
M2 Muscarinic Receptor Stimulation Induces Autophagy in Human Glioblastoma Cancer Stem Cells via mTOR Complex-1 Inhibition


 ,
,  , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Pharmacological Treatments
2.3. U251 Cell Line Transfection and Immunofluorescence Analysis
2.4. Cell Viability Assay
2.5. Protein Extraction and Western Blot Analysis
2.6. Statistical Analysis
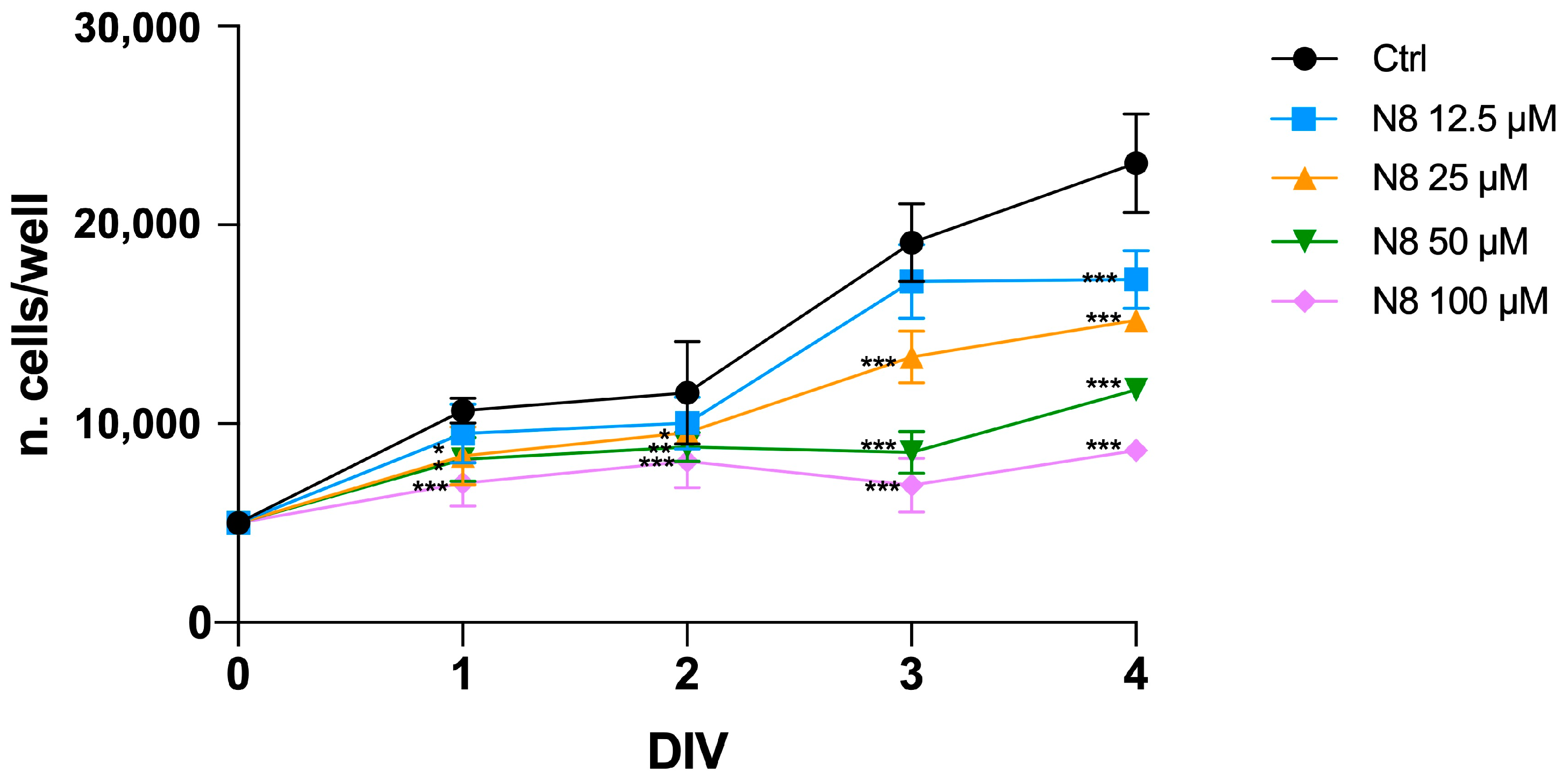
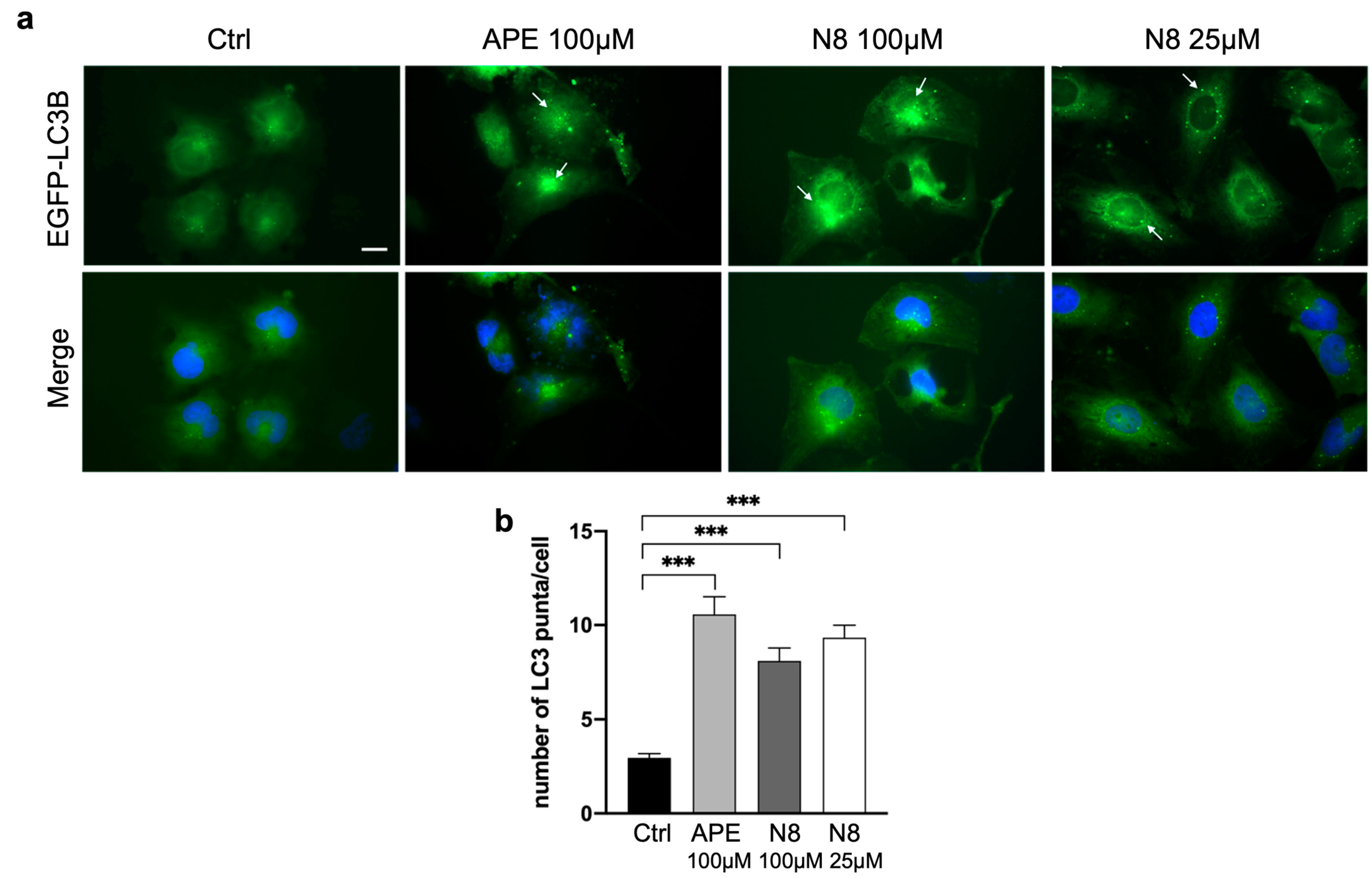
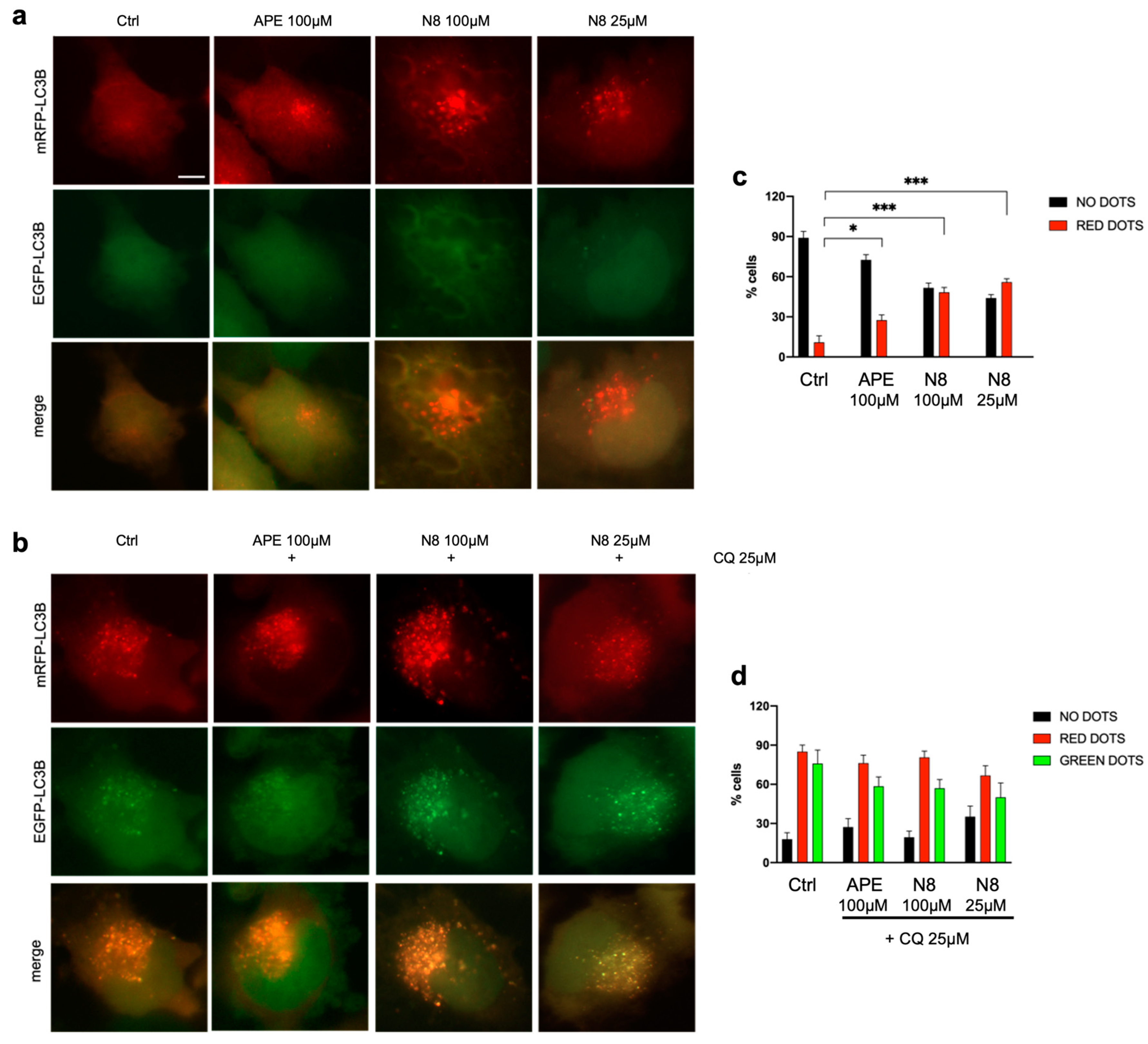
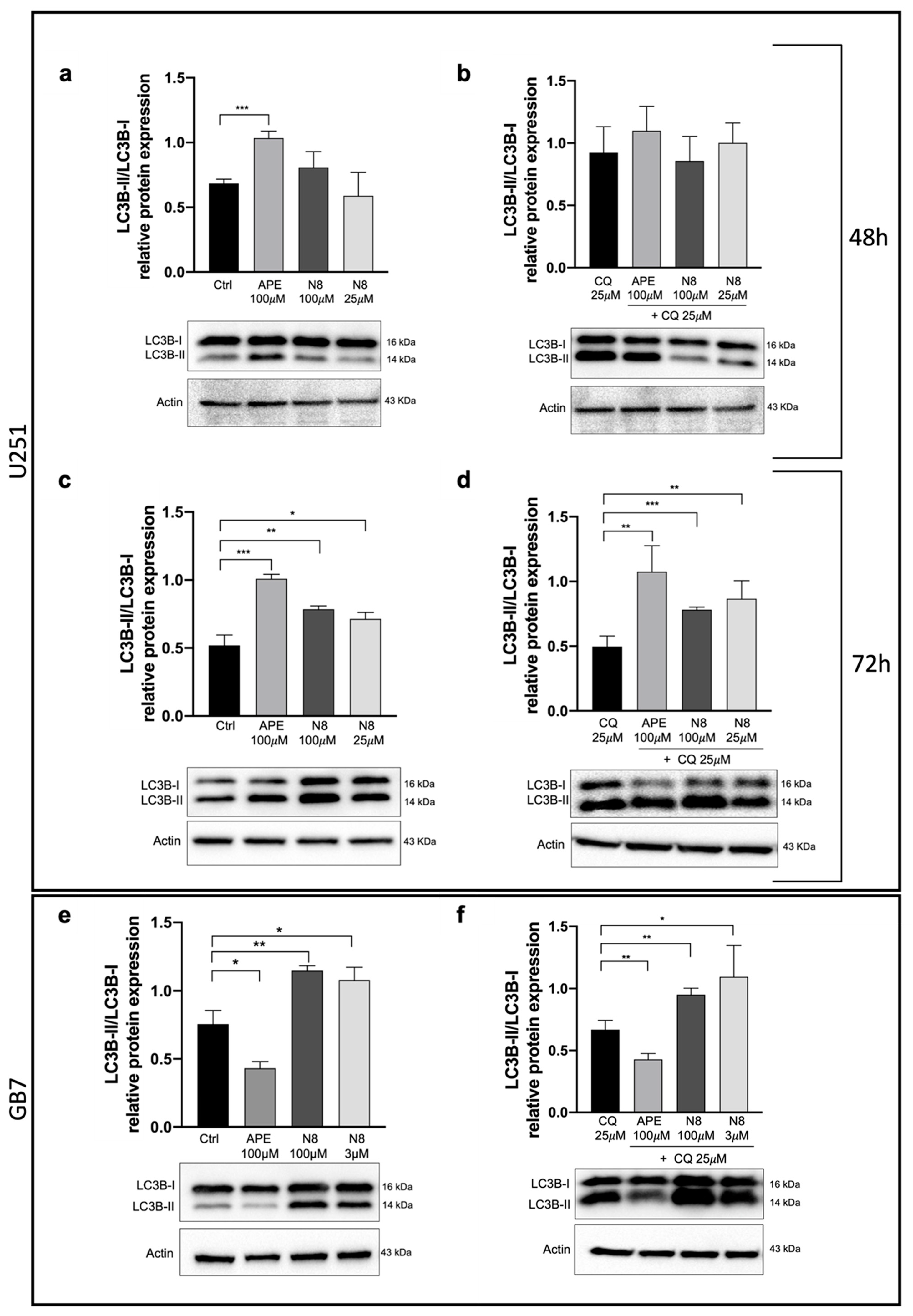
3. Results
3.1. Analysis of Autophagy in U251 Cell Line and GB7 Cells
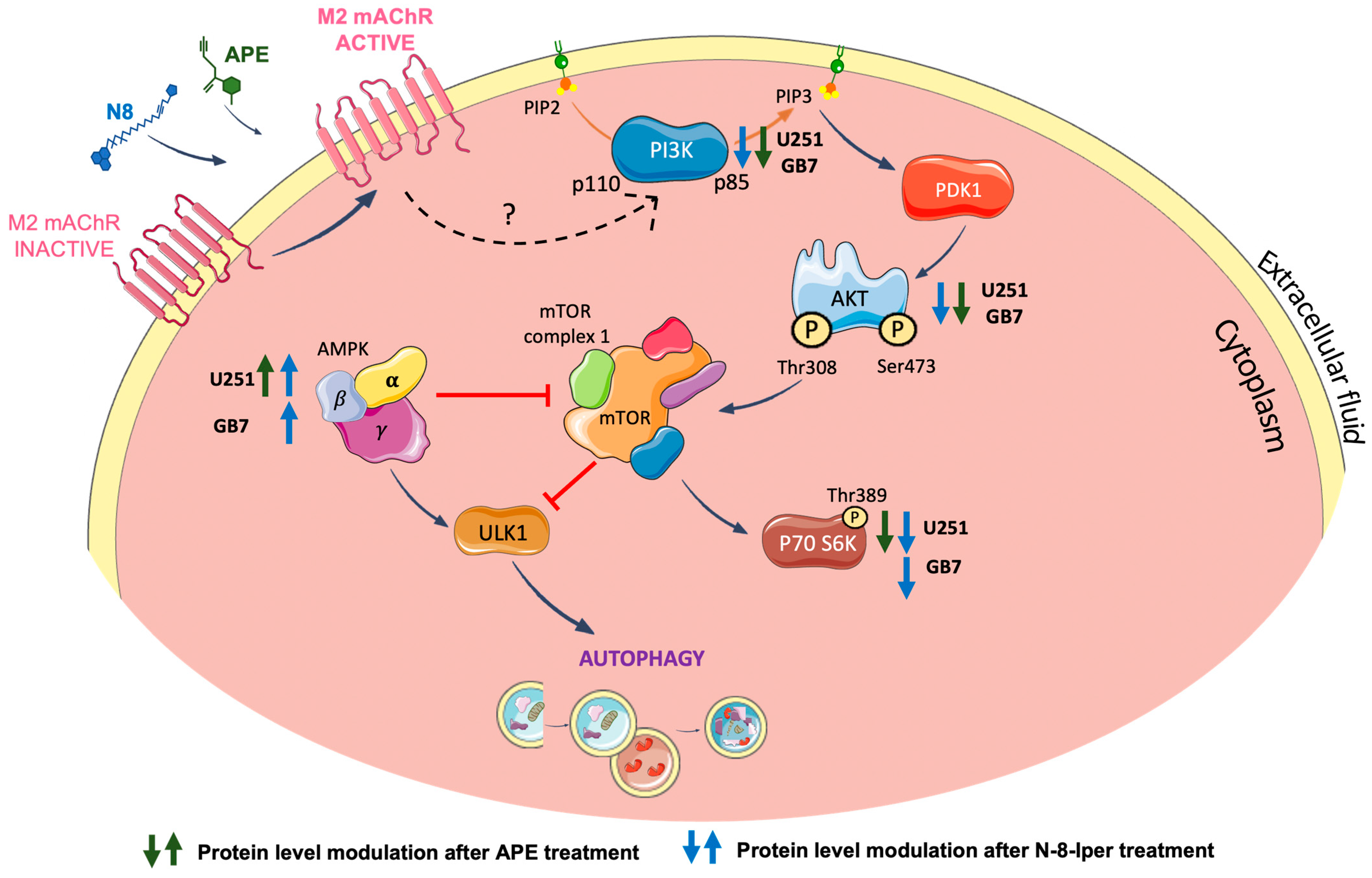
3.2. Analysis of TORC1 and AMPK Expression in U251 Cell Line and GB7 Cells
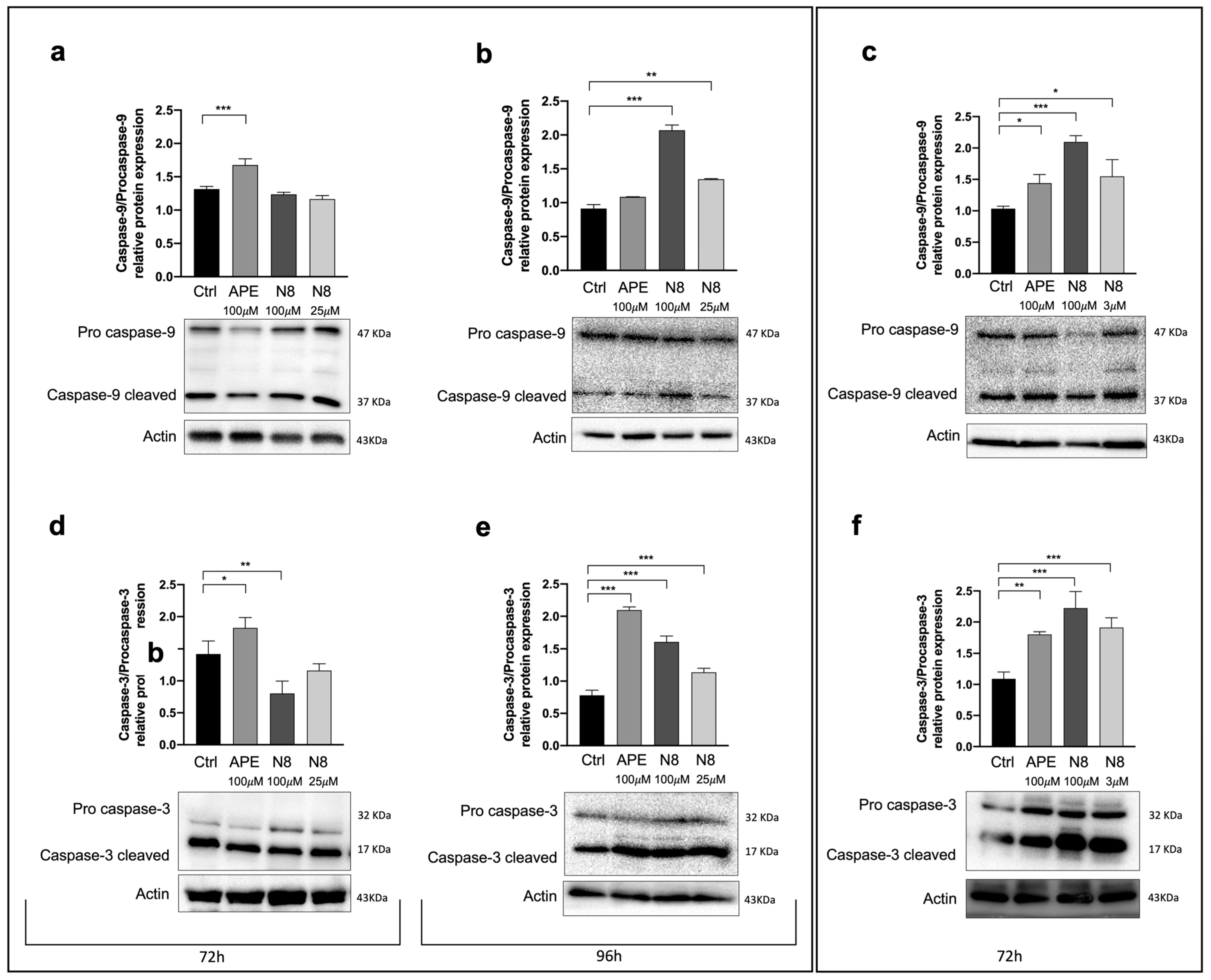
3.3. Analysis of Apoptosis Induction Following M2 mAChR Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a Molecular Target for Cancer Treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull. Du Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L. Autophagy: Supporting Cellular and Organismal Homeostasis by Self-Eating. Int. J. Biochem. Cell Biol. 2019, 111, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.-L. AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Mizushima, N. The Role of the Atg1/ULK1 Complex in Autophagy Regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.-Y.; et al. Autophagy. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 118, pp. 61–95. ISBN 978-0-12-407173-5. [Google Scholar]
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Vial, D.; McKeown-Longo, P.J. Role of EGFR Expression Levels in the Regulation of Integrin Function by EGF: EGFR LEVELS REGULATE INTEGRIN ACTIVATION. Mol. Carcinog. 2016, 55, 1118–1123. [Google Scholar] [CrossRef]
- Pirtoli, L.; Cevenini, G.; Tini, P.; Vannini, M.; Oliveri, G.; Marsili, S.; Mourmouras, V.; Rubino, G.; Miracco, C. The Prognostic Role of Beclin 1 Protein Expression in High-Grade Gliomas. Autophagy 2009, 5, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Patric, I.R.P.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation Silencing of ULK2, an Autophagy Gene, Is Essential for Astrocyte Transformation and Tumor Growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shinojima, N.; et al. Monitoring Autophagy in Glioblastoma with Antibody against Isoform B of Human Microtubule-Associated Protein 1 Light Chain 3. Autophagy 2008, 4, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Zhang, X.; Li, X.; Ren, Y.; Ma, X.; Li, X.; Wang, L. Association between AKT/mTOR Signalling Pathway and Malignancy Grade of Human Gliomas. J. Neurooncol. 2011, 103, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic Acetylcholine Receptors: Mutant Mice Provide New Insights for Drug Development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Sales, M.E.; Español, A.J.; Salem, A.R.; Pulido, P.M.; Sanchez, Y.; Sanchez, F. Role of Muscarinic Acetylcholine Receptors in Breast Cancer: Design of Metronomic Chemotherapy. CCP 2019, 14, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Taggi, M.; Kovacevic, A.; Capponi, C.; Falcinelli, M.; Cacciamani, V.; Vicini, E.; Canipari, R.; Tata, A.M. The Activation of M2 Muscarinic Receptor Inhibits Cell Growth and Survival in Human Epithelial Ovarian Carcinoma. J. Cell. Biochem. 2022, 123, 1440–1453. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Lu, A.; Arredondo, J.; Sauer, D.; Gravett, C.; Mark, G.P.; Grando, S.A.; Spindel, E.R. M3 Muscarinic Receptor Antagonists Inhibit Small Cell Lung Carcinoma Growth and Mitogen-Activated Protein Kinase Phosphorylation Induced by Acetylcholine Secretion. Cancer Res. 2007, 67, 3936–3944. [Google Scholar] [CrossRef]
- Lucianò, A.M.; Perciballi, E.; Fiore, M.; Del Bufalo, D.; Tata, A.M. The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 8433. [Google Scholar] [CrossRef]
- Calaf, G.M.; Crispin, L.A.; Muñoz, J.P.; Aguayo, F.; Bleak, T.C. Muscarinic Receptors Associated with Cancer. Cancers 2022, 14, 2322. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Cristofaro, I.; Di Bari, M.; Zasso, J.; Conti, L.; Tata, A.M. The Activation of M2 Muscarinic Receptor Inhibits Cell Growth and Survival in Human Glioblastoma Cancer Stem Cells. Int. Immunopharmacol. 2015, 29, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Bari, M.D.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 Receptor Activation Inhibits Cell Cycle Progression and Survival in Human Glioblastoma Cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Tombolillo, V.; Alessandrini, F.; Guerriero, C.; Fiore, M.; Asteriti, I.A.; Castigli, E.; Sciaccaluga, M.; Guarguaglini, G.; Degrassi, F.; et al. M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines. Cells 2021, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Fritz, J.J.; Wess, J.; Lah, J.J.; Levey, A.I. Deletion of M1 Muscarinic Acetylcholine Receptors Increases Amyloid Pathology in Vitro and in Vivo. J. Neurosci. 2010, 30, 4190–4196. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Kitazawa, M.; Caccamo, A.; Baglietto-Vargas, D.; Estrada-Hernandez, T.; Cribbs, D.H.; Fisher, A.; LaFerla, F.M. Loss of Muscarinic M1 Receptor Exacerbates Alzheimer’s Disease–Like Pathology and Cognitive Decline. Am. J. Pathol. 2011, 179, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Dencker, D.; Wörtwein, G.; Woldbye, D.P.D.; Cui, Y.; Davis, A.A.; Levey, A.I.; Schütz, G.; Sager, T.N.; Mørk, A.; et al. A Subpopulation of Neuronal M4 Muscarinic Acetylcholine Receptors Plays a Critical Role in Modulating Dopamine-Dependent Behaviors. J. Neurosci. 2010, 30, 2396–2405. [Google Scholar] [CrossRef] [PubMed]
- Español, A.J.; Salem, A.; Di Bari, M.; Cristofaro, I.; Sanchez, Y.; Tata, A.M.; Sales, M.E. The Metronomic Combination of Paclitaxel with Cholinergic Agonists Inhibits Triple Negative Breast Tumor Progression. Participation of M2 Receptor Subtype. PLoS ONE 2020, 15, e0226450. [Google Scholar] [CrossRef]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef]
- Bock, A.; Mohr, K. Dualsteric GPCR Targeting and Functional Selectivity: The Paradigmatic M(2) Muscarinic Acetylcholine Receptor. Drug Discov. Today Technol. 2013, 10, e245–e252. [Google Scholar] [CrossRef]
- Guerriero, C.; Matera, C.; Del Bufalo, D.; De Amici, M.; Conti, L.; Dallanoce, C.; Tata, A.M. The Combined Treatment with Chemotherapeutic Agents and the Dualsteric Muscarinic Agonist Iper-8-Naphthalimide Affects Drug Resistance in Glioblastoma Stem Cells. Cells 2021, 10, 1877. [Google Scholar] [CrossRef]
- Cristofaro, I.; Spinello, Z.; Matera, C.; Fiore, M.; Conti, L.; De Amici, M.; Dallanoce, C.; Tata, A.M. Activation of M2 Muscarinic Acetylcholine Receptors by a Hybrid Agonist Enhances Cytotoxic Effects in GB7 Glioblastoma Cancer Stem Cells. Neurochem. Int. 2018, 118, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, I.; Alessandrini, F.; Spinello, Z.; Guerriero, C.; Fiore, M.; Caffarelli, E.; Laneve, P.; Dini, L.; Conti, L.; Tata, A.M. Cross Interaction between M2 Muscarinic Receptor and Notch1/EGFR Pathway in Human Glioblastoma Cancer Stem Cells: Effects on Cell Cycle Progression and Survival. Cells 2020, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Baronchelli, S.; Bentivegna, A.; Redaelli, S.; Riva, G.; Butta, V.; Paoletta, L.; Isimbaldi, G.; Miozzo, M.; Tabano, S.; Daga, A.; et al. Delineating the Cytogenomic and Epigenomic Landscapes of Glioma Stem Cell Lines. PLoS ONE 2013, 8, e57462. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Merten, N.; Schrage, R.; Dallanoce, C.; Bätz, J.; Klöckner, J.; Schmitz, J.; Matera, C.; Simon, K.; Kebig, A.; et al. The Allosteric Vestibule of a Seven Transmembrane Helical Receptor Controls G-Protein Coupling. Nat. Commun. 2012, 3, 1044. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-Positive Structures Are Prominent in Autophagy-Deficient Cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Jia, B.; Xue, Y.; Yan, X.; Li, J.; Wu, Y.; Guo, R.; Zhang, J.; Zhang, L.; Li, Y.; Liu, Y.; et al. Autophagy Inhibitor Chloroquine Induces Apoptosis of Cholangiocarcinoma Cells via Endoplasmic Reticulum Stress. Oncol. Lett. 2018, 16, 3509–3516. [Google Scholar] [CrossRef]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The Role of Interaction between Autophagy and Apoptosis in Tumorigenesis (Review). Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.-P. Muscarinic Receptors and Ligands in Cancer. Am. J. Physiol.-Cell Physiol. 2009, 296, C221–C232. [Google Scholar] [CrossRef]
- Li, B.; Zhou, C.; Yi, L.; Xu, L.; Xu, M. Effect and Molecular Mechanism of mTOR Inhibitor Rapamycin on Temozolomide-induced Autophagic Death of U251 Glioma Cells. Oncol. Lett. 2017, 15, 2477–2484. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a Decisive Process for Cell Death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerriero, C.; Manfredelli, M.; Matera, C.; Iuzzolino, A.; Conti, L.; Dallanoce, C.; De Amici, M.; Trisciuoglio, D.; Tata, A.M. M2 Muscarinic Receptor Stimulation Induces Autophagy in Human Glioblastoma Cancer Stem Cells via mTOR Complex-1 Inhibition. Cancers 2024, 16, 25. https://doi.org/10.3390/cancers16010025
Guerriero C, Manfredelli M, Matera C, Iuzzolino A, Conti L, Dallanoce C, De Amici M, Trisciuoglio D, Tata AM. M2 Muscarinic Receptor Stimulation Induces Autophagy in Human Glioblastoma Cancer Stem Cells via mTOR Complex-1 Inhibition. Cancers. 2024; 16(1):25. https://doi.org/10.3390/cancers16010025
Chicago/Turabian StyleGuerriero, Claudia, Marianna Manfredelli, Carlo Matera, Angela Iuzzolino, Luciano Conti, Clelia Dallanoce, Marco De Amici, Daniela Trisciuoglio, and Ada Maria Tata. 2024. "M2 Muscarinic Receptor Stimulation Induces Autophagy in Human Glioblastoma Cancer Stem Cells via mTOR Complex-1 Inhibition" Cancers 16, no. 1: 25. https://doi.org/10.3390/cancers16010025
APA StyleGuerriero, C., Manfredelli, M., Matera, C., Iuzzolino, A., Conti, L., Dallanoce, C., De Amici, M., Trisciuoglio, D., & Tata, A. M. (2024). M2 Muscarinic Receptor Stimulation Induces Autophagy in Human Glioblastoma Cancer Stem Cells via mTOR Complex-1 Inhibition. Cancers, 16(1), 25. https://doi.org/10.3390/cancers16010025