
Fusobacterium & Co. at the Stem of Cancer: Microbe–Cancer Stem Cell Interactions in Colorectal Carcinogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction: The Physiology behind Cancer—Embryonic Development and Tissue Repair
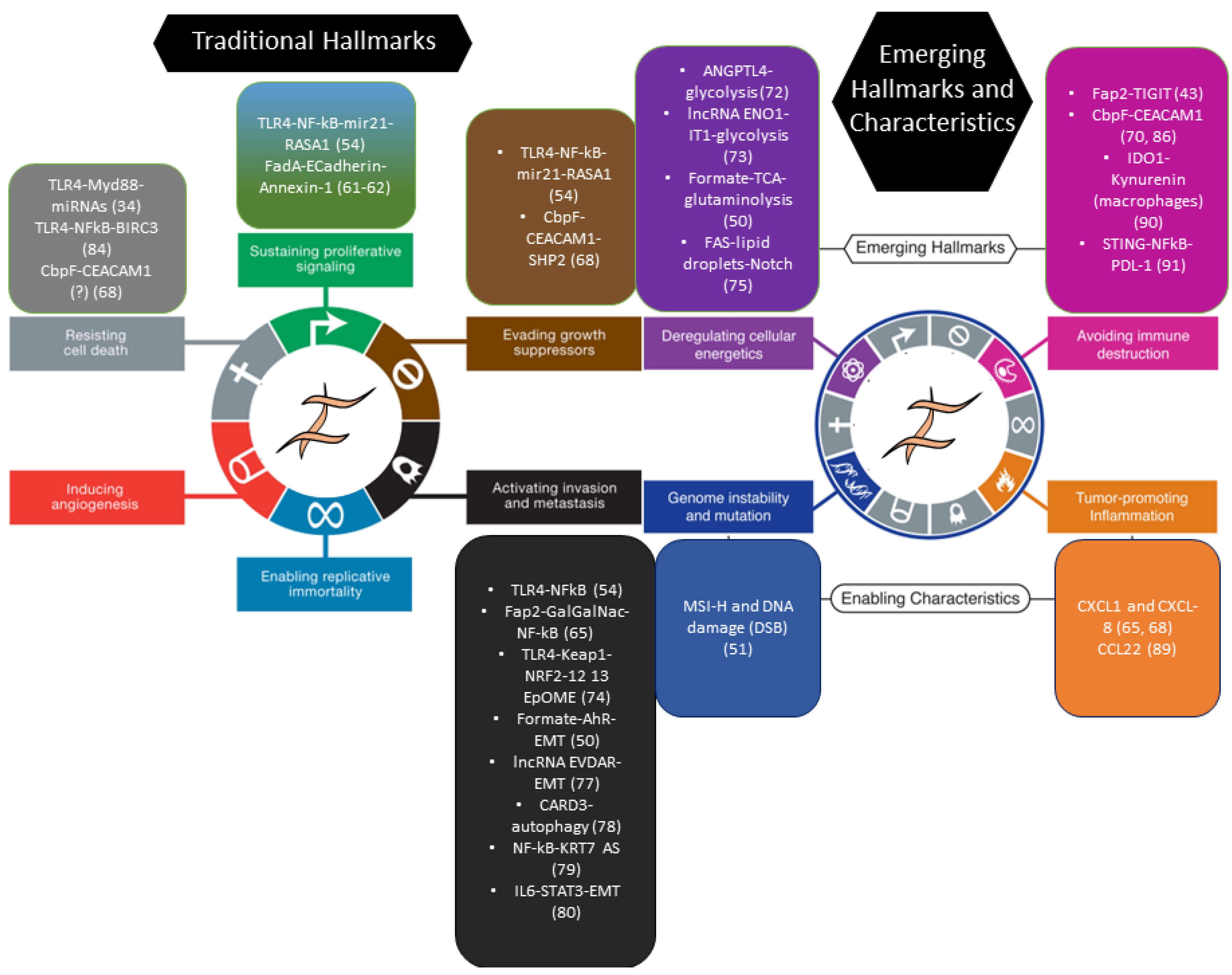
2. Bacterial Carcinogenesis in Colorectal Cancer
3. Fusobacterium nucleatum and Colorectal Cancer: Inflammation Meets Stemness
3.1. F. nucleatum and Human CRC
3.2. Mechanistic Studies In Vivo and In Vitro
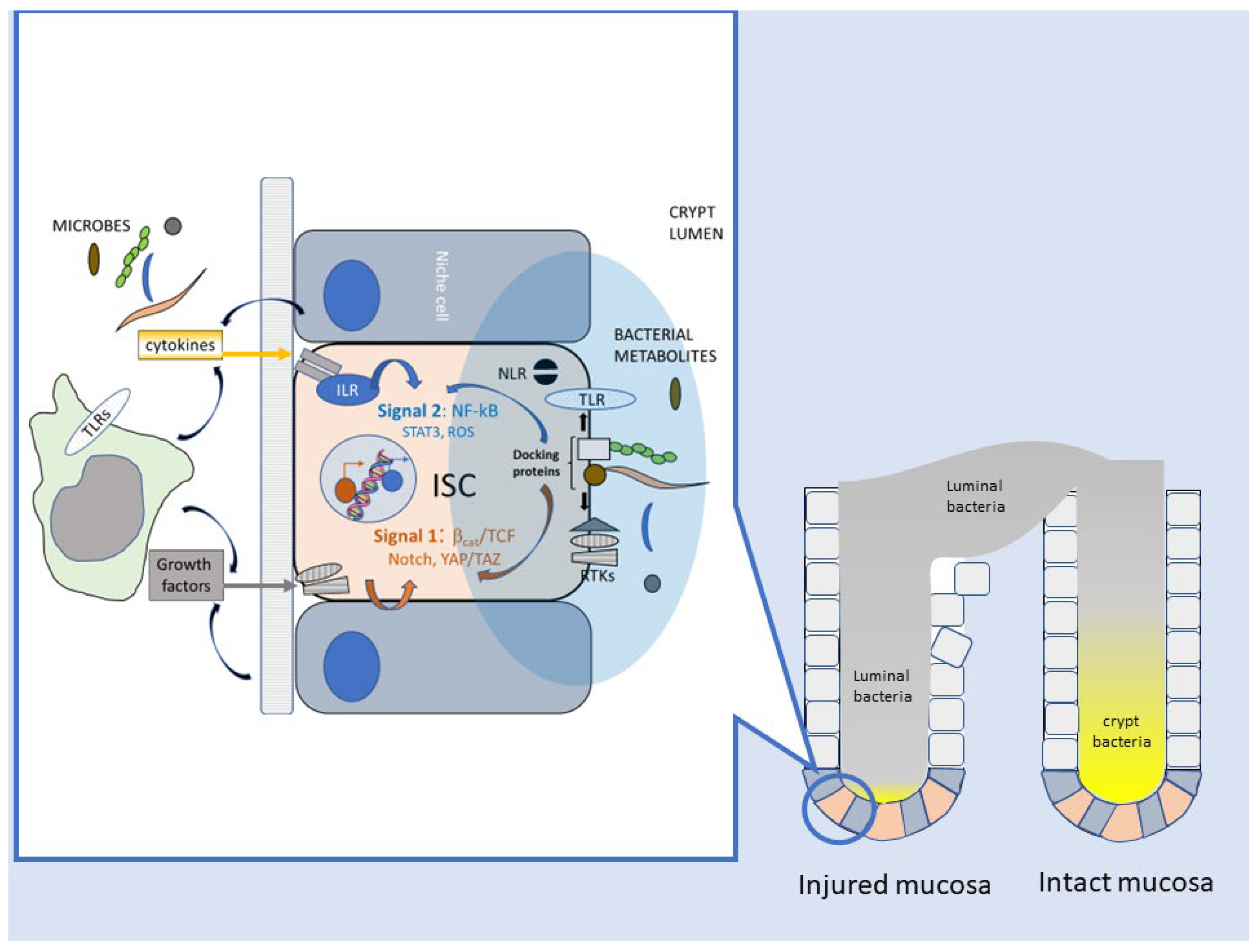
4. How Do Bacteria Talk to Intestinal Stem Cells (ISC)?
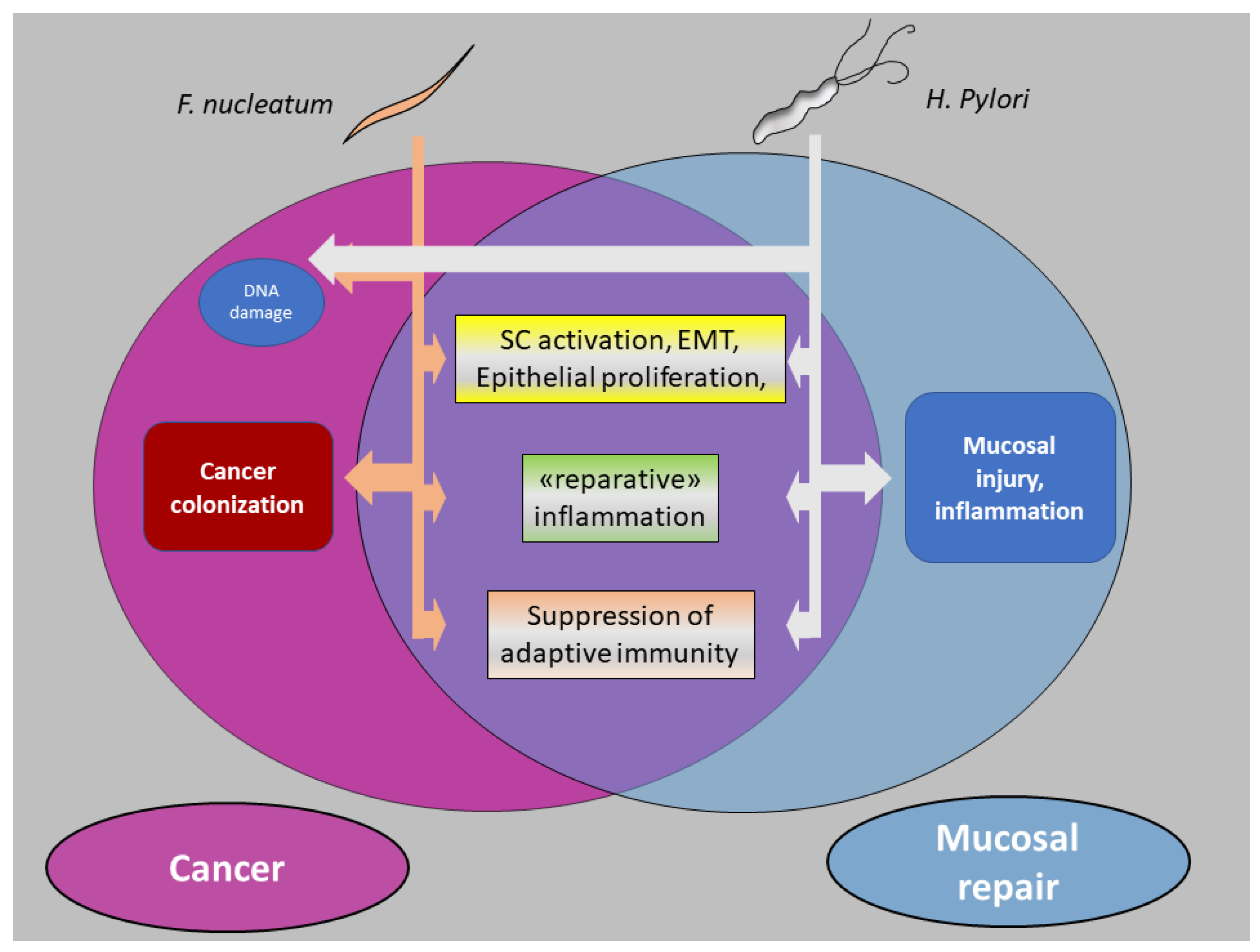
5. Bacteria and CRC: Intestinal Repair Gone Awry?
5.1. Enhancement of Stem-like Features in Epithelial Cells
5.2. Reparative Inflammatory Responses
5.3. Downregulation of Adaptive Immunity
6. Finale: Cancer from a Bug’s Perspective
7. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Manzo, G. Similarities Between Embryo Development and Cancer Process Suggest New Strategies for Research and Therapy of Tumors: A New Point of View. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an Overhealing Wound: An Old Hypothesis Revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer Stem Cell Definitions and Terminology: The Devil Is in the Details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Trosko, J.E. The Concept of “Cancer Stem Cells” in the Context of Classic Carcinogenesis Hypotheses and Experimental Findings. Life 2021, 11, 1308. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer Stem Cells: An Old Idea—A Paradigm Shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer Etiology. Variation in Cancer Risk among Tissues Can Be Explained by the Number of Stem Cell Divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Greig, J.M.; Ellis, C.J. Biological Agents. In Occupational Hygiene, 3rd ed.; Wiley Online Library: Hoboken, NJ, USA, 2008; Volume 100, pp. 344–359. [Google Scholar] [CrossRef]
- Hamid, H.K.S. Schistosoma Japonicum–Associated Colorectal Cancer: A Review. Am. J. Trop. Med. Hyg. 2019, 100, 501. [Google Scholar] [CrossRef]
- Aries, V.; Crowther, J.S.; Drasar, B.S.; Hill, M.J.; Williams, R.E. Bacteria and the Aetiology of Cancer of the Large Bowel. Gut 1969, 10, 334–335. [Google Scholar] [CrossRef]
- Elsland, D.; Neefjes, J. Bacterial Infections and Cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Sears, C.L.; Garrett, W.S. Microbes, Microbiota, and Colon Cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef]
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-Bugs, Their Microbial Partners, and the Link to Colon Cancer. J. Infect. Dis. 2011, 203, 306–311. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A Bacterial Driver-Passenger Model for Colorectal Cancer: Beyond the Usual Suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the Microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Higashi, H. Helicobacter Pylori CagA: A New Paradigm for Bacterial Carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [CrossRef]
- Song, X.; Xin, N.; Wang, W.; Zhao, C. Wnt/β-Catenin, an Oncogenic Pathway Targeted by H. Pylori in Gastric Carcinogenesis. Oncotarget 2015, 6, 35579. [Google Scholar] [CrossRef]
- Pöltl, L.; Kitsera, M.; Raffl, S.; Schild, S.; Cosic, A.; Kienesberger, S.; Unterhauser, K.; Raber, G.; Lembacher-Fadum, C.; Breinbauer, R.; et al. Microbiota-Derived Genotoxin Tilimycin Generates Colonic Stem Cell Mutations. Cell Rep. 2023, 42. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with Familial Adenomatous Polyposis Harbor Colonic Biofilms Containing Tumorigenic Bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter Pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum Infection Is Prevalent in Human Colorectal Carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic Analysis Identifies Association of Fusobacterium with Colorectal Carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome Analyses of Blood and Tissues Suggest Cancer Diagnostic Approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Huggan, P.J.; Murdoch, D.R. Fusobacterial Infections: Clinical Spectrum and Incidence of Invasive Disease. J. Infect. 2008, 57, 283–289. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A Microbial Signature for Crohn’s Disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Lee, S.A.; Liu, F.; Riordan, S.M.; Lee, C.S.; Zhang, L. Global Investigations of Fusobacterium nucleatum in Human Colorectal Cancer. Front. Oncol. 2019, 9, 566. [Google Scholar] [CrossRef]
- Janati, A.I.; Karp, I.; Laprise, C.; Sabri, H.; Emami, E. Detection of Fusobaterium Nucleatum in Feces and Colorectal Mucosa as a Risk Factor for Colorectal Cancer: A Systematic Review and Meta-Analysis. Syst. Rev. 2020, 9, 276. [Google Scholar] [CrossRef]
- Gethings-Behncke, C.; Coleman, H.G.; Jordao, H.W.T.; Longley, D.B.; Crawford, N.; Murray, L.J.; Kunzmann, A.T. Fusobacterium nucleatum in the Colorectum and Its Association with Cancer Risk and Survival: A Systematic Review and Meta-Analysis. Cancer Epidemiol. Biomark. Prev. 2020, 29, 539–548. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue and Patient Prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Yu, T.C.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef]
- Li, Y.Y.; Ge, Q.X.; Cao, J.; Zhou, Y.J.; Du, Y.L.; Shen, B.; Wan, Y.J.Y.; Nie, Y.Q. Association of Fusobacterium nucleatum Infection with Colorectal Cancer in Chinese Patients. World J. Gastroenterol. 2016, 22, 3227–3233. [Google Scholar] [CrossRef]
- Flanagan, L.; Schmid, J.; Ebert, M.; Soucek, P.; Kunicka, T.; Liska, V.; Bruha, J.; Neary, P.; Dezeeuw, N.; Tommasino, M.; et al. Fusobacterium nucleatum Associates with Stages of Colorectal Neoplasia Development, Colorectal Cancer and Disease Outcome. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1381–1390. [Google Scholar] [CrossRef]
- McCoy, A.N.; Araújo-Pérez, F.; Azcárate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium Is Associated with Colorectal Adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef]
- Amini, M.; Rezasoltani, S.; Pourhoseingholi, M.A.; Aghdaei, H.A.; Zali, M.R. Evaluating the Predictive Performance of Gut Microbiota for the Early-Stage Colorectal Cancer. BMC Gastroenterol. 2022, 22, 514. [Google Scholar] [CrossRef]
- Mima, K.; Cao, Y.; Chan, A.T.; Qian, Z.R.; Nowak, J.A.; Masugi, Y.; Shi, Y.; Song, M.; Da Silva, A.; Gu, M.; et al. Fusobacterium Nucleatum in Colorectal Carcinoma Tissue According to Tumor Location. Clin. Transl. Gastroenterol. 2016, 7, e200. [Google Scholar] [CrossRef]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.; Sugai, T.; An, B.; et al. Fusobacterium in Colonic Flora and Molecular Features of Colorectal Carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef]
- Mouradov, D.; Greenfield, P.; Li, S.; In, E.-J.; Storey, C.; Sakthianandeswaren, A.; Georgeson, P.; Buchanan, D.D.; Ward, R.L.; Hawkins, N.J.; et al. Onco-Microbial Community Profiling Identifies Clinico-Molecular and Prognostic Subtypes of Colorectal Cancer. Gastroenterology 2023. [Google Scholar] [CrossRef]
- Mima, K.; Sukawa, Y.; Nishihara, R.; Qian, Z.R.; Yamauchi, M.; Inamura, K.; Kim, S.A.; Masuda, A.; Nowak, J.A.; Nosho, K.; et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015, 1, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.A.; Cochrane, K. Tumor Potentiating Mechanisms of Fusobacterium nucleatum, A Multifaceted Microbe. Gastroenterology 2017, 152, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Serna, G.; Ruiz-Pace, F.; Hernando, J.; Alonso, L.; Fasani, R.; Landolfi, S.; Comas, R.; Jimenez, J.; Elez, E.; Bullman, S.; et al. Fusobacterium nucleatum Persistence and Risk of Recurrence after Preoperative Treatment in Locally Advanced Rectal Cancer. Ann. Oncol. 2020, 31, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Hoorn, S.T.; De Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 2022, 114, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Purcell, R.V.; Visnovska, M.; Biggs, P.J.; Schmeier, S.; Frizelle, F.A. Distinct Gut Microbiome Patterns Associate with Consensus Molecular Subtypes of Colorectal Cancer. Sci. Rep. 2017, 7, 11590. [Google Scholar] [CrossRef]
- Salvucci, M.; Crawford, N.; Stott, K.; Bullman, S.; Longley, D.B.; Prehn, J.H.M. Patients with Mesenchymal Tumours and High Fusobacteriales Prevalence Have Worse Prognosis in Colorectal Cancer (CRC). Gut 2022, 71, 1600–1612. [Google Scholar] [CrossRef]
- Ternes, D.; Tsenkova, M.; Pozdeev, V.I.; Meyers, M.; Koncina, E.; Atatri, S.; Schmitz, M.; Karta, J.; Schmoetten, M.; Heinken, A.; et al. The Gut Microbial Metabolite Formate Exacerbates Colorectal Cancer Progression. Nat. Metab. 2022, 4, 458–475. [Google Scholar] [CrossRef]
- Okita, Y.; Koi, M.; Takeda, K.; Ross, R.; Mukherjee, B.; Koeppe, E.; Stoffel, E.M.; Galanko, J.A.; McCoy, A.N.; Keku, T.O.; et al. Fusobacterium nucleatum Infection Correlates with Two Types of Microsatellite Alterations in Colorectal Cancer and Triggers DNA Damage. Gut Pathog. 2020, 12, 46. [Google Scholar] [CrossRef]
- Lo, C.H.; Wu, D.C.; Jao, S.W.; Wu, C.C.; Lin, C.Y.; Chuang, C.H.; Lin, Y.B.; Chen, C.H.; Chen, Y.T.; Chen, J.H.; et al. Enrichment of Prevotella Intermedia in Human Colorectal Cancer and Its Additive Effects with Fusobacterium nucleatum on the Malignant Transformation of Colorectal Adenomas. J. Biomed. Sci. 2022, 29, 88. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor−κB, and Up-Regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e24. [Google Scholar] [CrossRef]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A Human Colonic Commensal Promotes Colon Tumorigenesis via Activation of T Helper Type 17 T Cell Responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Yoshida, Y.; Ito, S.; Kamo, M.; Kezuka, Y.; Tamura, H.; Kunimatsu, K.; Kato, H. Production of Hydrogen Sulfide by Two Enzymes Associated with Biosynthesis of Homocysteine and Lanthionine in Fusobacterium nucleatum Subsp. Nucleatum ATCC 25586. Microbiology 2010, 156 Pt 7, 2260–2269. [Google Scholar] [CrossRef]
- Houghton, J.M.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric Cancer Originating from Bone Marrow-Derived Cells. Science 2004, 306, 1568–1571. [Google Scholar] [CrossRef]
- Stoddart, R.W. The Generation of Cancer: Initiation, Promotion, Progression and the Multiple Influences of the Environment. Nutr. Health 1983, 2, 153–162. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Cancer by Inducing Wnt/Β-catenin Modulator Annexin A1. EMBO Rep. 2019, 20, e47638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Z.; Gao, C.; Chen, P.; Chen, J.; Liu, W.; Xiao, S.; Lu, H. MiR-21 Plays a Pivotal Role in Gastric Cancer Pathogenesis and Progression. Lab. Investig. 2008, 88, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Wu, D.W.; Huang, C.C.; He, T.Y.; Chou, M.C.; Sheu, G.T.; Lee, H. MicroRNA-21 Promotes Tumour Malignancy via Increased Nuclear Translocation of β-Catenin and Predicts Poor Outcome in APC-Mutated but Not in APC-Wild-Type Colorectal Cancer. Carcinogenesis 2014, 35, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Casasanta, M.A.; Yoo, C.C.; Udayasuryan, B.; Sanders, B.E.; Umanã, A.; Zhang, Y.; Peng, H.; Duncan, A.J.; Wang, Y.; Li, L.; et al. Fusobacterium nucleatum Host-Cell Binding and Invasion Induces IL-8 and CXCL1 Secretion That Drives Colorectal Cancer Cell Migration. Sci. Signal. 2020, 13, eaba9157. [Google Scholar] [CrossRef]
- Abed, J.; Emgård, J.E.M.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium Persistence and Antibiotic Response in Colorectal Cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Cavallucci, V.; Palucci, I.; Fidaleo, M.; Mercuri, A.; Masi, L.; Emoli, V.; Bianchetti, G.; Fiori, M.E.; Bachrach, G.; Scaldaferri, F.; et al. Proinflammatory and Cancer-Promoting Pathobiont Fusobacterium nucleatum Directly Targets Colorectal Cancer Stem Cells. Biomolecules 2022, 12, 1256. [Google Scholar] [CrossRef]
- Brewer, M.L.; Dymock, D.; Brady, R.L.; Singer, B.B.; Virji, M.; Hill, D.J. Fusobacterium Spp. Target Human CEACAM1 via the Trimeric Autotransporter Adhesin CbpF. J. Oral Microbiol. 2019, 11, 1565043. [Google Scholar] [CrossRef]
- Galaski, J.; Shhadeh, A.; Umaña, A.; Yoo, C.C.; Arpinati, L.; Isaacson, B.; Berhani, O.; Singer, B.B.; Slade, D.J.; Bachrach, G.; et al. Fusobacterium nucleatum CbpF Mediates Inhibition of T Cell Function Through CEACAM1 Activation. Front. Cell. Infect. Microbiol. 2021, 11, 692544. [Google Scholar] [CrossRef]
- Wegwitz, F.; Lenfert, E.; Gerstel, D.; von Ehrenstein, L.; Einhoff, J.; Schmidt, G.; Logsdon, M.; Brandner, J.; Tiegs, G.; Beauchemin, N.; et al. CEACAM1 Controls the EMT Switch in Murine Mammary Carcinoma in Vitro and in Vivo. Oncotarget 2016, 7, 63730–63746. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, R.; Zhou, C.; Yu, H.; Luo, W.; Zhu, J.; Liu, J.; Zhang, Z.; Xie, N.; Peng, X.; et al. ANGPTL4-Mediated Promotion of Glycolysis Facilitates the Colonization of Fusobacterium nucleatum in Colorectal Cancer. Cancer Res. 2021, 81, 6157–6170. [Google Scholar] [CrossRef]
- Hong, J.; Guo, F.; Lu, S.Y.; Shen, C.; Ma, D.; Zhang, X.; Xie, Y.; Yan, T.; Yu, T.; Sun, T.; et al. Nucleatum Targets LncRNA ENO1-IT1 to Promote Glycolysis and Oncogenesis in Colorectal Cancer. Gut 2021, 70, 2123–2137. [Google Scholar] [CrossRef]
- Kong, C.; Yan, X.; Zhu, Y.; Zhu, H.; Luo, Y.; Liu, P.; Ferrandon, S.; Kalady, M.F.; Gao, R.; He, J.; et al. Fusobacterium nucleatum Promotes the Development of Colorectal Cancer by Activating a Cytochrome P450/Epoxyoctadecenoic Acid Axis via Tlr4/Keap1/Nrf2 Signaling. Cancer Res. 2021, 81, 485–498. [Google Scholar] [CrossRef]
- Liu, H.; Du, J.; Chao, S.; Li, S.; Cai, H.; Zhang, H.; Chen, G.; Liu, P.; Bu, P. Fusobacterium nucleatum Promotes Colorectal Cancer Cell to Acquire Stem Cell-Like Features by Manipulating Lipid Droplet-Mediated Numb Degradation. Adv. Sci. 2022, 9, 2105222. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, Y.; Wang, J.; Lai, X.; Xu, Y.; Zhang, X.; Zhu, X.; Ruan, C.; Huang, Y. Clinicopathological Differences of High Fusobacterium nucleatum Levels in Colorectal Cancer: A Review and Meta-Analysis. Front. Microbiol. 2022, 13, 3715. [Google Scholar] [CrossRef]
- Lu, X.; Xu, Q.; Tong, Y.; Zhang, Z.; Dun, G.; Feng, Y.; Tang, J.; Han, D.; Mao, Y.; Deng, L.; et al. Long Non-Coding RNA EVADR Induced by Fusobacterium nucleatum Infection Promotes Colorectal Cancer Metastasis. Cell Rep. 2022, 40, 111127. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Zhang, J.; Cao, P.; Su, W.; Deng, Y.; Zhan, N.; Fu, X.; Huang, Y.; Dong, W. Fusobacterium nucleatum Promotes Metastasis in Colorectal Cancer by Activating Autophagy Signaling via the Upregulation of CARD3 Expression. Theranostics 2020, 10, 323–339. [Google Scholar] [CrossRef]
- Chen, S.; Su, T.; Zhang, Y.; Lee, A.; He, J.; Ge, Q.; Wang, L.; Si, J.; Zhuo, W.; Wang, L. Fusobacterium nucleatum Promotes Colorectal Cancer Metastasis by Modulating KRT7-AS/KRT7. Gut Microbes 2020, 11, 511–525. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, C.; Yue, C.; Liu, X. Fusobacterium nucleatum Produces Cancer Stem Cell Characteristics via EMT-Resembling Variations. Int. J. Clin. Exp. Pathol. 2020, 13, 1819–1828. [Google Scholar]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Chen, P.; Hsu, W.-H.; Han, J.; Xia, Y.; Depinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef] [PubMed]
- Musella, M.; Guarracino, A.; Manduca, N.; Galassi, C.; Ruggiero, E.; Potenza, A.; Maccafeo, E.; Manic, G.; Mattiello, L.; Soliman Abdel Rehim, S.; et al. Type I IFNs Promote Cancer Cell Stemness by Triggering the Epigenetic Regulator KDM1B. Nat. Immunol. 2022, 23, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, Y.; Weng, W.; Guo, B.; Cai, G.; Ma, Y.; Cai, S. Fusobacterium nucleatum Promotes Chemoresistance to 5-Fluorouracil by Upregulation of BIRC3 Expression in Colorectal Caner. J. Exp. Clin. Cancer Res. 2019, 38, 14. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zhang, J.; Wu, X.; Chen, M.; Huang, H.; Zeng, S.; Xiang, Z.; Li, X.; Dong, W. Fusobacterium nucleatum Extracellular Vesicles Promote Experimental Colitis by Modulating Autophagy via the MiR-574-5p/CARD3 Axis. Inflamm. Bowel Dis. 2023, 29, 9–26. [Google Scholar] [CrossRef]
- Gur, C.; Maalouf, N.; Shhadeh, A.; Berhani, O.; Singer, B.B.; Bachrach, G.; Mandelboim, O. Fusobacterium nucleatum Supresses Anti-Tumor Immunity by Activating CEACAM1. Oncoimmunology 2019, 8, e1581531. [Google Scholar] [CrossRef]
- Volonté, A.; Di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-Initiating Cells from Colorectal Cancer Patients Escape from T Cell–Mediated Immunosurveillance In Vitro through Membrane-Bound IL-4. J. Immunol. 2014, 192, 523–532. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Ochoa, M.C.; Villalba, M.; Olivera, I.; Cirella, A.; Eguren-Santamaria, I.; Berraondo, P.; Schalper, K.A.; de Andrea, C.E.; et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin. Cancer Res. 2021, 27, 2383–2393. [Google Scholar] [CrossRef]
- Wang, H.; Luo, K.; Guan, Z.; Li, Z.; Xiang, J.; Ou, S.; Tao, Y.; Ran, S.; Ye, J.; Ma, T.; et al. Identification of the Crucial Role of CCL22 in F. Nucleatum-Related Colorectal Tumorigenesis That Correlates With Tumor Microenvironment and Immune Checkpoint Therapy. Front. Genet. 2022, 13, 83. [Google Scholar] [CrossRef]
- Xue, Y.; Xiao, H.; Guo, S.; Xu, B.; Liao, Y.; Wu, Y.; Zhang, G. Indoleamine 2,3-Dioxygenase Expression Regulates the Survival and Proliferation of Fusobacterium nucleatum in THP-1-Derived Macrophages. Cell Death Dis. 2018, 9, 355. [Google Scholar] [CrossRef]
- Gao, Y.; Bi, D.; Xie, R.; Li, M.; Guo, J.; Liu, H.; Guo, X.; Fang, J.; Ding, T.; Zhu, H.; et al. Fusobacterium nucleatum Enhances the Efficacy of PD-L1 Blockade in Colorectal Cancer. Signal Transduct. Target. Ther. 2021, 6, 398. [Google Scholar] [CrossRef]
- Karin, M.; Clevers, H. Reparative Inflammation Takes Charge of Tissue Regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Whyte, J.L.; Smith, A.A.; Helms, J.A. Wnt Signaling and Injury Repair. Cold Spring Harb. Perspect. Biol. 2012, 4, a008078. [Google Scholar] [CrossRef]
- Radtke, F.; Clevers, H. Self-Renewal and Cancer of the Gut: Two Sides of a Coin. Science 2005, 307, 1904–1909. [Google Scholar] [CrossRef]
- Clevers, H. The Intestinal Crypt, A Prototype Stem Cell Compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Lemaitre, B. Gut Homeostasis in a Microbial World: Insights from Drosophila Melanogaster. Nat. Rev. Microbiol. 2013, 11, 615–626. [Google Scholar] [CrossRef]
- Xu, N.; Wang, S.Q.; Tan, D.; Gao, Y.; Lin, G.; Xi, R. EGFR, Wingless and JAK/STAT Signaling Cooperatively Maintain Drosophila Intestinal Stem Cells. Dev. Biol. 2011, 354, 31–43. [Google Scholar] [CrossRef]
- Cordero, J.B.; Stefanatos, R.K.; Scopelliti, A.; Vidal, M.; Sansom, O.J. Inducible Progenitor-Derived Wingless Regulates Adult Midgut Regeneration in Drosophila. EMBO J. 2012, 31, 3901–3917. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Chakrabarti, S.; Lemaitre, B. Invasive and Indigenous Microbiota Impact Intestinal Stem Cell Activity through Multiple Pathways in Drosophila. Genes Dev. 2009, 23, 2333–2344. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Kuraishi, T.; Lemaitre, B. Drosophila EGFR Pathway Coordinates Stem Cell Proliferation and Gut Remodeling Following Infection. BMC Biol. 2010, 8, 152. [Google Scholar] [CrossRef]
- Biteau, B.; Hochmuth, C.E.; Jasper, H. JNK Activity in Somatic Stem Cells Causes Loss of Tissue Homeostasis in the Aging Drosophila Gut. Cell Stem Cell 2008, 3, 442–455. [Google Scholar] [CrossRef]
- Patel, P.H.; Dutta, D.; Edgar, B.A. Niche Appropriation by Drosophila Intestinal Stem Cell Tumours. Nat. Cell Biol. 2015, 17, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nagy, P.; Bonfini, A.; Houtz, P.; Bing, X.L.; Yang, X.; Buchon, N. Microbes Affect Gut Epithelial Cell Composition through Immune-Dependent Regulation of Intestinal Stem Cell Differentiation. Cell Rep. 2022, 38, 110572. [Google Scholar] [CrossRef] [PubMed]
- Abrams, G.D. Microbial Effects on Mucosal Structure and Function. Am. J. Clin. Nutr. 1977, 30, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of Commensal Microflora by Toll-like Receptors Is Required for Intestinal Homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Medzhitov, R. Regulation of Spontaneous Intestinal Tumorigenesis through the Adaptor Protein MyD88. Science 2007, 317, 124–127. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Fukata, M.; Chen, A.; Klepper, A.; Krishnareddy, S.; Vamadevan, A.S.; Thomas, L.S.; Xu, R.; Inoue, H.; Arditi, M.; Dannenberg, A.J.; et al. Cox-2 Is Regulated by Toll-Like Receptor-4 (TLR4) Signaling: Role in Proliferation and Apoptosis in the Intestine. Gastroenterology 2006, 131, 862–877. [Google Scholar] [CrossRef]
- Santaolalla, R.; Sussman, D.A.; Ruiz, J.R.; Davies, J.M.; Pastorini, C.; España, C.L.; Sotolongo, J.; Burlingame, O.; Bejarano, P.A.; Philip, S.; et al. TLR4 Activates the β-Catenin Pathway to Cause Intestinal Neoplasia. PLoS ONE 2013, 8, e63298. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt Signalling in Stem Cells and Cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa E Melo, F.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt Activity Defines Colon Cancer Stem Cells and Is Regulated by the Microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like Receptor 4 Is Expressed on Intestinal Stem Cells and Regulates Their Proliferation and Apoptosis via the P53 Up-Regulated Modulator of Apoptosis. J. Biol. Chem. 2012, 287, 37296. [Google Scholar] [CrossRef]
- Van der Post, S.; Birchenough, G.M.H.; Held, J.M. NOX1-Dependent Redox Signaling Potentiates Colonic Stem Cell Proliferation to Adapt to the Intestinal Microbiota by Linking EGFR and TLR Activation. Cell Rep. 2021, 35, 108949. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH Oxidases: An Overview from Structure to Innate Immunity-Associated Pathologies. Cell. Mol. Immunol. 2014, 12, 5–23. [Google Scholar] [CrossRef]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS Production and NF-ΚB Activation Triggered by RAC1 Facilitate WNT-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef]
- Nigro, G.; Rossi, R.; Commere, P.-H.; Jay, P.; Sansonetti, P.J. The Cytosolic Bacterial Peptidoglycan Sensor Nod2 Affords Stem Cell Protection and Links Microbes to Gut Epithelial Regeneration. Cell Host Microbe 2014, 15, 792–798. [Google Scholar] [CrossRef]
- Levy, A.; Stedman, A.; Deutsch, E.; Donnadieu, F.; Virgin, H.W.; Sansonetti, P.J.; Nigro, G. Innate Immune Receptor NOD2 Mediates LGR5+ Intestinal Stem Cell Protection against ROS Cytotoxicity via Mitophagy Stimulation. Proc. Natl. Acad. Sci. USA 2020, 117, 1994–2003. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. New Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef]
- Udden, S.M.N.; Peng, L.; Gan, J.L.; Shelton, J.M.; Malter, J.S.; Hooper, L.V.; Zaki, M.H. NOD2 Suppresses Colorectal Tumorigenesis via Downregulation of the TLR Pathways. Cell Rep. 2017, 19, 2756–2770. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.-J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth Cells as a Site of Origin for Intestinal Inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Pani, G.; Bedogni, B.; Colavitti, R.; Anzevino, R.; Borrello, S.; Galeotti, T. Cell Compartmentalization in Redox Signaling. IUBMB Life 2001, 52, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T. Toll-like Receptor Signalling in the Intestinal Epithelium: How Bacterial Recognition Shapes Intestinal Function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Pédron, T.; Mulet, C.; Dauga, C.; Frangeul, L.; Chervaux, C.; Grompone, G.; Sansonettia, P.J. A Crypt-Specific Core Microbiota Resides in the Mouse Colon. mBio 2012, 3, e00116-12. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Mulet, C.; De Castro, C.; Molinaro, A.; Saffarian, A.; Nigro, G.; Bérard, M.; Clerc, M.; Pedersen, A.B.; Sansonetti, P.J.; et al. Lipopolysaccharide from Crypt-Specific Core Microbiota Modulates the Colonic Epithelial Proliferation-to-Differentiation Balance. mBio 2017, 8, e01680-17. [Google Scholar] [CrossRef]
- Konjar, Š.; Pavšič, M.; Veldhoen, M. Regulation of Oxygen Homeostasis at the Intestinal Epithelial Barrier Site. Int. J. Mol. Sci. 2021, 22, 9170. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Kumar, S.; Green, B.; et al. Gut Microbial Metabolism Drives Transformation of Msh2-Deficient Colon Epithelial Cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe 2018, 24, 833–846.e6. [Google Scholar] [CrossRef]
- Rodríguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between Metabolic Identities in the Intestinal Crypt Supports Stem Cell Function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362.e5. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 Impacts Colitis by Altering Gut Microbiota Metabolism of Tryptophan into Aryl Hydrocarbon Receptor Ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Lamas, B.; Hernandez-Galan, L.; Galipeau, H.J.; Constante, M.; Clarizio, A.; Jury, J.; Breyner, N.M.; Caminero, A.; Rueda, G.; Hayes, C.L.; et al. Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is Decreased in Celiac Disease Leading to Intestinal Inflammation. Sci. Transl. Med. 2020, 12, eaba0624. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; DeLuca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 Promotes Intestinal-Stem-Cell-Mediated Epithelial Regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl Hydrocarbon Receptor/Cytochrome P450 1A1 Pathway Mediates Breast Cancer Stem Cells Expansion through PTEN Inhibition and β-Catenin and Akt Activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef]
- Jin, U.H.; Michelhaugh, S.K.; Polin, L.A.; Shrestha, R.; Mittal, S.; Safe, S. Omeprazole Inhibits Glioblastoma Cell Invasion and Tumor Growth. Cancers 2020, 12, 2097. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; De Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A Gp130–Src–YAP Module Links Inflammation to Epithelial Regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef]
- Yilmaz, Ö.H.; Katajisto, P.; Lamming, D.W.; Gültekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. MTORC1 in the Paneth Cell Niche Couples Intestinal Stem-Cell Function to Calorie Intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef]
- Igarashi, M.; Guarente, L. MTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450. [Google Scholar] [CrossRef]
- Manik, M.K.; Shi, Y.; Li, S.; Zaydman, M.A.; Damaraju, N.; Eastman, S.; Smith, T.G.; Gu, W.; Masic, V.; Mosaiab, T.; et al. Cyclic ADP Ribose Isomers: Production, Chemical Structures, and Immune Signaling. Science 2022, 377, eadc8969. [Google Scholar] [CrossRef] [PubMed]
- Weagley, J.S.; Zaydman, M.; Venkatesh, S.; Sasaki, Y.; Damaraju, N.; Yenkin, A.; Buchser, W.; Rodionov, D.A.; Osterman, A.; Ahmed, T.; et al. Products of Gut Microbial Toll/Interleukin-1 Receptor Domain NADase Activities in Gnotobiotic Mice and Bangladeshi Children with Malnutrition. Cell Rep. 2022, 39, 110738. [Google Scholar] [CrossRef] [PubMed]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 Immunity in Tissue Repair and Fibrosis. Nat. Rev. Immunol. 2017, 18, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lee, D.G.; Kim, H.S.; Lee, Y.S.; Kim, S.; Cha, S.Y.; Ota, I.; Kim, N.H.; Cha, Y.H.; Yang, D.H.; Lee, Y.; et al. Helicobacter Pylori CagA Promotes Snail-Mediated Epithelial–mesenchymal Transition by Reducing GSK-3 Activity. Nat. Commun. 2014, 5, 4423. [Google Scholar] [CrossRef]
- Lu, R.; Wu, S.; Zhang, Y.G.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.L.; Zhou, Z.; Bissonnette, M.; et al. Enteric Bacterial Protein AvrA Promotes Colonic Tumorigenesis and Activates Colonic Beta-Catenin Signaling Pathway. Oncogenesis 2014, 3, e105. [Google Scholar] [CrossRef]
- Sigal, M.; Rothenberg, M.E.; Logan, C.Y.; Lee, J.Y.; Honaker, R.W.; Cooper, R.L.; Passarelli, B.; Camorlinga, M.; Bouley, D.M.; Alvarez, G.; et al. Helicobacter Pylori Activates and Expands Lgr5+ Stem Cells through Direct Colonization of the Gastric Glands. Gastroenterology 2015, 148, 1392–1404.e21. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Wizenty, J.; Müllerke, S.; Kolesnichenko, M.; Heuberger, J.; Lin, M.; Fischer, A.; Mollenkopf, H.; Berger, H.; Tacke, F.; Sigal, M. Gastric Stem Cells Promote Inflammation and Gland Remodeling in Response to Helicobacter Pylori via Rspo3-Lgr4 Axis. EMBO J. 2022, 41, e109996. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Krappmann, D. Controlling NF-ΚB Activation in T Cells by Costimulatory Receptors. Cell Death Differ. 2006, 13, 834–842. [Google Scholar] [CrossRef]
- Fournier, B.M.; Parkos, C.A. The Role of Neutrophils during Intestinal Inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The Regulatory Roles of Neutrophils in Adaptive Immunity. Cell Commun. Signal. 2019, 17, 147. [Google Scholar] [CrossRef]
- Li, D. Understanding the Protective Effect of Helicobacter Pylori Eradication on Gastric Cancer After a Quarter Century: New Insights From an Old Trial. Gastroenterology 2022, 163, 42–44. [Google Scholar] [CrossRef]
- Périchon, B.; Lichtl-Häfele, J.; Bergsten, E.; Delage, V.; Trieu-Cuot, P.; Sansonetti, P.; Sobhani, I.; Dramsi, S. Detection of Streptococcus Gallolyticus and Four Other CRC-Associated Bacteria in Patient Stools Reveals a Potential “Driver” Role for Enterotoxigenic Bacteroides Fragilis. Front. Cell. Infect. Microbiol. 2022, 12, 186. [Google Scholar] [CrossRef]
- Peixoto, A.; Fernandes, E.; Gaiteiro, C.; Lima, L.; Azevedo, R.; Soares, J.; Cotton, S.; Parreira, B.; Neves, M.; Amaro, T.; et al. Hypoxia Enhances the Malignant Nature of Bladder Cancer Cells and Concomitantly Antagonizes Protein O -Glycosylation Extension. Oncotarget 2016, 7, 63138–63157. [Google Scholar] [CrossRef]
- Nauta, T.D.; van Hinsbergh, V.W.M.; Koolwijk, P. Hypoxic Signaling During Tissue Repair and Regenerative Medicine. Int. J. Mol. Sci. 2014, 15, 19791. [Google Scholar] [CrossRef]
- Mendes, R.T.; Nguyen, D.; Stephens, D.; Pamuk, F.; Fernandes, D.; Hasturk, H.; Van Dyke, T.E.; Kantarci, A. Hypoxia-Induced Endothelial Cell Responses—Possible Roles during Periodontal Disease. Clin. Exp. Dent. Res. 2018, 4, 241–248. [Google Scholar] [CrossRef]
- Rogan, M.R.; Patterson, L.L.; Wang, J.Y.; McBride, J.W. Bacterial Manipulation of Wnt Signaling: A Host-Pathogen Tug-of-Wnt. Front. Immunol. 2019, 10, 2390. [Google Scholar] [CrossRef]
- Sáenz, J.B.; Mills, J.C. Acid and the Basis for Cellular Plasticity and Reprogramming in Gastric Repair and Cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Nascakova, Z.; Mihai, A.I.; Cheng, P.F.; Levesque, M.P.; Lampart, S.; Hurwitz, R.; Pfannkuch, L.; Dobrovolna, J.; Jacobs, M.; et al. The ALPK1/TIFA/NF-ΚB Axis Links a Bacterial Carcinogen to R-Loop-Induced Replication Stress. Nat. Commun. 2020, 11, 5117. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.Y.; Kim, N.; Park, Y.S.; Hwang, J.H.; Kim, J.W.; Jeong, S.H.; Lee, D.H.; Jung, H.C.; Song, I.S. Progression of Atrophic Gastritis and Intestinal Metaplasia Drives Helicobacter Pylori out of the Gastric Mucosa. Dig. Dis. Sci. 2006, 51, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.N.; Ganeshkumar, N.; Kolenbrander, P.E. Helicobacter Pylori Adheres Selectively to Fusobacterium spp. Oral Microbiol. Immunol. 1998, 13, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-Y.; Kuo, W.-L.; Hsu, W.-T.; Tung, S.-Y.; Li, C.; Hsieh, Y.-Y.; Kuo, W.-L.; Hsu, W.-T.; Tung, S.-Y.; Li, C. Fusobacterium nucleatum-Induced Tumor Mutation Burden Predicts Poor Survival of Gastric Cancer Patients. Cancers 2022, 15, 269. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The Human Tumor Microbiome is Composed of Tumor Type-Specific Intracellular Bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Udayasuryan, B.; Nguyen, T.T.D.; Umaña, A.; Roberts, L.M.; Ahmad, R.N.; Sobol, P.; Jones, S.D.; Munson, J.M.; Slade, D.J.; Verbridge, S.S. Fusobacterium nucleatum Infection Induces Pancreatic Cancer Cell Proliferation and Migration through Regulation of Host Cytokine Signaling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Parhi, L.; Alon-Maimon, T.; Sol, A.; Nejman, D.; Shhadeh, A.; Fainsod-Levi, T.; Yajuk, O.; Isaacson, B.; Abed, J.; Maalouf, N.; et al. Breast Cancer Colonization by Fusobacterium nucleatum Accelerates Tumor Growth and Metastatic Progression. Nat. Commun. 2020, 11, 3259. [Google Scholar] [CrossRef]
- Brennan, C.A.; Nakatsu, G.; Comeau, C.A.G.; Drew, D.A.; Glickman, J.N.; Schoen, R.E.; Chan, A.T.; Garrett, W.S. Aspirin Modulation of the Colorectal Cancer-Associated Microbe Fusobacterium nucleatum. mBio 2021, 12, e00547-21. [Google Scholar] [CrossRef]
- Lu, S.S.M.; Mohammed, Z.; Häggström, C.; Myte, R.; Lindquist, E.; Gylfe, Å.; Van Guelpen, B.; Harlid, S. Antibiotics Use and Subsequent Risk of Colorectal Cancer: A Swedish Nationwide Population-Based Study. JNCI J. Natl. Cancer Inst. 2022, 114, 38–46. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pani, G. Fusobacterium & Co. at the Stem of Cancer: Microbe–Cancer Stem Cell Interactions in Colorectal Carcinogenesis. Cancers 2023, 15, 2583. https://doi.org/10.3390/cancers15092583
Pani G. Fusobacterium & Co. at the Stem of Cancer: Microbe–Cancer Stem Cell Interactions in Colorectal Carcinogenesis. Cancers. 2023; 15(9):2583. https://doi.org/10.3390/cancers15092583
Chicago/Turabian StylePani, Giovambattista. 2023. "Fusobacterium & Co. at the Stem of Cancer: Microbe–Cancer Stem Cell Interactions in Colorectal Carcinogenesis" Cancers 15, no. 9: 2583. https://doi.org/10.3390/cancers15092583