Metabolic Alterations in Multiple Myeloma: From Oncogenesis to Proteasome Inhibitor Resistance


Abstract
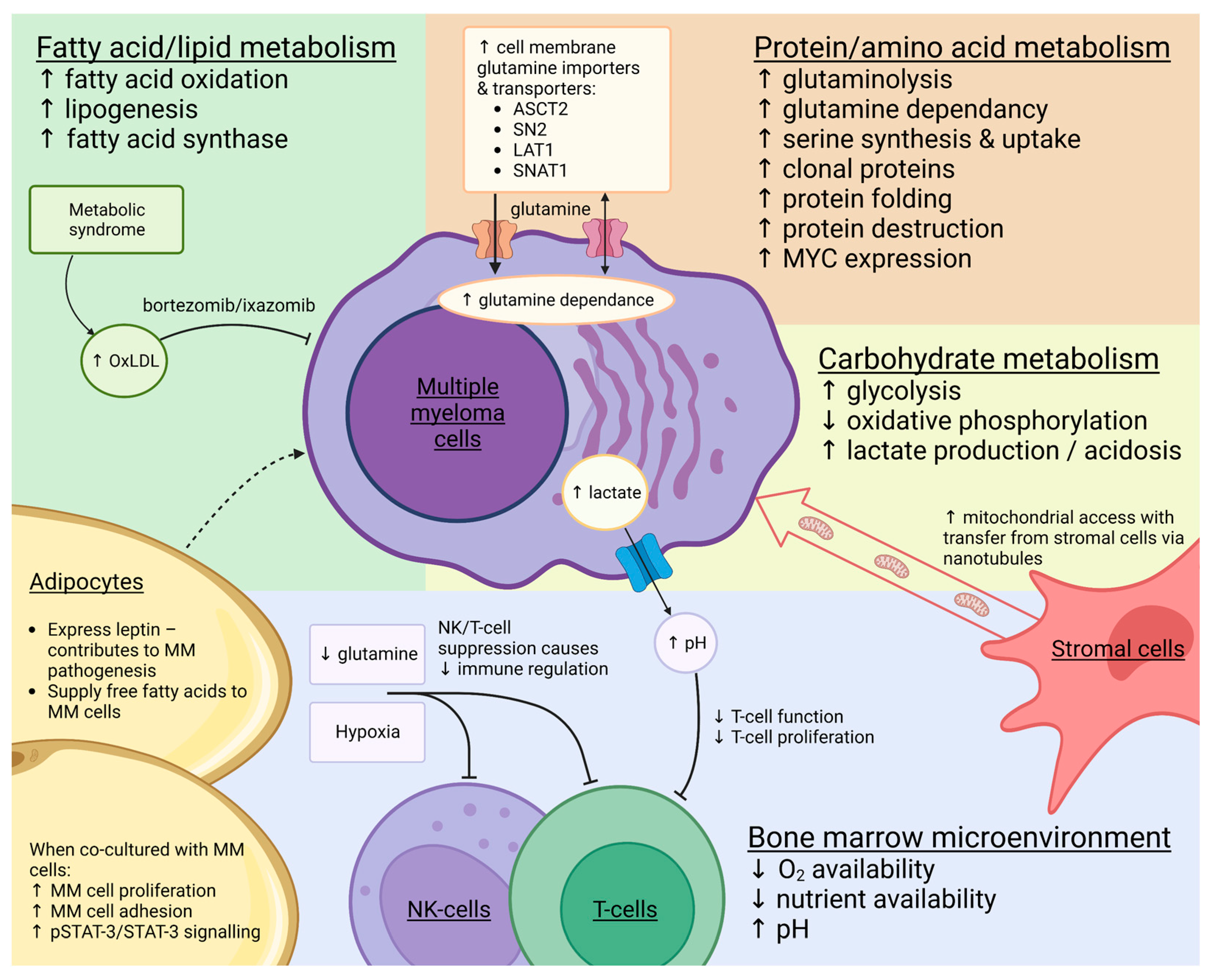
:Simple Summary
Abstract
1. Introduction
2. Proteasome Inhibition
3. Carbohydrate Metabolism
3.1. The Warburg Effect
3.2. Hypoxic Microenvironment
3.3. Glucose Transporters
3.4. Oxidative Phosphorylation
3.5. Lactate
3.6. Pentose Phosphate Pathway
4. Protein/Amino Acid Metabolism
4.1. Glutamine Metabolism
4.2. Serine Synthesis
4.3. Other Amino Acids
5. Fatty Acid/Lipid Metabolism
Lipid Metabolism and PI Resistance
6. Nicotinamide Metabolic Pathways
7. Gene Expression Signatures
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zweegman, S.; Palumbo, A.; Bringhen, S.; Sonneveld, P. Age and aging in blood disorders: Multiple myeloma. Haematologica 2014, 99, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Kuang, H.; Ke, J.; Pi, M.; Yang, D.H. Metabolic Reprogramming Induces Immune Cell Dysfunction in the Tumor Microenvironment of Multiple Myeloma. Front. Oncol. 2020, 10, 591342. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: Structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef]
- Wang, J.; Fang, Y.; Fan, R.A.; Kirk, C.J. Proteasome Inhibitors and Their Pharmacokinetics, Pharmacodynamics, and Metabolism. Int. J. Mol. Sci. 2021, 22, 11595. [Google Scholar] [CrossRef]
- Kühnel, A.; Blau, O.; Nogai, K.A.; Blau, I.W. The Warburg effect in Multiple Myeloma and its microenvironment. Med. Res. Arch. 2017, 5, 9. [Google Scholar]
- El Arfani, C.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E. Metabolic Features of Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1200. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, S.; Wada, N.; Kawano, Y.; Okuno, Y.; Kikukawa, Y.; Endo, S.; Nishimura, N.; Ueno, N.; Mitsuya, H.; Hata, H. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp. Hematol. Oncol. 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsi, E.; Perrone, G.; Terragna, C.; Martello, M.; Zamagni, E.; Tachetti, P.; Pantani, L.; Brioli, A.; Dico, A.F.; Zannetti, B.A.; et al. HIF-1α inhibition blocks the cross talk between multiple myeloma plasma cells and tumour microenvironment. Exp. Cell Res. 2014, 328, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Borsi, E.; Perrone, G.; Tarragna, C.; Martello, M.; Dico, A.F.; Solaini, G.; Baracca, A.; Sgarbi, G.; Pasquinelli, G.; Valente, S.; et al. Hypoxia inducible factor-1 alpha as a therapeutic target in multiple myeloma. Oncotarget 2014, 5, 1779–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storti, P.; Bolzoni, M.; Donofrio, G.; Aisoldi, I.; Guasco, D.; Toscani, D.; Martella, E.; Lazzaretti, M.; Mancini, C.; Agnelli, L.; et al. Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo by inhibiting angiogenesis and bone destruction. Leukaemia 2013, 27, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzieri, D.; Paul, B.; Kang, Y. Metabolic alterations and the potential for targeting metabolic pathways in the treatment of multiple myeloma. J. Cancer Metastasis Treat. 2019, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: Implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, G.; Holzhutter, H.G.; Bogyo, M.; Kairies, N.; Groll, M.; de Giuli, R.; Emch, S.; Groettrup, M. How an inhibitor of the HIV-I protease modulates proteasome activity. J. Biol. Chem. 1999, 274, 35734–35740. [Google Scholar] [CrossRef] [Green Version]
- Andre, P.; Groettrup, M.; Klenerman, P.; de Giuli, R.; Booth, B.L., Jr.; Cerundolo, V.; Bonneville, M.; Jotereau, F.; Zinkernagel, R.M.; Lotteau, V. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc. Natl. Acad. Sci. USA 1998, 95, 13120–13124. [Google Scholar] [CrossRef] [Green Version]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, N.; Palmer, J.; Synold, T.W.; Salehian, B.; Rosenweig, M.; Sanchez, J.F.; Hammond, S.N.; Adekola, K.; Tomarchio, V.; Chowdhury, A. Toxicities Associated with Metformin/Ritonavir Combination Treatment in Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2020, 20, e667–e672. [Google Scholar] [CrossRef]
- Ikeda, S.; Abe, F.; Matsuda, Y.; Kitadate, A.; Takahashi, N.; Tagawa, H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci. 2020, 111, 4088–4101. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Matsuda, T.; Seki, S.; Tomonari, Y.; Koizumi, S.; Nagatakiya, M.; Katsuyama, M.; Yamamoto, Y.; Tsurushima, K.; et al. Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-kappaB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells. Biomedicines 2021, 9, 33. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [Green Version]
- Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Wissam Saab, M.; Tavazzi, B.; et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules 2020, 10, 696. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in Adenosinergic Pathways and Metabolic Re-programming in Human Multiple Myeloma Cells: In-tandem Insights from Basic Science to Therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, R.; Matulis, S.M.; Wei, C.; Nooka, A.K.; Von Hollen, H.E.; Lonial, S.; Boise, L.H.; Shanmugam, M. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene 2016, 35, 3955–3964. [Google Scholar] [CrossRef] [Green Version]
- Gonsalves, W.I.; Ramakrishnan, V.; Hitosugi, T.; Ghosh, T.; Jevremovic, D.; Dutta, T.; Sakrikar, D.; Petterson, X.M.; Wellik, L.; Kumar, S.K.; et al. Glutamine-derived 2-hydroxyglutarate is associated with disease progression in plasma cell malignancies. JCI Insight 2018, 3, e94543. [Google Scholar] [CrossRef] [Green Version]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effenberger, M.; Bommert, K.S.; Kunz, V.; Kruk, J.; Leich, E.; Rudelius, M.; Bargou, R.; Bommert, K. Glutaminase inhibition in multiple myeloma induces apoptosis via MYC degradation. Oncotarget 2017, 8, 85858–85867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prelowska, M.K.; Mehlich, D.; Ugurlu, M.T.; Kedzierska, H.; Cwiek, A.; Kosnik, A.; Kaminska, K.; Marusiak, A.A.; Nowis, D. Inhibition of the L-glutamine transporter ASCT2 sensitizes plasma cell myeloma cells to proteasome inhibitors. Cancer Lett. 2021, 507, 13–25. [Google Scholar] [CrossRef]
- Maneix, L.; Sweeney, M.A.; Lee, S.; Iakova, P.; Moree, S.E.; Sahin, E.; Lulla, P.; Yellapragada, S.V.; Tsai, F.T.F.; Catic, A. The Mitochondrial Protease LonP1 Promotes Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers 2021, 13, 843. [Google Scholar] [CrossRef]
- Elsaadi, S.; Steiro, I.; Abdollahi, P.; Vandsemb, E.N.; Yang, R.; Slordahl, T.S.; Ro, T.B.; Menu, E.; Sponaas, A.M.; Borset, M. Targeting phosphoglycerate dehydrogenase in multiple myeloma. Exp. Hematol. Oncol. 2021, 10, 3. [Google Scholar] [CrossRef]
- Steiner, N.; Muller, U.; Hajek, R.; Sevcikova, S.; Borjan, B.; Johrer, K.; Gobel, G.; Pircher, A.; Gunsilius, E. The metabolomic plasma profile of myeloma patients is considerably different from healthy subjects and reveals potential new therapeutic targets. PLoS ONE 2018, 13, e0202045. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Kato, S.; Nesline, M.K.; Conroy, J.M.; DePietro, P.; Pabla, S.; Kurzrock, R. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat. Rev. 2022, 110, 102461. [Google Scholar] [CrossRef]
- Oudaert, I.; Satilmis, H.; Vlummens, P.; De Brouwer, W.; Maes, A.; Hose, D.; De Bruyne, E.; Ghesquiere, B.; Vanderkerken, K.; De Veirman, K.; et al. Pyrroline-5-Carboxylate Reductase 1: A novel target for sensitizing multiple myeloma cells to bortezomib by inhibition of PRAS40-mediated protein synthesis. J. Exp. Clin. Cancer Res. 2022, 41, 45. [Google Scholar] [CrossRef]
- Chen, J.; Zaal, E.A.; Berkers, C.R.; Ruijtenbeek, R.; Garssen, J.; Redegeld, F.A. Omega-3 Fatty Acids DHA and EPA Reduce Bortezomib Resistance in Multiple Myeloma Cells by Promoting Glutathione Degradation. Cells 2021, 10, 2287. [Google Scholar] [CrossRef]
- Bullwinkle, E.M.; Parker, M.D.; Bonan, N.F.; Falkenberg, L.G.; Davison, S.P.; DeCicco-Skinner, K.L. Adipocytes contribute to the growth and progression of multiple myeloma: Unraveling obesity related differences in adipocyte signaling. Cancer Lett. 2016, 380, 114–121. [Google Scholar] [CrossRef]
- Masarwi, M.; DeSchiffart, A.; Ham, J.; Reagan, M.R. Multiple Myeloma and Fatty Acid Metabolism. JBMR Plus 2019, 3, e10173. [Google Scholar] [CrossRef] [Green Version]
- Polusani, S.R.; Cortez, V.; Esparza, J.; Nguyen, H.N.; Fan, H.; Velagaleti, G.V.N.; Butler, M.J.; Kinney, M.C.; Oyajobi, B.O.; Habib, S.L.; et al. Oxidatively modified low-density lipoproteins are potential mediators of proteasome inhibitor resistance in multiple myeloma. Int. J. Cancer 2021, 148, 3032–3040. [Google Scholar] [CrossRef]
- Sanfilippo, K.M.; Keller, J.; Gage, B.F.; Luo, S.; Wang, T.F.; Moskowitz, G.; Gumbel, J.; Blue, B.; O’Brian, K.; Carson, K.R. Statins Are Associated With Reduced Mortality in Multiple Myeloma. J. Clin. Oncol. 2016, 34, 4008–4014. [Google Scholar] [CrossRef] [Green Version]
- Gronich, N.; Drucker, L.; Shapiro, H.; Radnay, J.; Yarkoni, S.; Lishner, M. Simvastatin induces death of multiple myeloma cell lines. J. Investig. Med. 2004, 52, 335–344. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Baumann, P.; Bumeder, I.; Meinhardt, G.; Straka, C.; Emmerich, B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur. J. Haematol. 2007, 79, 240–243. [Google Scholar] [CrossRef]
- Jurczyszyn, A.; Czepiel, J.; Gdula-Argasinska, J.; Czapkiewicz, A.; Biesiada, G.; Drozdz, M.; Perucki, W.; Castillo, J.J. Erythrocyte membrane fatty acids in multiple myeloma patients. Leuk. Res. 2014, 38, 1260–1265. [Google Scholar] [CrossRef]
- Hossen, M.A.; Nagata, Y.; Waki, M.; Ide, Y.; Takei, S.; Fukano, H.; Romero-Perez, G.A.; Tajima, S.; Yao, I.; Ohnishi, K.; et al. Decreased level of phosphatidylcholine (16:0/20:4) in multiple myeloma cells compared to plasma cells: A single-cell MALDI-IMS approach. Anal. Bioanal. Chem. 2015, 407, 5273–5280. [Google Scholar] [CrossRef] [Green Version]
- Lipchick, B.C.; Utley, A.; Han, Z.; Moparthy, S.; Yun, D.H.; Bianchi-Smiraglia, A.; Wolff, D.W.; Fink, E.; Liu, L.; Furdui, C.M.; et al. The fatty acid elongase ELOVL6 regulates bortezomib resistance in multiple myeloma. Blood Adv. 2021, 5, 1933–1946. [Google Scholar] [CrossRef]
- Xu, G.; Huang, S.; Peng, J.; Gao, X.; Li, M.; Yu, S.; Liu, Z.; Qie, P.; Wang, Y.; Yu, S.; et al. Targeting lipid metabolism in multiple myeloma cells: Rational development of a synergistic strategy with proteasome inhibitors. Br. J. Pharmacol. 2021, 178, 4741–4757. [Google Scholar] [CrossRef]
- Besse, L.; Besse, A.; Mendez-Lopez, M.; Vasickova, K.; Sedlackova, M.; Vanhara, P.; Kraus, M.; Bader, J.; Ferreira, R.B.; Castellano, R.K.; et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019, 104, e415–e419. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.K.; Li, M.; Tea, M.N.; Pitman, M.R.; Toubia, J.; Wang, P.P.; Anderson, D.; Creek, D.J.; Orlowski, R.Z.; Gliddon, B.L.; et al. Resensitising proteasome inhibitor-resistant myeloma with sphingosine kinase 2 inhibition. Neoplasia 2022, 24, 1–11. [Google Scholar] [CrossRef]
- Zaal, E.A.; de Grooth, H.J.; Oudaert, I.; Langerhorst, P.; Levantovsky, S.; van Slobbe, G.J.J.; Jansen, J.W.A.; Menu, E.; Wu, W.; Berkers, C.R. Targeting coenzyme Q10 synthesis overcomes bortezomib resistance in multiple myeloma. Mol. Omics 2022, 18, 19–30. [Google Scholar] [CrossRef]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.Y.; Wang, Y.; Wang, W.D.; Wei, X.L.; Gale, R.P.; Li, J.Y.; Zhang, Q.Y.; Shu, L.L.; Li, L.; Li, J.; et al. A prognostic survival model based on metabolism-related gene expression in plasma cell myeloma. Leukemia 2021, 35, 3212–3222. [Google Scholar] [CrossRef]
- Wang, R.; Bu, W.; Yang, Y. Identification of Metabolism-Related Genes Influencing Prognosis of Multiple Myeloma Patients. J. Healthc. Eng. 2021, 2021, 6574491. [Google Scholar] [CrossRef]
- Kakoo, A.; Al-Attar, M.; Rasheed, T. Exonic variants in multiple myeloma patients associated with relapsed/ refractory and response to bortezomib regimens. Saudi J. Biol. Sci. 2022, 29, 610–614. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Zada, M.; Wang, S.Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Metabolic Pathway | Target | Compound | Reference |
|---|---|---|---|
| Glycolysis | PDK1 | dichloroacetate | [12] |
| Glucose transport | GLUT4 | Ritonavir | [19,21] |
| Hexokinase 2 | 3-bromopyruvate/ | ||
| 2-deoxyglucose | [8,23] | ||
| SGK1 | GSK650394 | [24] | |
| Hypoxia | HIF1A | EZN-2968 | [13,14] |
| Lactate transport | MCT1 | α-cyano-4-hydroxycinnamic | [17] |
| Glutamine metabolism | glutaminase | Compound-968 | [33] |
| glutamine | L-asparaginase | [8] | |
| ASCT2 | GPNA/CB-839 | [34,35,36] | |
| Serine synthesis | PGDH | NCT-503 | [38] |
| Proline metabolism | PYCR1 | paragline | [41] |
| Glutathione metabolism | Docosahexaenoic acid/ Eicosapentaenoic acid | [42] | |
| Lipid metabolism | LDL | simvastatin | [47,48] |
| orlistat/etomoxir | [17] | ||
| Lovastatin/fenofibrate | [52] | ||
| Sphingomyelin synthase | D609 | [53] | |
| K145 | [54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weir, P.; Donaldson, D.; McMullin, M.F.; Crawford, L. Metabolic Alterations in Multiple Myeloma: From Oncogenesis to Proteasome Inhibitor Resistance. Cancers 2023, 15, 1682. https://doi.org/10.3390/cancers15061682
Weir P, Donaldson D, McMullin MF, Crawford L. Metabolic Alterations in Multiple Myeloma: From Oncogenesis to Proteasome Inhibitor Resistance. Cancers. 2023; 15(6):1682. https://doi.org/10.3390/cancers15061682
Chicago/Turabian StyleWeir, Philip, David Donaldson, Mary Frances McMullin, and Lisa Crawford. 2023. "Metabolic Alterations in Multiple Myeloma: From Oncogenesis to Proteasome Inhibitor Resistance" Cancers 15, no. 6: 1682. https://doi.org/10.3390/cancers15061682