is large (our experiment consisted of nearly 1000 observations), since the residual distribution will approximate to normality [
24,
25].
Regarding which experimental conditions were different from each other, all possible pairwise comparisons were made based on combinations between levels for the two explanatory variables considered: treatment and cellular line. At first, the expected value for each experimental condition was estimated from a linear mixed model. Secondly, the differences between each pair of expected values for each experimental condition were estimated; p-values were corrected using the Tukey method with a significance level of 0.05; and experimental conditions were grouped according to a pairwise comparison, i.e., if there were no differences between two experimental conditions, they belonged to the same group. In the graphs, this is shown by letters, which indicate the group to which each experimental condition belonged. The last step was based on graph theory and was carried out using the Bron–Kerbosch algorithm to find all maximal cliques.
All data analyses and data collection processes were performed in the R environment [
26]; the lmer [
27] and nlme [
28] packages were used for creating linear mixed models, lmer [
27] and boot [
29,
30] packages for performing bootstrap methods, and multcomp [
31] and igraph [
32] for pairwise comparison and experimental condition grouping.
2.10. Data Analysis: Transcriptomic Experiment Based on Microarrays
First at all, the .CEL files, one per sample, were read with R software (oligo and affy packages). Then, samples were subjected to quality control through different methods, such that those samples which did not pass the quality control were removed from the posterior analysis. At first, the distribution of each sample and the possible presence of any batch effect or the need to normalize was tested through box plots and kernel estimators from non-normalized data. Then, pattern matching was conducted. To achieve this, it was necessary to normalize (quantile) and transform (log
2) the datasets to make the samples comparable between them. Next, a principal component analysis (PCA) across all samples was conducted, and outlier detection was carried out by determining the robust Mahalanobis distance within each class (group of interest, e.g., observations from NCM460 cells treated with DFMO), where the input variables were the first principal components such that they explained at least 70% (PC70%) of the variability [
33]. Another pattern-matching method employed was hierarchical cluster analysis with the Ward method, PC70%, and the Euclidean distance. Finally, probe-level models were employed from which both the normalized unscaled standard error (NUSE) and the relative log expression (RLE) were extracted and evaluated [
34].
After quality control, the dataset was preprocessed, i.e., the dataset background was corrected, normalized, and finally summarized, all through the RMA method [
35]. Subsequently, data were filtered by employing a non-specific filter implemented by the gene filter R function, and we kept those genes (probes) whose interquartile ranges (across all samples) were larger than the median of all interquartile ranges. Furthermore, we removed all those genes whose ALIAS annotation was unknown. Nevertheless, since we were concerned about a near 80 calcium gene set, all of them were retained in the dataset after the filtering process. In this way, the dataset passed from 138,745 to 12,933 probes. Afterward, differential expression analysis was carried out through linear models, specifically, through linear models for the microarray data (limma) method [
36]. Regarding the variables in the model, the fixed factors were cellular line (levels: NCM460 and HT29) and treatment (levels: control and DFMO), whereas the random factor was day, i.e., the day on which the experimental units were processed, taking into account that each day a replicate corresponding to each experimental condition was processed. Then, the linear model selected was:
where Day is a random factor. Finally, to control I-type error rates, we had to employ the false discovery rate (FDR) [
37]. Differences were considered significant at FDR < 0.05.
4. Discussion
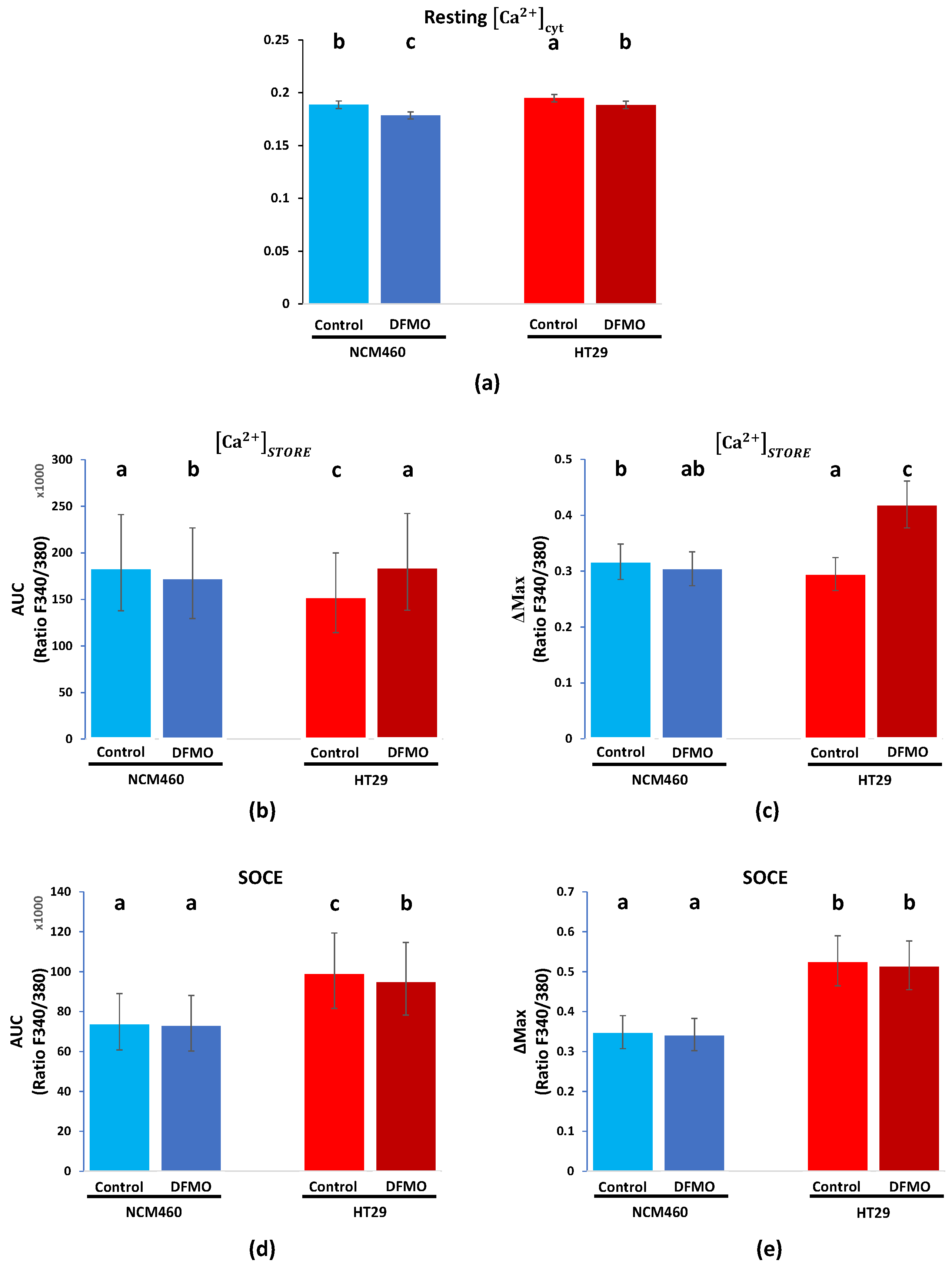
We investigated whether polyamine synthesis inhibition using DFMO as an ODC suicide inhibitor was able to reverse Ca
2+ remodeling in cancer cells. To this end, HT29 and NCM460 cells were used as well-established cell models representing CRC and normal colonic cells, respectively [
16]. CRC cells displayed significantly enhanced levels of resting intracellular [Ca
2+] and SOCE relative to normal cells. Likewise, CRC cells showed decreased Ca
2+ store content relative to normal cells. These findings are similar to previous results reported in [
16,
17]. We also showed here that SW480 colon cancer cells displayed enhanced SOCE and decreased Ca
2+ stores compared to NCM460 cells, thus resembling the same intracellular Ca
2+ remodeling as HT29 colon cancer cells. Although these changes were observed in cell lines representative of normal and tumor cells and should be confirmed in primary normal and tumor cells, changes in intracellular [Ca
2+] homeostasis, collectively referred to as Ca
2+ remodeling, could be part of the phenotypic changes associated with carcinogenesis; they may contribute to the hallmarks of cancer displayed by CRC cells, including enhanced cell proliferation and resistance to cell death [
18]. We found that the incubation of cells with DFMO, a treatment that abolishes polyamine biosynthesis and decreases levels of intracellular polyamines, also significantly decreased both resting Ca
2+ and SOCE, whereas it increased the Ca
2+ store content in CRC cells, thus reversing the hypothetical Ca
2+ remodeling associated with CRC. These results are similar to our recently reported results [
19]. In contrast, in normal cells, the effects of DFMO were less important. These data suggested that polyamine synthesis inhibition may reverse, at least partially, the hypothetical Ca
2+ remodeling associated with CRC, having minor effects on normal cells, where the ODC expression is low. Conversely, our results suggested that the excess polyamine synthesis observed in c-myc-related cancers may contribute to the hypothetical Ca
2+ remodeling associated with CRC.
The transcriptomic analysis of normal and CRC cells treated with DFMO showed that polyamine synthesis inhibition was able to significantly modify a large fraction (nearly 25%) of all gene transcripts studied in CRC cells. In striking contrast, in normal colonic cells, the same treatment affected less than 0.5% of the transcripts. Therefore, DFMO treatment was highly specific for cancer cells and had a nearly negligible effect on normal colonic cells. Again, we could explain these results by the fact that normal cells do not express ODC or express very low ODC levels unless this gene is induced during epithelial restitution, for instance. In this scenario, DFMO treatment probably induces only off-target effects or nearly negligible effects due to polyamine synthesis inhibition. These data also suggested that polyamines may induce rather dramatic effects on the transcriptome and the differential expression of nearly 25% of all the transcripts studied.
In this work, we focused on the transcriptional effects on genes directly involved in intracellular Ca
2+ homeostasis. We analyzed the effects of DFMO treatment (polyamine synthesis inhibition) on the transcription of the 10 voltage-gated Ca
2+ channels, the 23 genes known to be involved in SOCE, the 28 TRP channels, the 6 Ca
2+ release channels of the ER, the 12 Ca
2+ pumps and exchangers, and the 11 genes known to be involved in mitochondrial Ca
2+ transport.
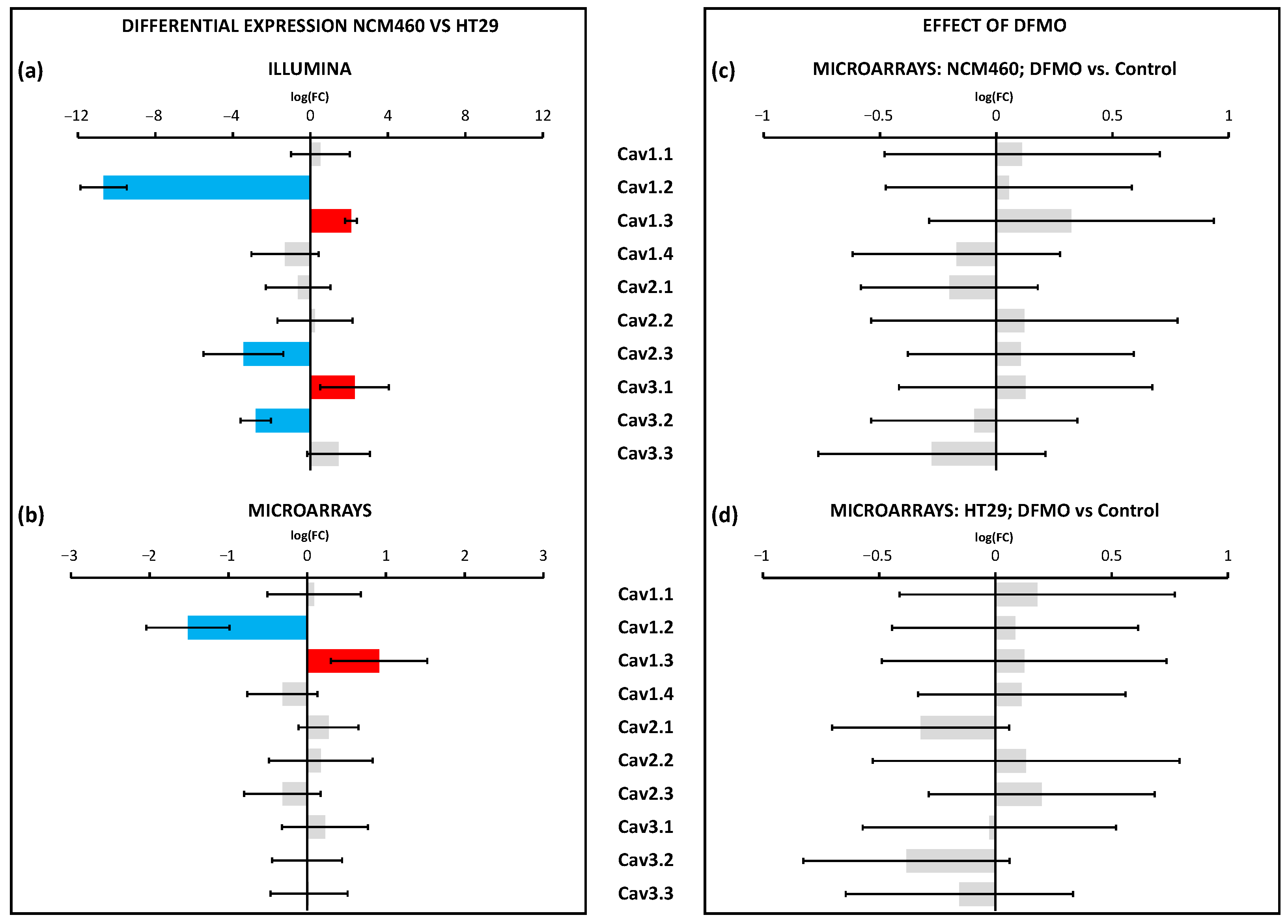
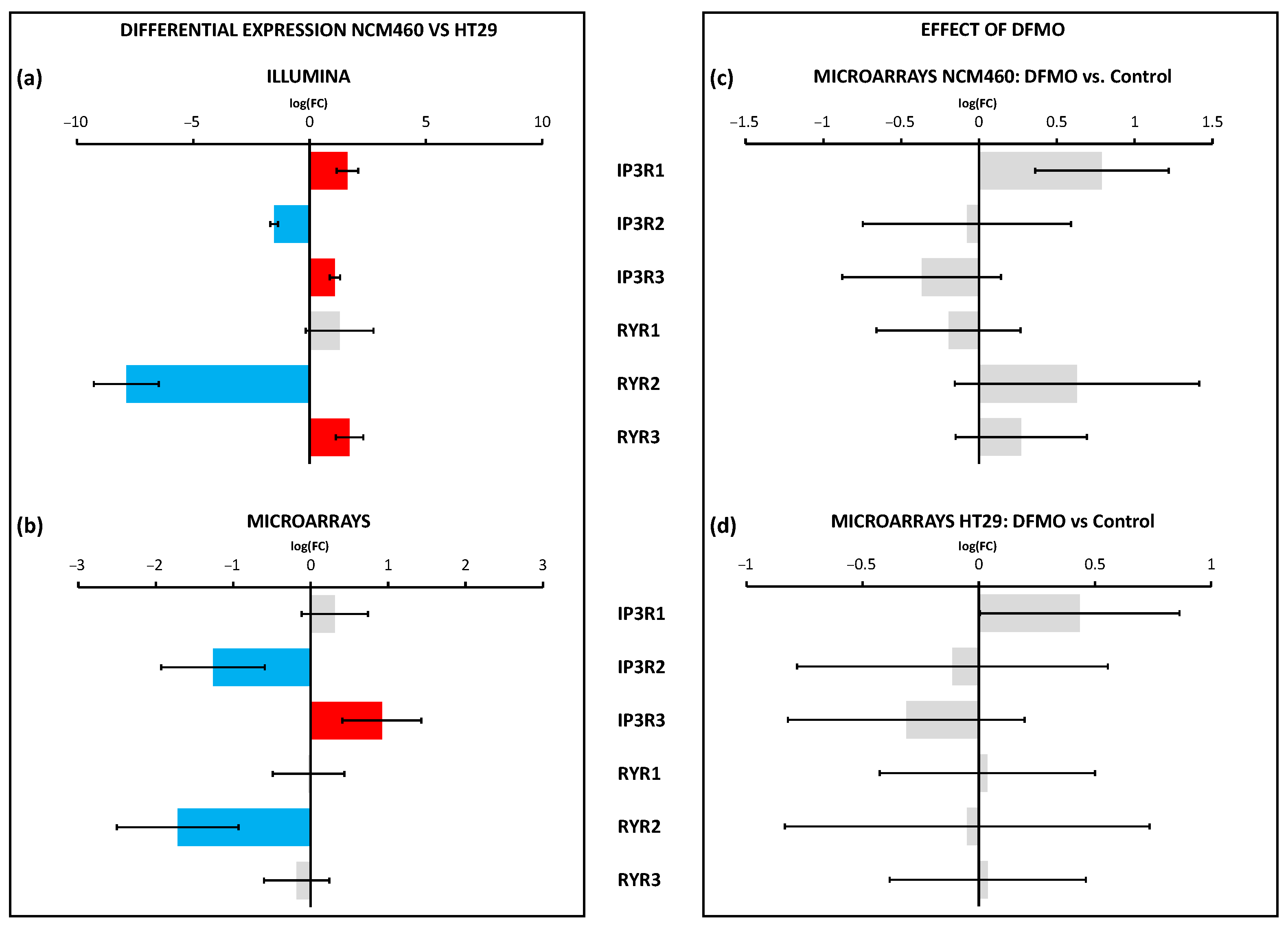
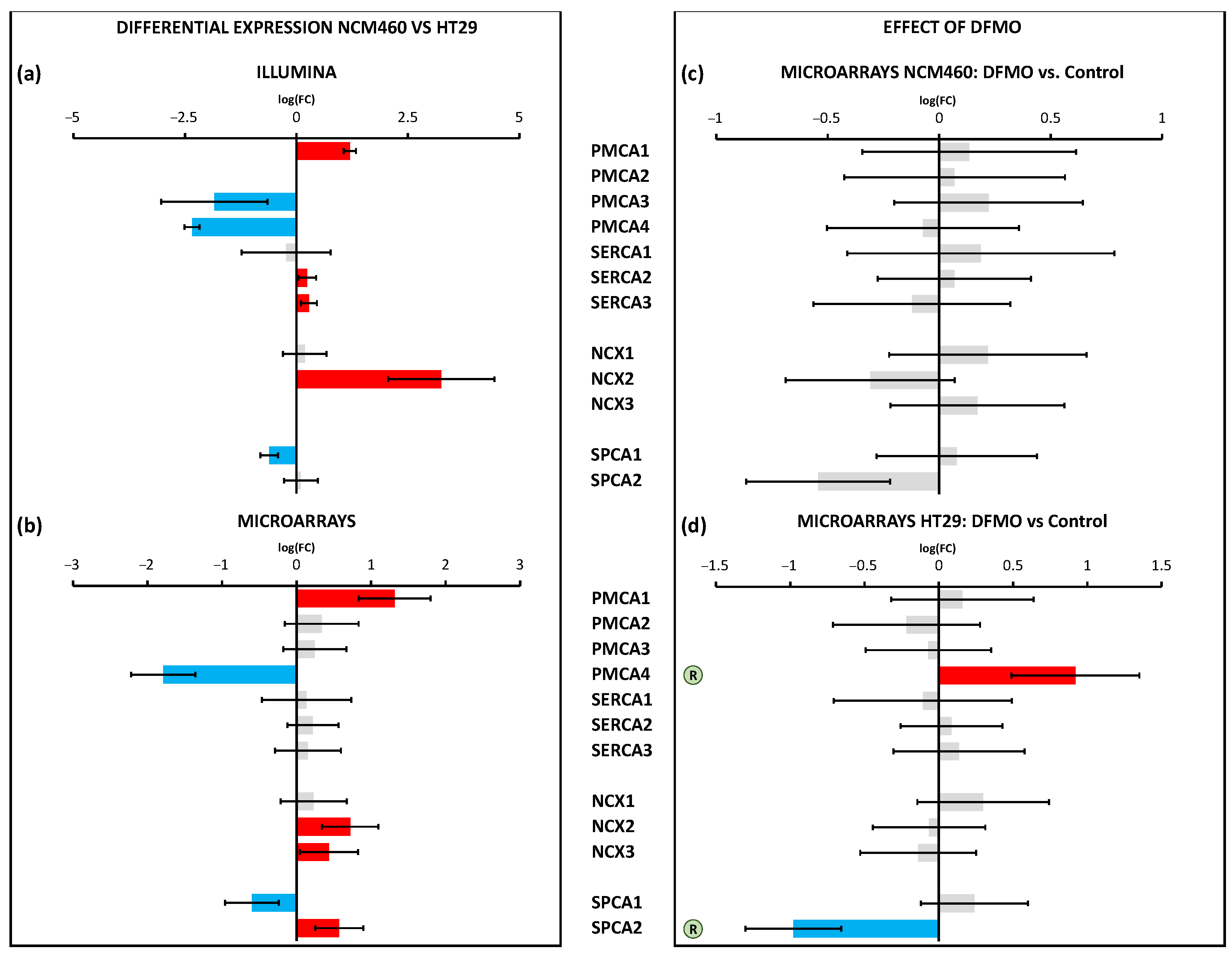
Figure 12 summarizes the changes in the Ca
2+ transport systems in cancer cells relative to normal cells and the effects of DFMO. We used two independent approaches for transcriptomic analysis, i.e., Illumina next-generation RNA sequencing (RNAseq) and microarrays. Illumina data include sequence data that are critical in cases where polymorphisms or mutations are the main target. Microarrays only provide data on the relative transcription level of selected sequences. We selected microarrays instead of RNAseq for our analysis of the effects of DFMO treatment for several reasons. First, because microarray sensitivity is lower than that of RNAseq, we expected that the differentially expressed genes would be those more significantly influenced by DFMO treatment, leading to less artefactual DEGs. Second, the computational and preprocessing methods of transcriptomic outcomes are simpler with microarrays, i.e., RNAseq outcomes warrant more sophisticated and more computationally powerful methods than microarray outcomes. Finally, transcriptomic microarrays are generally more affordable.
Regarding voltage-gated Ca
2+ channels, the data obtained using Illumina were quite similar but not identical to our previous results obtained using Ion Torrent technology [
18]. Microarray analysis showed that only two transcripts were differentially expressed in cancer cells, whereas changes in three other transcripts were only significant according to Illumina and not microarray. Specifically, we found that Cav1.2 was downregulated, whereas Cav1.3 was upregulated in CRC cells. Polyamine synthesis inhibition had no effect on the expression of any of the voltage-gated Ca
2+ channels, either in the CRC cells or in the normal cells. Accordingly, these data indicated that, although the previous results suggested the differential expression of certain voltage-gated Ca
2+ channels in CRC, they could not be attributed to excess polyamine synthesis in CRC.
In marked contrast, a number of molecular players involved in SOCE (ORAI1,2,3 and STIM1,2) and its modulatory proteins were differentially expressed in CRC cells relative to normal cells. Moreover, the transcription levels of SOCE molecular players were deeply influenced by polyamine synthesis inhibition. However, these changes were only observed in CRC and not in normal colon cells. Specifically, we found that DFMO treatment induced the increased expression of Stim1 and Stim2 transcripts in HT29 cells. These data are similar to our previous results using qRT-PCR [
19]. However, at the protein level, DFMO treatment decreased Stim1 expression in HT29 cells without changing Stim2 expression [
19]. In addition, DFMO treatment increased the expression of a number of SOCE modulators, including CRACR2A, ORMDL3, and several septins. In nearly all these cases, except for SEPTIN9, the change induced by DFMO reversed the change associated with cancer. These data suggested that a number of SOCE modulators are downregulated in CRC, leading to a loss in SOCE modulation that might contribute to the enhanced SOCE in cancer cells. However, in contrast to the microarray data, the qPCR data did not confirm that DFMO increased either SEPTIN6 or ORMDL3. Although qPCR is considered the gold-standard for testing differential expression, the larger number of primers used for the microarrays (20 instead of just one for qPCR) may explain this difference. Further research is required, therefore, to confirm the effects of the polyamine depletion of SOCE modulators.
SPCA2, a secretory pathway Ca
2+ ATPase that has recently been reported to interact and modulate Orai1 channels to activate them independently of store depletion [
38], was overexpressed in CRC cells. Interestingly, we found that SPCA2 was downregulated after DFMO treatment in HT29 colon cancer cells but not in normal cells. These data were also confirmed by qPCR analysis. Therefore, these data suggested that overexpressed SPCA2 in CRC cells might contribute to enhance resting [Ca
2+] and Orai1 activation independently of store depletion in CRC, a process that is limited by polyamine synthesis inhibition. In addition, it has recently been reported that the epigenetic modulation of SPCA2 reverses the epithelial-to-mesenchymal transition in breast cancer cells [
39]. It is tempting to speculate that overexpressed SPCLA2 in CRC cells due to polyamine excess may play a similar, previously unrecognized role in CRC as well.
TRP channels were also differentially expressed in CRC cells relative to normal cells. In general, both the Illumina and microarray technologies showed the decreased expression of a number of different TRP channels in CRC cells, particularly TRPML1,2,3 and TRPP2. However, CRC cells also showed the enhanced expression of a few TRP channels, including the TRPC5, TRPV6, and TRPP1 channels, that may have contributed to the appearance of non-selective store-operated currents characteristic of CRC cells [
16]. qPCR data confirmed these results for TRPV6 and TRPP1 but not for TRPC5. This finding for TRPC5 could have been due to the extremely low number of copies of TRPC5 transcripts. Again, polyamine synthesis inhibition had no effect on the transcription of TRP channels in normal cells. In contrast, in CRC cells, polyamine synthesis inhibition decreased the expression of the TRPC1 and 5, TRPV6, and TRPP1 channels, whereas it increased TRPP2 gene transcription. The pPCR data also confirmed the effects of DFMO on the expression of TRPV6 and TRPP1. We reported previously that DFMO treatment decreased the expression of TRPC1 in HT29 cells at the protein level [
19]. Further research is required to validate these additional changes in the TRP channel expression at the protein level.
The data on TRP channel remodeling induced by DFMO treatment strongly suggested that polyamines may influence cancer hallmarks acting on the transcriptional level of these TRP channels. For instance, TRPV6, the epithelial Ca
2+ channel modulated by the vitamin D receptor, has also been previously related to CRC and other forms of cancer [
40]. The cases of TRPP1 and TRPP2 are intriguing, as these are channel complexes involved in sensing shear stress in epithelia that are present in the ER. The results indicated an increased TRPP1/TRPP2 ratio in CRC cells, a feature that was also reversed by polyamine synthesis inhibition. Further research is required to understand the role of the TRPP1 and 2 channels in colon cancer and their relation to polyamine synthesis.
The Ca
2+ release channels of the ER, including IP
3 and ryanodine receptors, were differentially expressed in cancer cells and may also contribute to cancer hallmarks. However, none of these Ca
2+ release channels were modulated by polyamine synthesis inhibition, either in normal or CRC cells, at least at the transcriptional level, thus excluding the possibility that changes in the transcription of these channels are mediated by ODC overexpression. Interestingly, as mentioned above, the TRPP1 and TRPP2 channels can also be found at the ER membranes, where they could play a role as leak channels. Specifically, TRPP2 is a calcium-permeant transient receptor potential (TRP) cation channel expressed primarily on the ER membrane and primary cilia of all cell and tissue types [
41]. TRPP2 mutations lead to autosomal-dominant polycystic kidney disease. Recent data indicate that TRPP2 is involved in susceptibility to cell death induced by stress [
41]. Accordingly, changes in the TRPP1/TRPP2 ratio related to CRC and polyamine synthesis could be involved in Ca
2+ store content, Ca
2+ transfer from the ER to mitochondria, and sensitivity to stress. Again, further research is required to validate this possibility.
Regarding Ca
2+ extrusion systems such as Ca
2+ pumps and transporters, the data showed the differential expression of several transport systems, including the enhanced expression of the pumps PMCA1 and SPCA2 and the exchanger NCX2, along with the decreased transcription of the pumps PMCA4 and SPCA1. Again, polyamine synthesis inhibition had no effect on the transcription of any of these calcium extrusion systems in normal cells. However, in CRC cells, polyamine synthesis inhibition reversed the changes in the transcription of PMCA4 and SPCA2 linked to CRC. These results were confirmed by qPCR analysis. As stated above, SPCA2 may interact and modulate Orai1 to activate Orai1 independently of both Stim1 and Ca
2+ store depletion, thus leading to store-independent Ca
2+ entry. PMCA4, the plasma membrane Ca
2+ ATPase linked previously to CRC [
42], was downregulated in CRC, likely contributing to enhanced resting intracellular [Ca
2+]. This gene returned to normal levels after DFMO treatment, thus probably contributing to decreased resting Ca
2+ levels in treated cells.
Finally, mitochondrial Ca
2+ transport systems were differentially expressed in CRC cells. Most changes involved the decreased expression of the molecular players involved in the control of the mitochondrial Ca
2+ uniporter (MCU) and the Ca
2+ channel involved in mitochondrial Ca
2+ uptake. Once more, polyamine synthesis inhibition had no effect on the transcription levels of the mitochondrial Ca
2+ transport systems in normal cells. However, in cancer cells, polyamine synthesis inhibition increased the expression of both MCU and VDAC3. qPCR analysis also confirmed the increased expression of VDAC3 in DFMO-treated HT29 cells. We used western blotting to confirm that MCU expression was also enhanced at the protein level in HT29 cells treated with DFMO (
Supplementary Figure S3). Therefore, the modulation of the transcription of these two particularly relevant mitochondrial channels in CRC could be mediated by ODC and polyamines. Interestingly, these changes may contribute to explaining the reversal of the cancer hallmarks related to susceptibility to apoptosis. In other words, the enhanced resistance to apoptosis characteristic of CRC cells could be mediated, at least partially, by changes in the expression of MCU and VDAC3 due to ODC activation.
In summary, our results indicated that a few genes involved in Ca2+ transport appeared to be specifically modulated by polyamines in CRC cells and could be responsible for most changes in intracellular Ca2+ homeostasis in CRC cells. Thus, an excess of polyamines could induce the downregulation of SOCE modulatory genes (STIM1,2), the plasma membrane Ca2+ ATPase PMCA4 Ca2+ pump, and the mitochondrial Ca2+ channels MCU and VDAC3, contributing to dysregulated SOCE and enhanced resting Ca2+ and mitochondrial Ca2+ uptake in CRC. Conversely, polyamines could also induce the overexpression of TRPV6 channels, the Orai1 activator SPCA2, and the exchange of TRPP1 channels by TRPP2, thus likely contributing to enhanced Ca2+ entry and decreased Ca2+ store content in CRC. These findings may provide new insights into the role of polyamines in Ca2+ remodeling in cancer. Further research in animal models and/or tumor samples from patients is required to validate our findings.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}