
Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives

 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Landscape
2.1. Carcinogenesis and Drivers
2.2. Immunology of HCC
2.3. Molecular and Immune HCC Classes
3. First-Line Treatment
3.1. Tyrosine Kinase Inhibitors
3.2. Immune Checkpoint Inhibitors and Combination Regimens
3.3. Other Treatment Options under Investigation
4. Second-Line and Beyond
4.1. Tyrosine Kinase Inhibitors
4.2. Immune Checkpoint Inhibitors
4.3. Other Immune Checkpoint Inhibitors under Investigation
5. Sequencing Treatment
6. Biomarkers
6.1. Biomarkers for Immunotherapy
6.1.1. PD-L1 Expression
6.1.2. Tumor Mutational Burden and Microsatellite Instability
6.1.3. Other Possible Biomarkers: Circulating Tumor Cells, Gut Microbiota and WNT/β-Catenin Signaling
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D. Cancer statistics. ACS J. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- White, D.L.; Thrift, A.P.; Kanwal, F.; Davila, J.; El-Serag, H.B. Incidence of Hepatocellular Carcinoma in All 50 United States, From 2000 Through 2012. Gastroenterology 2017, 152, 812–820.e815. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Liang, K.Y.; Chang, A.S.; Chang, Y.C.; Lu, S.N.; Liaw, Y.F.; Chang, W.Y.; Sheen, M.C.; Lin, T.M. Effects of hepatitis B virus, alcohol drinking, cigarette smoking and familial tendency on hepatocellular carcinoma. Hepatology 1991, 13, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Di Bisceglie, A.M.; Bruix, J.; Kramer, B.S.; Lencioni, R.; Zhu, A.X.; Sherman, M.; Schwartz, M.; Lotze, M.; Talwalkar, J.; et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J. Natl. Cancer Inst. 2008, 100, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e1224. [Google Scholar] [CrossRef] [Green Version]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef]
- Llovet, J.M.; Pinyol, R.; Kelley, R.K.; El-Khoueiry, A.; Reeves, H.L.; Wang, X.W.; Gores, G.J.; Villanueva, A. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat. Cancer 2022, 3, 386–401. [Google Scholar] [CrossRef]
- Nault, J.C.; Calderaro, J.; di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e1323. [CrossRef] [Green Version]
- Giraud, J.; Chalopin, D.; Blanc, J.F.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.Z.; Liu, Z.W.; Zhang, J.Y.; Yang, Y.P.; Tien, P.; Wang, F.S. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangro, B.; Sarobe, P. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montironi, C.; Castet, F. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsunaga, S.; Ohno, I.; Hashimoto, Y.; Takahashi, H.; Watanabe, K.; Umemoto, K.; Okusaka, T. Systemic Chemotherapy for Advanced Hepatocellular Carcinoma: Past, Present, and Future. Diseases 2015, 3, 360–381. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Rimassa, L.; Cheng, A.L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef]
- Kudo, M. Durvalumab plus tremelimumab in unresectable hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2022, 11, 592–596. [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Macarulla, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. RATIONALE 301 study: Tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Future Oncol. 2019, 15, 1811–1822. [Google Scholar] [CrossRef] [Green Version]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracián, A.C.; Acosta-Rivera, M.; Choo, S.P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.; et al. CheckMate 040 cohort 5: A phase I/II study of nivolumab in patients with advanced hepatocellular carcinoma and Child-Pugh B cirrhosis. J. Hepatol. 2021, 75, 600–609. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Lyu, N.; Wang, X.; Li, J.B.; Lai, J.F.; Chen, Q.F.; Li, S.L. Arterial Chemotherapy of Oxaliplatin Plus Fluorouracil Versus Sorafenib in Advanced Hepatocellular Carcinoma: A Biomolecular Exploratory, Randomized, Phase III Trial (FOHAIC-1). J. Clin. Oncol. 2022, 40, 468–480. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.Y.; Ren, Z.; et al. LBA34 Primary results from the phase III LEAP-002 study: Lenvatinib plus pembrolizumab versus lenvatinib as first-line (1L) therapy for advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2022, 33, S1401. [Google Scholar] [CrossRef]
- Qin, S.; Chan, L.S.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. LBA35 Camrelizumab (C) plus rivoceranib (R) vs. sorafenib (S) as first-line therapy for unresectable hepatocellular carcinoma (uHCC): A randomized, phase III trial. Ann. Oncol. 2022, 33, S1401–S1402. [Google Scholar] [CrossRef]
- Peng, Z.; Fan, W.; Zhu, B. Lenvatinib Combined With Transarterial Chemoembolization as First-Line Treatment for Advanced Hepatocellular Carcinoma: A Phase III, Randomized Clinical Trial (LAUNCH). J. Clin. Oncol. 2023, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K. Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Kennedy, E.B. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Tai, D.; Chan, S.L.; Huang, Y.H.; Choo, S.P.; Hsu, C.; Cheung, T.T.; Lin, S.M.; Yong, W.P.; Lee, J.; et al. Systemic Treatment of Advanced Unresectable Hepatocellular Carcinoma after First-Line Therapy: Expert Recommendations from Hong Kong, Singapore, and Taiwan. Liver Cancer 2022, 11, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; Toni, E.N.D.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Breedis, C.; Young, G. The blood supply of neoplasms in the liver. Am. J. Pathol. 1954, 30, 969–977. [Google Scholar]
- Ueshima, K.; Ogasawara, S.; Ikeda, M.; Yasui, Y.; Terashima, T.; Yamashita, T.; Obi, S.; Sato, S.; Aikata, H.; Ohmura, T.; et al. Hepatic Arterial Infusion Chemotherapy versus Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Liver Cancer 2020, 9, 583–595. [Google Scholar] [CrossRef]
- Lyu, N.; Kong, Y.; Mu, L.; Lin, Y.; Li, J.; Liu, Y.; Zhang, Z.; Zheng, L.; Deng, H.; Li, S.; et al. Hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin vs. sorafenib for advanced hepatocellular carcinoma. J. Hepatol. 2018, 69, 60–69. [Google Scholar] [CrossRef]
- Lyu, N.; Lin, Y.; Kong, Y.; Zhang, Z.; Liu, L.; Zheng, L.; Mu, L.; Wang, J.; Li, X.; Pan, T.; et al. FOXAI: A phase II trial evaluating the efficacy and safety of hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin for advanced hepatocellular carcinoma. Gut 2018, 67, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Park, J.W.; Kim, J.H.; An, M.; Kong, S.Y.; Nam, B.H.; Choi, J.I.; Kim, H.B.; Lee, W.J.; Kim, C.M. Association between increment of serum VEGF level and prognosis after transcatheter arterial chemoembolization in hepatocellular carcinoma patients. Cancer Sci. 2008, 99, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, R.K.; Ryoo, B.Y.; Merle, P.; Park, J.W.; Bolondi, L.; Chan, S.L.; Lim, H.Y.; Baron, A.D.; Parnis, F.; Knox, J.; et al. Second-line cabozantinib after sorafenib treatment for advanced hepatocellular carcinoma: A subgroup analysis of the phase 3 CELESTIAL trial. ESMO Open 2020, 5, e000714. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.K.; Assenat, E.; Lim, H.Y.; Boige, V.; et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: Results from the randomized phase III BRISK-PS study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef]
- Qin, S.; Li, Q.; Gu, S.; Chen, X.; Lin, L.; Wang, Z.; Xu, A.; Chen, X.; Zhou, C.; Ren, Z.; et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELP): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 559–568. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chen, Z.; Fang, W. Pembrolizumab Versus Placebo as Second-Line Therapy in Patients From Asia With Advanced Hepatocellular Carcinoma: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2023, 41, 1434–1443. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef] [PubMed]
- He, A.R.; Weiss, G.J.; Falchook, G.; Yee, N.S.; Gil-Martin, M.; Shahda, S.; Moreno, V.; Brana, I.; Crittenden, M.; Formenti, S.; et al. Cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with advanced or metastatic hepatocellular carcinoma (HCC): Data from an expansion cohort (EC) in a phase I study. Ann. Oncol. 2018, 29, x26. [Google Scholar] [CrossRef]
- Shen, L.; Guo, J.; Zhang, Q.; Pan, H.; Yuan, Y.; Bai, Y.; Liu, T.; Zhou, Q.; Zhao, J.; Shu, Y.; et al. Tislelizumab in Chinese patients with advanced solid tumors: An open-label, non-comparative, phase 1/2 study. J. Immunother. Cancer 2020, 8, e000437. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Ramjeesingh, R.; Liu, D.; Tam, V.C.; Knox, J.J.; Card, P.B.; Meyers, B.M. Optimizing Survival and the Changing Landscape of Targeted Therapy for Intermediate and Advanced Hepatocellular Carcinoma: A Systematic Review. J. Natl. Cancer Inst. 2021, 113, 123–136. [Google Scholar] [CrossRef]
- Storandt, M.H.; Gile, J.J. Cabozantinib Following Immunotherapy in Patients with Advanced Hepatocellular Carcinoma. Cancers 2022, 14, 5173. [Google Scholar] [CrossRef]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef]
- Zhang, D.; Liao, X. Pan-TRK Immunohistochemistry and NTRK Gene Fusions in Primary Carcinomas of the Liver. Appl. Immunohistochem. Mol. Morphol. AIMM 2022, 30, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated With Response to Regorafenib in Patients With Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.X.; Finn, R.S.; Kang, Y.K.; Yen, C.J.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Motomura, K.; Ohno, I.; et al. Serum alpha-fetoprotein and clinical outcomes in patients with advanced hepatocellular carcinoma treated with ramucirumab. Br. J. Cancer 2021, 124, 1388–1397. [Google Scholar] [CrossRef]
- Girardi, D.M.; Pacífico, J.P.M.; Guedes de Amorim, F.P.L.; Dos Santos Fernandes, G.; Teixeira, M.C.; Pereira, A.A.L. Immunotherapy and Targeted Therapy for Hepatocellular Carcinoma: A Literature Review and Treatment Perspectives. Pharmaceuticals 2020, 14, 28. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.L.; Yau, T.; Furuse, J.; Park, J.W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Hou, M.-M.; Rau, K.-M.; Kang, Y.-K.; Lee, J.-S.; Pan, H.; Yuan, Y.; Yu, C.; Zhang, Y.; Ma, X.; Wu, X.; et al. 77 Association between programmed death-ligand 1 (PD-L1) expression and gene signatures of response or resistance to tislelizumab monotherapy in hepatocellular carcinoma (HCC). J. ImmunoTherapy Cancer 2020, 8, A47–A48. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Abou-Alfa Ghassan, K.; Lau, G.; Kudo, M.; Chan Stephen, L.; Kelley Robin, K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni Enrico, N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- He, Y.; Lu, M.; Che, J.; Chu, Q.; Zhang, P.; Chen, Y. Biomarkers and Future Perspectives for Hepatocellular Carcinoma Immunotherapy. Front. Oncol. 2021, 11, 716844. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Felden, J.; Garcia-Lezana, T.; Schulze, K.; Losic, B.; Villanueva, A. Liquid biopsy in the clinical management of hepatocellular carcinoma. Gut 2020, 69, 2025–2034. [Google Scholar] [CrossRef]
- Winograd, P.; Hou, S.; Court, C.M. Hepatocellular Carcinoma-Circulating Tumor Cells Expressing PD-L1 Are Prognostic and Potentially Associated With Response to Checkpoint Inhibitors. Hepatol. Commun. 2020, 4, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ying, J. Gut Microbiota and Immunotherapy. Front. Microbiol. 2022, 13, 945887. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, R.; Gao, J.; Cui, Q.; Wang, Q. Strategies to Improve the Antitumor Effect of Immunotherapy for Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 783236. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Galle, P.R. The Current Landscape of Clinical Trials for Systemic Treatment of HCC. Cancers 2021, 13, 1962. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Zhang, J.; Li, A.; Niu, M.; Yan, Y.; Jiao, Y.; Luo, S.; Zhou, P.; Wu, K. The construction, expression, and enhanced anti-tumor activity of YM101: A bispecific antibody simultaneously targeting TGF-β and PD-L1. J. Hematol. Oncol. 2021, 14, 27. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, J.J.; Coffey, H.; Corcoran, E.; Tsai, J.; Huang, C.L.; Ichikawa, K.; Prajapati, S.; Hao, M.H.; Bailey, S.; Wu, J.; et al. H3B-6527 Is a Potent and Selective Inhibitor of FGFR4 in FGF19-Driven Hepatocellular Carcinoma. Cancer Res. 2017, 77, 6999–7013. [Google Scholar] [CrossRef] [PubMed] [Green Version]


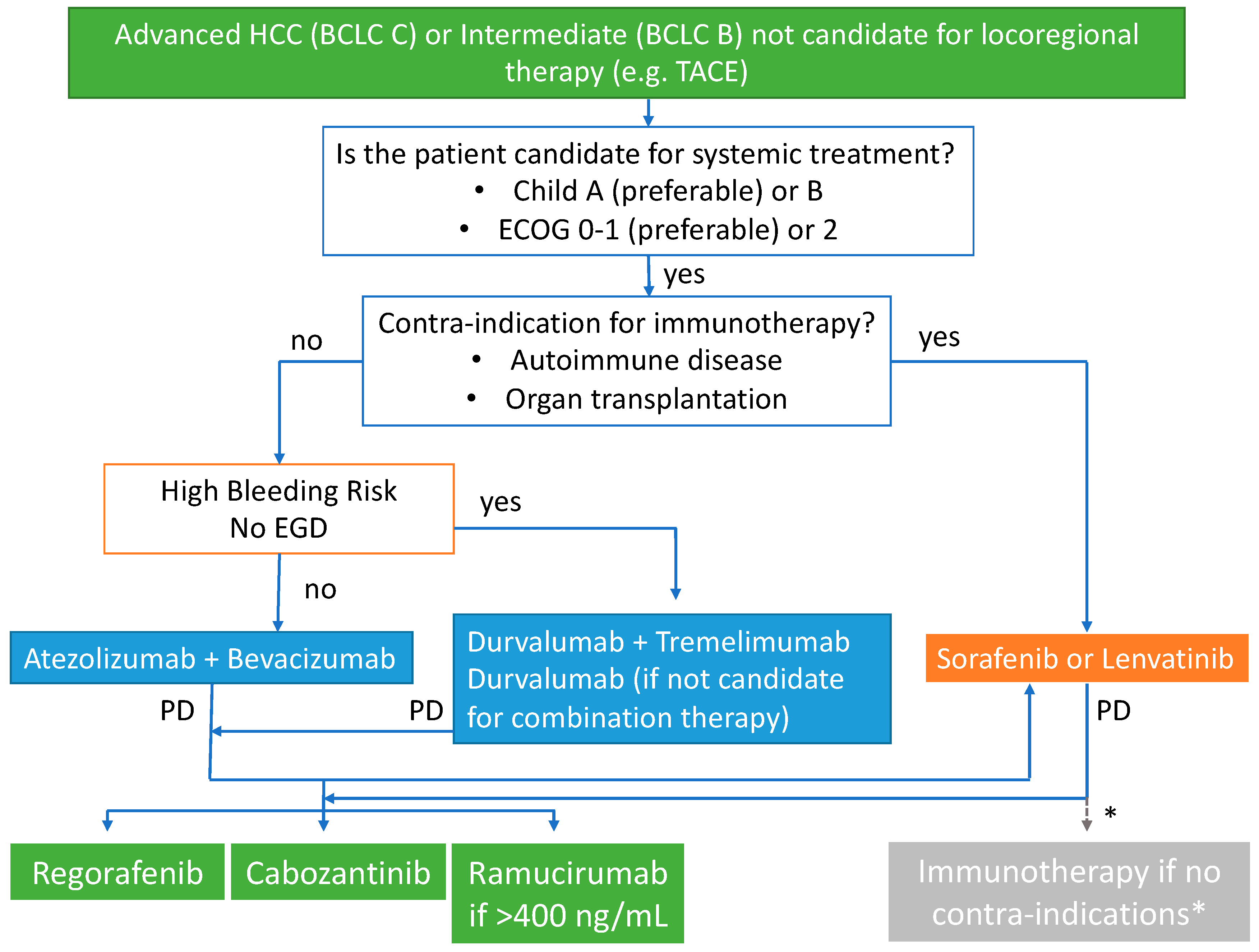{kind=link}
| Study (Year) | Phase | N | Population | Geographical Region | Drug | Median Overall Survival | Median Progression-Free Survival | Objective Response Rate |
|---|---|---|---|---|---|---|---|---|
| REFLECT trial (2018) [31] | III noninferiority | 954 | Unresectable HCC and no prior systemic therapy (99% Child-Turcotte-Pugh class A) | 29% (white); 69% (Asian); 2% (other) | Lenvatinib vs. sorafenib | 13.6 mo for lenvatinib vs. 12.3 mo for sorafenib (HR: 0.92, 95% CI: 0.79–1.06) | 7.4 mo for lenvatinib vs. 3.7 mo for sorafenib (HR: 0.66; p < 0.0001) | 24.1% for lenvatinib vs. 9.2% for sorafenib (p < 0.0001) |
| IMbrave 150 trial (2021) [32,33] | III | 336 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 40% (Asians, excluding Japan); 60% (rest of the world) | Atezolizumab-bevacizumab vs. sorafenib | 19.2 mo for atezolizumab-bevacizumab vs. 13.4 mo for sorafenib (HR: 0.66; p < 0.001) | 6.9 mo for atezolizumab-bevacizumab vs. 4.3 mo for sorafenib (HR: 0.65; p < 0.001) | 30% to atezolizumab-bevacizumab vs. 11% to sorafenib |
| COSMIC-321 trial (2022) [34] | III | 837 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 29.3% (Asians); 70.7% (Other) | Cabozantinib-atezolizumab vs. sorafenib | 15.4 mo for cabozantinib-atezolizumab vs. 15.5 mo for sorafenib (HR: 0.90, p = 0.44) | 6.8 mo for cabozantinib-atezolizumab vs. 4.2 mo for sorafenib (HR: 0.63, p = 0.0012) | 13% to cabozantinib-atezolizumab vs. 6% to sorafenib |
| HIMALAYA trial (2022) [35] | III | 1171 | Unresectable HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 40.9% (Asians, excluding Japan); 59.1% (rest of the world) | Durvalumab-tremelimumab or durvalumab vs. sorafenib | 16.43 mo for STRIDE vs. 13.77 for sorafenib (HR: 0.78; p = 0.0035) | 3.78 mo for STRIDE and 3.65 mo for durvalumab vs. 4.07 for sorafenib (HR: 0.90; p = 0.0035 and HR: 1.02, p = 0.0674) | 20.1% to STRIDE, 17% to durvalumab vs. 5.1 to sorafenib |
| RATIONALE-301 trial (2022) [36] | III | 674 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 63.1% (Asians, excluding Japan); 11.4 (Japan); 25.5% (rest of the world) | Tislelizumab vs. sorafenib | 15.9 mo for tislelizumab vs. 14.1 mo for sorafenib (HR: 0.8) | 2.2 mo for tislelizumab vs. 3.6 mo for sorafenib (HR: 1.1) | 14.3% to tislelizumab vs. 5.4% to sorafenib |
| CheckMate 459 (2019) [37] | III | 743 | Unresectable Child-Pugh A HCC naïve to systemic treatment | 40% (Asian); 60% (United States, Canada or Europe) | Nivolumab vs. sorafenib | 16.4 mo for nivolumab vs. 14.7 mo for sorafenib (HR: 0.85; p = 0.0752) | 3.7 mo for nivolumab vs. 3.8 mo for sorafenib | 15% for nivolumab and 7% to sorafenib |
| CheckMate-040: cohort B (2021) [38] | I/II | 49 | Unresectable or metastatic HCC, Child-Pugh liver function score B, with or without prior systemic therapy | 55% (Asian); 41% (white); 2% (black); 2% (other) | Nivolumab single arm | 9.8 mo for sorafenib naïve patients and 7.4 mo for previously treated patients | 3.4 mo for sorafenib naïve patients and 2.2 mo for previously treated patients | 12% |
| KEYNOTE-524 trial (2022) [39] | Ib | 104 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 51% (white); 28% (Asian); 2% (black); 5% (other); 14% (missing) | Lenvatinib-pembrolizumab single arm | 22 mo | 9.3 mo per mRECIST; 8.6 per RECIST v1.1 | 46% per mRECIST; 36% per RECIST v1.1 |
| FOHAIC-1 (2021) [40] | III | 262 | Locally advanced or unresectable HCC with or without extrahepatic oligometastasis, Child-Pugh liver function score ≤ 7 | HAIC (FOLFOX) vs. sorafenib | 13.9 mo for HAIC vs. 8.2 mo for sorafenib (HR: 0.408, p < 0.001) | 7.8 mo for HAIC vs. 4.3 mo for sorafenib (HR: 0.451, p < 0.001) | 31.5% to HAIC and 1.5% to sorafenib per RECIST; 35.4% to HAIC and 5.3% to sorafenib per mRECIST (p < 0.001) | |
| LEAP-002 (2021) [41] | III | 794 | Primary treatment-naive HCC, non-amenable to curative therapy, Child-Pugh A | 30.7% (Asian without Japan) vs. 69.3% (western regions and Japan) | lenvantinib plus pembrolizumab vs. lenvantinib | 21.2 mo for lenvatinib and pembrolizumab vs. 19 mo for lenvatinib (HR: 0.84; p = 0.0227) | 8.2 mo for lenvatinib and pembrolizumab vs. 8.1 mo for lenvatinib (HR: 0.834; p = 0.0466) | 26.1% for lenvatinib and pembrolizumab and 17.5% for lenvatinib per RECIST 1.1; 40.6% for lenvatinib and pembrolizumab and 34.1% for lenvatinib per mRECIST |
| Qin, et al. (2022) [42] | III | 543 | Unresectable or metastatic HCC primary treatment naive, BCLC satage B, Child-Pugh A | 82.7% (Asian) vs. 17.3% (non-Asian) | Camrelizumab + rivoceranib vs. sorafenib | 22.1 mo for canrelizumab + rivoceranib vs. 15.2 mo for sorafenib (HR: 0.62; 95% CI: 0.49–0.80) | 5.6 mo for canrelizumab + rivoceranib vs. 3.7 mo for sorafenib (HR: 0.52; 95% CI: 0.41–0.65) | 25.4% for camrelizumab and rivoceranib and 5.9% for sorafenib per RECIST 1.1; 33.1% for camrelizumab and rivoceranib and 10% for sorafenib per mRECIST |
| LAUNCH (2022) [43] | 338 | Primary treatment-naive or initial recurrent advanced HCC after surgery without adjuvant treatment, Child-Pugh class A | 100% (Asian—China) | LEN-TACE vs. lenvatinib | 17.8 mo forLEN-TACE vs. 11.5 mo for lenvatinib (HR: 0.33; p < 0.001) | 10.6 mo forLEN-TACE vs. 6.4 mo for lenvatinib (HR: 0.36; p < 0.001) | 45.9% to LEN_TACE and 20.8% to lenvatinib per RECIST; 54.1% to LEN-TACE and 25% to lenvatinib per mRECIST (p < 0.001) |
| Study (Year) | Phase | N | Population | Drug | Median Overall Survival | Median Progression-Free Survival | Objective Response Rate |
|---|---|---|---|---|---|---|---|
| RESORCE trial (2017) [56] | III | 573 | Advanced HCC that progressed after first-line treatment with sorafenib, Child-Pugh A | Regorafenib vs. placebo | 10.6 mo for regorafenib vs. 7.8 mo for placebo (HR: 0.63; p < 0.0001) | 3.1 mo for regorafenib vs. 1.5 mo for placebo (HR: 0.46; p < 0.0001) | 11% for regorafenib vs. 4% for placebo (p = 0.0047) |
| CELESTIAL trial (2018) [57] | III | 707 | Advanced and progressing HCC and not worse than Child-Pugh A | Cabozantinib vs. placebo | 10.2 mo for cabozantinib vs. 8.0 mo for placebo (HR: 0.76; p = 0.005) | 5.2 mo for cabozantinib vs. 1.9 mo for placebo (HR: 0.44; p < 0.001) | 4% for cabozantinib vs. less than 1% for placebo (p = 0.009) |
| REACH trial (2015) [58] | III | 565 | Advanced HCC following first-line therapy with sorafenib and Child-Pugh A | Ramucirumab vs. placebo | 9.2 mo for ramucirumab vs. 7.6 mo for placebo (HR: 0.87; p = 0.14). | 2.8 mo for ramucirumab vs. 2.1 mo for placebo (HR 0.63; p < 0.0001) | 7% for ramucirumab vs. < 1% for placebo (p < 0.0001) |
| REACH-2 trial (2019) [59] | III | 292 | Advanced HCC, Child-Pugh class A, and serum AFP ≥ 400 ng/mL in patients who had disease progression under first-line sorafenib | Ramucirumab vs. placebo | 8.5 mo for ramucirumab vs. 7.3 mo for placebo (HR: 0.71; p = 0.0199 | 2.8 mo for ramucirumab vs. 1.6 mo for placebo (HR: 0.452; p < 0. 0001) | 5% for ramucirumab vs. 1% for placebo (p = 0.1697) |
| BRISK-PS study [60] | III | 395 | Advanced HCC who progressed on/after or were intolerant to sorafenib | Brivanib vs. placebo | 9.4 mo for brivanib vs. 8.2 mo for placebo (HR: 0.89; p = 3307) | 4.2 mo for brivanib vs. 2.7 mo for placebo (HR, 0.56; p < 0.001) | 10% for brivanib vs. 2% for placebo (odds ratio, 5.72). |
| Qiu Li et al. (2020) [61] | III | 393 | Advanced HCC after failure of sorafenib and oxaliplatin-based chemotherapy and Child-Pugh liver function class A or B ≤ 7 points | Apatinib vs. placebo | 8.7 mo for apatinib vs. 6.8 mo for placebo (HR: 0.785; p = 0.0476) | 4.5 mo for apatinib vs. 1.9 mo for placebo (HR: 0.471; p < 0.0001) | 10.7% for ramucirumab vs. 1.5% for placebo |
| CheckMate 040 (2020) [62] | I/II | 148 | Advanced HCC patients who were treatment-naive or received sorafenib previously | Nivolumab and ipilimumab | Arm A: 22.8 mo Arm B: 12.5 mo ArmC: 12.7 mo | Arm A: 32% Arm B: 27% Arm C: 29% | |
| KeyNote 240 (2019) [63] | III | 413 | Child-Pugh A HCC patients, after progression or intolerance to sorafenib | Pembrolizumab vs. BSC | NR | 13.8 months | 18.3 versus 4.4%. |
| KeyNote 394 (2022) [64] | III | 453 | Second-line therapy for previously treated advanced HCC | Pembrolizumab vs. placebo | 14.6 vs. 13.0 months | 2.6 vs. 2.3 months | 12.7% vs. 1.3%, |
| Kelley et al.. (2020) [65] | I/II | 332 | Advanced HCC patients who progressed on, were intolerant to or refused sorafenib | Durvalumab + tremelimumab | T300 + D: 18.7 mo Durvalumab: 13.6 mo Tremelimumab: 15.1 mo T75 + D: 11.3 mo | T300 + D: 2.17 mo Durvalumab: 2.07 mo Tremelimumab: 2.69 mo T75 + D: 1.87 mo | T300 + D: 24.0% Durvalumab: 106% Tremelimumab: 7.2% T75 + D: 9.5% |
| Xu et al. (2019) [66] | I | 18 | HCC patients, Child-Pugh A, and previously treated with sorafenib | SHR-1210 + apatinib | NR | 5.8 mo | 50% |
| Qin et al. (2020) [67] | II | 217 | Advanced HCC, Child-Pugh A or B7 after sorafenib failure or intolerance to first-line systemic therapy | Camrelizumab | 13.8 mo | 2.1 mo | 14.7% |
| He et al. (2018) [68] | Ib | 26 | Advanced HCC, Child-Pugh A after failure or intolerance to first-line systemic therapy | Cemiplimab | 3.7 mo | 19.2% | |
| Shen et al. (2020) [69] | I/II | 300 | Advanced or metastatic solid tumors, including HCC, in patients who have progressed since their last standard antitumor treatment, had no available (or refused) standard treatment, or become intolerant to treatment | Tislelizumab | immature for HCC | 4.0 mo |
| Drugs | Phase | Setting | Endpoint | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| SBRT followed by sintilimab vs. SBRT | II/III | Palliative—1st line | Primary: 24-week PFS rate Secondary: PFS; OS; ORR; DCR; DOR | NCT04167293 |
| Lenvatinib + pembrolizumab +TACE vs. TACE | III | Palliative—1st line | Primary: PFS; OS Secondary: ORR; DCR; DOR; TTP; AEs | NCT04246177 |
| Arm A: TACE + durvalumab; Arm B: TACE + durvalumab + bevacizumab; Arm C: TACE | III | Palliative—1st line | Primary: PFS (Arm B vs. Arm C) Secondary: PFS (Arm A vs. Arm C); OS | NCT03778957 |
| Nivolumab + Ipilimumab vs. sorafenib or lenvatinib | III | Palliative—1st line | Primary: OS Secondary: ORR; DOR; TTSD | NCT04039607 |
| Finotonlimab (anti PD1) + SCT510 (bavacizumab) vs. Sorafenib | II/III | Palliative—1st line | Primary: OS Secondary: ORR; PFS | NCT04560894 |
| Toripalimab + Lenvatinib vs. Lenvatinib | III | Palliative—1st line | Primary: OS, PFS Secondary: ORR; DOR; TTP | NCT04523493 |
| Nofazinlimab (CS1003) + Lenvatinib vs. Lenvatinib | III | Palliative—1st line | Primary: OS, PFS | NCT04194775 |
| Atezolizumab + Lenvatinib or Sorafenib vs. Lenvatinib or Sorafenib | III | Palliative—2nd line | Primary: OS Secondary: ORR; PFS; DOR; TTP | NCT04770896 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girardi, D.M.; Sousa, L.P.; Miranda, T.A.; Haum, F.N.C.; Pereira, G.C.B.; Pereira, A.A.L. Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers 2023, 15, 1680. https://doi.org/10.3390/cancers15061680
Girardi DM, Sousa LP, Miranda TA, Haum FNC, Pereira GCB, Pereira AAL. Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers. 2023; 15(6):1680. https://doi.org/10.3390/cancers15061680
Chicago/Turabian StyleGirardi, Daniel M., Lara P. Sousa, Thiago A. Miranda, Fernanda N. C. Haum, Gabriel C. B. Pereira, and Allan A. L. Pereira. 2023. "Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives" Cancers 15, no. 6: 1680. https://doi.org/10.3390/cancers15061680
APA StyleGirardi, D. M., Sousa, L. P., Miranda, T. A., Haum, F. N. C., Pereira, G. C. B., & Pereira, A. A. L. (2023). Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers, 15(6), 1680. https://doi.org/10.3390/cancers15061680