

Therapeutic and Diagnostic Potential of Folic Acid Receptors and Glycosylphosphatidylinositol (GPI) Transamidase in Prostate Cancer

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Tissue Sections
2.3. Quantification of RNA Transcripts
2.3.1. RNA Isolation
2.3.2. cDNA Synthesis
2.3.3. qRT-PCR
2.4. Quantification of Proteins
2.4.1. Protein Isolation
2.4.2. Determination of Protein Concentration
2.4.3. SDS Page
2.4.4. Antibodies
2.4.5. Western Blot
2.4.6. Immunofluorescence Staining
Cell Culture
Tissue
2.5. Transfection
2.5.1. Preparation of Standard Lipoplexes—Lipofectamine 3000
2.5.2. Functionalization of Lipoplexes
2.5.3. Transfection and Immunofluorescence Staining
2.6. Microscopy
2.7. Flow Cytometry
2.7.1. FACS Canto II
2.7.2. LSRFortessa
2.8. Image Analysis
2.9. Statistical Analysis
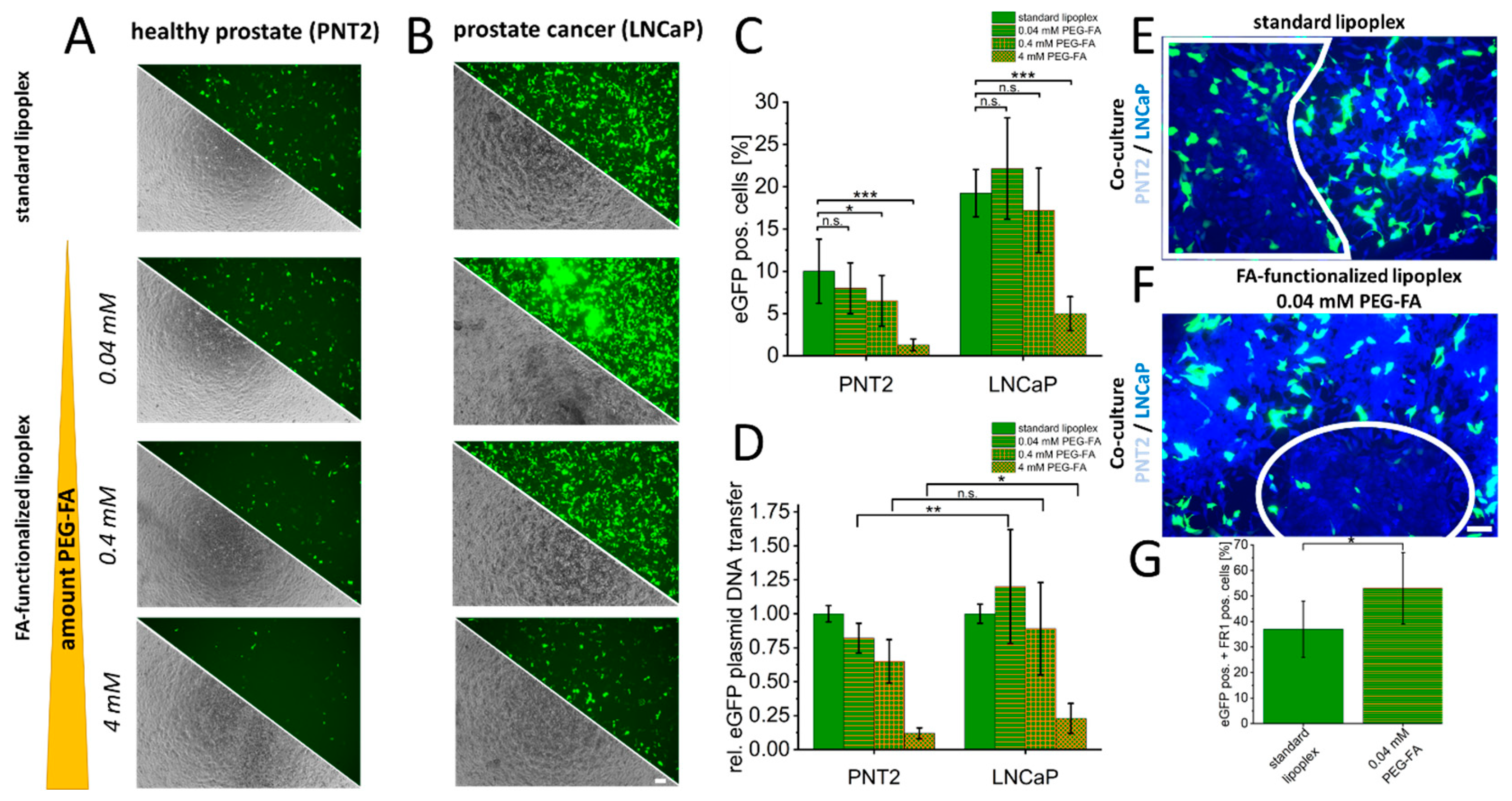
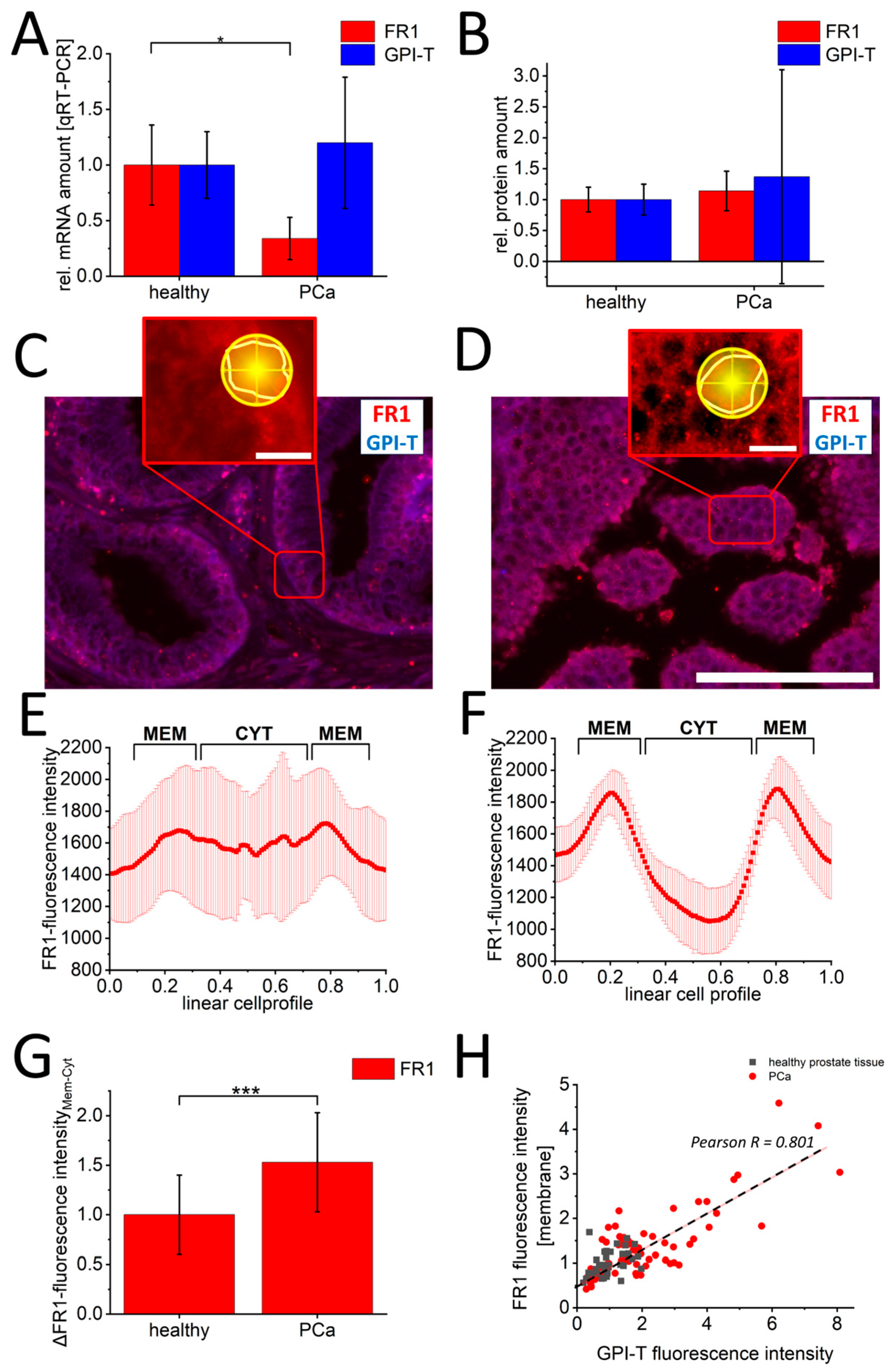
3. Results
4. Discussion
4.1. Expression Pattern of Folate Receptor in Prostate Cancer
4.2. Regulation of Folate Receptors in Prostate Cancer
4.3. Clinical Utility of Prostate Cancer Folate Receptor Activation
4.4. Limitations and Clinical Significance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Descotes, J.L. Diagnosis of prostate cancer. Asian J. Urol. 2019, 6, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Tsechelidis, I.; Vrachimis, A. PSMA PET in Imaging Prostate Cancer. Front. Oncol. 2022, 12, 831429. [Google Scholar] [CrossRef] [PubMed]
- Herawi, M.; Kahane, H.; Cavallo, C.; Epstein, J.I. Risk of prostate cancer on first re-biopsy within 1 year following a diagnosis of high grade prostatic intraepithelial neoplasia is related to the number of cores sampled. J. Urol. 2006, 175, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Zendejas, A.P.; Scavuzzo, A.; Jimenez-Rios, M.A.; Alvarez-Gomez, R.M.; Montiel-Manriquez, R.; Castro-Hernandez, C.; Jimenez-Davila, M.A.; Perez-Montiel, D.; Gonzalez-Barrios, R.; Jimenez-Trejo, F.; et al. The promising role of new molecular biomarkers in prostate cancer: From coding and non-coding genes to artificial intelligence approaches. Prostate Cancer Prostatic Dis. 2022, 25, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Javaid, A.; Amjad, F.; Youssif, T.A.; Afzal, S. An overview of prostate cancer (PCa) diagnosis: Potential role of miRNAs. Transl. Oncol. 2022, 26, 101542. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D. Combination therapy in metastatic hormone-sensitive prostate cancer: Is three a crowd? Ther. Adv. Med. Oncol. 2022, 14, 17588359221086827. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.R.; Thompson, T.C. Considering the potential for gene-based therapy in prostate cancer. Nat. Rev. Urol. 2021, 18, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef]
- Martin-Sabroso, C.; Torres-Suarez, A.I.; Alonso-Gonzalez, M.; Fernandez-Carballido, A.; Fraguas-Sanchez, A.I. Active Targeted Nanoformulations via Folate Receptors: State of the Art and Future Perspectives. Pharmaceutics 2021, 14, 14. [Google Scholar] [CrossRef]
- Zhao, R.; Diop-Bove, N.; Goldman, I.D. Enhanced receptor-mediated endocytosis and cytotoxicity of a folic acid-desacetylvinblastine monohydrazide conjugate in a pemetrexed-resistant cell line lacking folate-specific facilitative carriers but with increased folate receptor expression. Mol. Pharmacol. 2014, 85, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Gerlach, S.; Hoffmann, C.; Richter, N.; Hersch, N.; Csiszar, A.; Merkel, R.; Hoffmann, B. PEGylation and folic-acid functionalization of cationic lipoplexes-Improved nucleic acid transfer into cancer cells. Front. Bioeng. Biotechnol. 2022, 10, 1066887. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.; Elkhoury, K.; Velot, E.; Bianchi, A.; Acherar, S.; Francius, G.; Tamayol, A.; Grandemange, S.; Arab-Tehrany, E. Functionalized liposomes for targeted breast cancer drug delivery. Bioact. Mater. 2023, 24, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Yao, V.; Berkman, C.E.; Choi, J.K.; O’Keefe, D.S.; Bacich, D.J. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate 2010, 70, 305–316. [Google Scholar] [CrossRef]
- Bistulfi, G.; Diegelman, P.; Foster, B.A.; Kramer, D.L.; Porter, C.W.; Smiraglia, D.J. Polyamine biosynthesis impacts cellular folate requirements necessary to maintain S-adenosylmethionine and nucleotide pools. FASEB J. 2009, 23, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Marley, J. Campbell-Walsh Urology, 9th Edition (E-dition). Int. J. Urol. Nurs. 2007, 1, 94–95. [Google Scholar] [CrossRef]
- Patil, Y.; Shmeeda, H.; Amitay, Y.; Ohana, P.; Kumar, S.; Gabizon, A. Targeting of folate-conjugated liposomes with co-entrapped drugs to prostate cancer cells via prostate-specific membrane antigen (PSMA). Nanomedicine 2018, 14, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Alserihi, R.F.; Mohammed, M.R.S.; Kaleem, M.; Khan, M.I.; Sechi, M.; Sanna, V.; Zughaibi, T.A.; Abuzenadah, A.M.; Tabrez, S. Development of (−)-epigallocatechin-3-gallate-loaded folate receptor-targeted nanoparticles for prostate cancer treatment. Nanotechnol. Rev. 2022, 11, 298–311. [Google Scholar] [CrossRef]
- Singh, S.K.; Lillard, J.W., Jr.; Singh, R. Reversal of drug resistance by planetary ball milled (PBM) nanoparticle loaded with resveratrol and docetaxel in prostate cancer. Cancer Lett. 2018, 427, 49–62. [Google Scholar] [CrossRef]
- Lian, S.; Yang, L.; Feng, Q.; Wang, P.; Wang, Y.; Li, Z. Folate-Receptor Positive Circulating Tumor Cell Is a Potential Diagnostic Marker of Prostate Cancer. Front. Oncol. 2021, 11, 708214. [Google Scholar] [CrossRef]
- Nagpal, J.K.; Dasgupta, S.; Jadallah, S.; Chae, Y.K.; Ratovitski, E.A.; Toubaji, A.; Netto, G.J.; Eagle, T.; Nissan, A.; Sidransky, D.; et al. Profiling the expression pattern of GPI transamidase complex subunits in human cancer. Mod. Pathol. 2008, 21, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Gamage, D.G.; Hendrickson, T.L. GPI transamidase and GPI anchored proteins: Oncogenes and biomarkers for cancer. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 446–464. [Google Scholar] [CrossRef] [PubMed]
- Leung, F.; Dimitromanolakis, A.; Kobayashi, H.; Diamandis, E.P.; Kulasingam, V. Folate-receptor 1 (FOLR1) protein is elevated in the serum of ovarian cancer patients. Clin. Biochem. 2013, 46, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- van Dam, G.M.; Themelis, G.; Crane, L.M.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Sarantopoulos, A.; de Jong, J.S.; Arts, H.J.; van der Zee, A.G.; et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: First in-human results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, Y.; Yamaoka, T.; Ohba, M.; Miura, S.; Masuda, H.; Sangai, T.; Takimoto, M.; Nakamura, S.; Tsurutani, J. Novel Anti-FOLR1 Antibody-Drug Conjugate MORAb-202 in Breast Cancer and Non-Small Cell Lung Cancer Cells. Antibodies 2021, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, Y.V.; Davila, J.; Ortiz-Rivera, J.; Inyushin, M.; Almodovar, L.; Mayol, M.; Morales-Cruz, M.; Cruz-Montanez, A.; Barcelo-Bovea, V.; Griebenow, K.; et al. Targeted Delivery of Nanoparticulate Cytochrome C into Glioma Cells Through the Proton-Coupled Folate Transporter. Biomolecules 2019, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Boshnjaku, V.; Shim, K.W.; Tsurubuchi, T.; Ichi, S.; Szany, E.V.; Xi, G.; Mania-Farnell, B.; McLone, D.G.; Tomita, T.; Mayanil, C.S. Nuclear localization of folate receptor alpha: A new role as a transcription factor. Sci. Rep. 2012, 2, 980. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Yu, D. Chapter 8—Immunofluorescence. In Basic Science Methods for Clinical Researchers; Jalali, M., Saldanha, F.Y.L., Jalali, M., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 135–150. [Google Scholar]
- Rogers, S.L.; Rogers, G.C. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat. Protoc. 2008, 3, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.; Brandsma, M.; Janssen, G.M.; Dijk, J.; Moller, W. Immunofluorescence studies of human fibroblasts demonstrate the presence of the complex of elongation factor-1 beta gamma delta in the endoplasmic reticulum. J. Cell Sci. 1996, 109 Pt 5, 1113–1117. [Google Scholar] [CrossRef]
- Pang, Y.; Young, C.Y.F.; Yuan, H. MicroRNAs and prostate cancer. Acta Biochim. Biophys. Sin. 2010, 42, 363–369. [Google Scholar] [CrossRef]
- Ribas, J.; Lupold, S.E. The transcriptional regulation of miR-21, its multiple transcripts, and their implication in prostate cancer. Cell Cycle 2010, 9, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Ru, P.; Steele, R.; Newhall, P.; Phillips, N.J.; Toth, K.; Ray, R.B. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol. Cancer Ther. 2012, 11, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Steele, R.; Mott, J.L.; Ray, R.B. MBP-1 upregulates miR-29b that represses Mcl-1, collagens, and matrix-metalloproteinase-2 in prostate cancer cells. Genes Cancer 2010, 1, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Zhang, J.; Huang, G.; Zhang, Y.; Zhao, M.; Zhang, Y.; Xie, J. microRNA-29b inhibits cell growth and promotes sensitivity to oxaliplatin in colon cancer by targeting FOLR1. Biofactors 2020, 46, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar] [PubMed]
- Wibowo, A.S.; Singh, M.; Reeder, K.M.; Carter, J.J.; Kovach, A.R.; Meng, W.; Ratnam, M.; Zhang, F.; Dann, C.E., 3rd. Structures of human folate receptors reveal biological trafficking states and diversity in folate and antifolate recognition. Proc. Natl. Acad. Sci. USA 2013, 110, 15180–15188. [Google Scholar] [CrossRef] [PubMed]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Loura, L.M.; Prieto, M. FRET in Membrane Biophysics: An Overview. Front. Physiol. 2011, 2, 82. [Google Scholar] [CrossRef]
- Zhang, X.X.; Ni, B.; Li, Q.; Hu, L.P.; Jiang, S.H.; Li, R.K.; Tian, G.A.; Zhu, L.L.; Li, J.; Zhang, X.L.; et al. GPAA1 promotes gastric cancer progression via upregulation of GPI-anchored protein and enhancement of ERBB signalling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 214. [Google Scholar] [CrossRef]
- Cao, J.; Wang, P.; Chen, J.; He, X. PIGU overexpression adds value to TNM staging in the prognostic stratification of patients with hepatocellular carcinoma. Hum. Pathol. 2019, 83, 90–99. [Google Scholar] [CrossRef]
- Wu, G.; Guo, Z.; Chatterjee, A.; Huang, X.; Rubin, E.; Wu, F.; Mambo, E.; Chang, X.; Osada, M.; Sook Kim, M.; et al. Overexpression of glycosylphosphatidylinositol (GPI) transamidase subunits phosphatidylinositol glycan class T and/or GPI anchor attachment 1 induces tumorigenesis and contributes to invasion in human breast cancer. Cancer Res. 2006, 66, 9829–9836. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fujiwara, Y.; Yonemori, K.; Koyama, T.; Sato, J.; Tamura, K.; Shimomura, A.; Ikezawa, H.; Nomoto, M.; Furuuchi, K.; et al. First-in-Human Phase 1 Study of MORAb-202, an Antibody-Drug Conjugate Comprising Farletuzumab Linked to Eribulin Mesylate, in Patients with Folate Receptor-alpha-Positive Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3905–3915. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Itamochi, H. Profile of farletuzumab and its potential in the treatment of solid tumors. OncoTargets Ther. 2016, 9, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Wilson, L.; Azarenko, O.; Zhu, X.; Lewis, B.M.; Littlefield, B.A.; Jordan, M.A. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 2010, 49, 1331–1337. [Google Scholar] [CrossRef]
- TCGA Research Network. Available online: https://www.cancer.gov/tcga (accessed on 10 April 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer | Content | Manufacturer (Order ID.) | Amount |
|---|---|---|---|
| RIPA lysis buffer | TRIS HCl | Carl Roth, Karlsruhe, Germany (9090.2) | 2.42 g |
| NaCl | Carl Roth (3957.1) | 8.76 g | |
| Nonidet P 40-Replacement product solution | Merck (74388) | 20 mL | |
| SDS pellets | Carl Roth (CN30.2) | 1 g | |
| Deoxycholic acid sodium salt | Carl Roth (3484.2) | 5 g | |
| Demineralized water | Fill to 1 L | ||
| RIPA lysis buffer + inhibitors | Protease inhibitor | Merck (11873580001) | 1/2 tablet |
| Phosphatase inhibitor | Merck (P0044) | 50 µL | |
| RIPA lysis buffer | Fill to 5 mL |
| Secondary Antibody | StarBright Blue 700 Goat Anti-Rabbit IgG (Bio-Rad, 12004161) | StarBright Blue 520 Goat Anti-Mouse IgG (Bio-Rad, 12005866) | Goat-Anti Rabbit IgG Alexa Fluor 488 (Abcam, ab150116, Cambridge, UK) | Goat-Anti Mouse IgG Alexa Fluor 594 (Abcam, ab150077) | Goat Anti-Rabbit IgG Alexa Fluor 594 (Thermo Scientific, A-11012) | |
|---|---|---|---|---|---|---|
| Primary Antibody | ||||||
| GPI-T—PIGK (N-Term) (antikörper-online, ABIN389064) Occludin (invitrogen, 1529359A) | WB (Section 2.4.5): 1:10,000 | - | IF-c (Section 2.4.6) *: 1:400 IF-t (Section 2.4.6): 1:400 | - | IF-c-t (Section 2.5.3): 1:400 | |
| WB (Section 2.4.5): 1:1000 | IF-c (Section 2.4.6) *: 1:100 IF-t (Section 2.4.6): 1:50 | IF-c-t (Section 2.5.3): 1:100 | ||||
| FR1—FOLR1 (AA 41-227) (antikörper-online, ABIN5611335) | - | WB (Section 2.4.5): 1:10,000 | - | IF-c (Section 2.4.6): 1:400 IF-t (Section 2.4.6): 1:400 | - | |
| WB (Section 2.4.5): 1:1000 | IF-c (Section 2.4.6): 1:100 IF-t (Section 2.4.6): 1:50 | |||||
| Experiment | Channel | Magnification | Exposure Time [ms] | Gain |
|---|---|---|---|---|
| Immunofluorescence of cell culture (Section 2.4.6) | GFP | 40×/20× | 150 | 10 |
| RFP | 40×/20× | 150 | 10 | |
| DAPI | 40× | 10 | 10 | |
| Immunofluorescence of tissue (Section 2.4.6) | GFP | 40× | 538 | 10 |
| RFP | 40× | 4180 | 30 | |
| DAPI | 40× | 513 | 10 | |
| Transfection (Section 2.5.1/Section 2.5.2) | GFP | 4× | 18 | 50 |
| Transfection and immunofluorescence staining (Section 2.5.3) | GFP | 10× | 18 | 37 |
| RFP | 10× | 164 | 89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, M.; Ermler, T.F.; Hoffmann, F.; Alexa, R.; Kranz, J.; Steinke, N.; Leypold, S.; Gaisa, N.T.; Saar, M. Therapeutic and Diagnostic Potential of Folic Acid Receptors and Glycosylphosphatidylinositol (GPI) Transamidase in Prostate Cancer. Cancers 2024, 16, 2008. https://doi.org/10.3390/cancers16112008
Hoffmann M, Ermler TF, Hoffmann F, Alexa R, Kranz J, Steinke N, Leypold S, Gaisa NT, Saar M. Therapeutic and Diagnostic Potential of Folic Acid Receptors and Glycosylphosphatidylinositol (GPI) Transamidase in Prostate Cancer. Cancers. 2024; 16(11):2008. https://doi.org/10.3390/cancers16112008
Chicago/Turabian StyleHoffmann, Marco, Thomas Frank Ermler, Felix Hoffmann, Radu Alexa, Jennifer Kranz, Nathalie Steinke, Sophie Leypold, Nadine Therese Gaisa, and Matthias Saar. 2024. "Therapeutic and Diagnostic Potential of Folic Acid Receptors and Glycosylphosphatidylinositol (GPI) Transamidase in Prostate Cancer" Cancers 16, no. 11: 2008. https://doi.org/10.3390/cancers16112008
APA StyleHoffmann, M., Ermler, T. F., Hoffmann, F., Alexa, R., Kranz, J., Steinke, N., Leypold, S., Gaisa, N. T., & Saar, M. (2024). Therapeutic and Diagnostic Potential of Folic Acid Receptors and Glycosylphosphatidylinositol (GPI) Transamidase in Prostate Cancer. Cancers, 16(11), 2008. https://doi.org/10.3390/cancers16112008