Simple Summary
This study explores hypoxia-inducible factors (HIFs) in glioblastoma development, progression, and treatment. Reviewing 104 relevant studies, it highlights diverse global contributions, with China leading at 23.1%. The most productive year was 2019, contributing 11.5% of the studies. Key factors studied included HIF1α, HIF2α, osteopontin, and cavolin-1, involving pathways such as GLUT1, GLUT3, VEGF, PI3K-Akt-mTOR, and ROS. HIF expression correlates with glioblastoma progression, survival, neovascularization, glucose metabolism, migration, and invasion. Overcoming treatment resistance and the lack of biomarkers is crucial for integrating HIF-related therapies into glioblastoma treatment to improve patient outcomes.
Abstract
Background: The study aims to investigate the role of hypoxia-inducible factors (HIFs) in the development, progression, and therapeutic potential of glioblastomas. Methodology: The study, following PRISMA guidelines, systematically examined hypoxia and HIFs in glioblastoma using MEDLINE (PubMed), Web of Science, and Scopus. A total of 104 relevant studies underwent data extraction. Results: Among the 104 studies, global contributions were diverse, with China leading at 23.1%. The most productive year was 2019, accounting for 11.5%. Hypoxia-inducible factor 1 alpha (HIF1α) was frequently studied, followed by hypoxia-inducible factor 2 alpha (HIF2α), osteopontin, and cavolin-1. Commonly associated factors and pathways include glucose transporter 1 (GLUT1) and glucose transporter 3 (GLUT3) receptors, vascular endothelial growth factor (VEGF), phosphoinositide 3-kinase (PI3K)-Akt-mechanistic target of rapamycin (mTOR) pathway, and reactive oxygen species (ROS). HIF expression correlates with various glioblastoma hallmarks, including progression, survival, neovascularization, glucose metabolism, migration, and invasion. Conclusion: Overcoming challenges such as treatment resistance and the absence of biomarkers is critical for the effective integration of HIF-related therapies into the treatment of glioblastoma with the aim of optimizing patient outcomes.
1. Introduction
Glioblastoma is a highly aggressive grade 4 glioma with an annual incidence of approximately six cases per 100,000 persons in older adults and a 15–20% proportion of all brain tumors in pediatric patients. In children, glioblastoma is highly invasive and leads to an 80% recurrence rate within two years of treatment. Survival rates are dismal, with less than 2% of the adults surviving more than three years after diagnosis [1,2,3,4].
Recent advances favor molecular analysis for the prognosis of glioblastoma, especially in younger patients where molecular factors are more important than histological grading. Biomarkers such as isocitrate dehydrogenase (IDH) mutations and O6-methylguanine DNA methyltransferase (MGMT) methylation status support prognosis [5]. However, the final diagnosis of glioblastoma depends on the surgical biopsy, which is crucial for the detection of hypoxic tumor niches manifested by vascular proliferation and tissue necrosis. Hypoxia, which is prevalent in solid tumors such as glioblastoma, is due to reduced oxygen levels, which are particularly dangerous in the oxygen-dependent brain [6]. Tumor progression exacerbates hypoxia and leads to uncontrolled neovascularization that perpetuates the cycle of inadequate oxygen supply. Hypoxia-induced angiogenesis is typical of the progression that occurs in escalation-grade astrocytomas and is characterized by central necrosis and pseudo-palisades on magnetic resonance imaging (MRI) scans [7].
Hypoxia-inducible factor (HIF) emerges as a key molecule in promoting neovascularization in hypoxic niches, which is critical for tumor progression [8]. To date, the involvement of the hypoxic microenvironment in carcinogenesis has been extensively validated across various tumor types [9], particularly in pancreatic cancer, wherein hypoxic conditions have been shown to facilitate metastasis and drug resistance [10]. The HIF1α and HIF-1β subunits form an active heterodimer that initiates the transcription of over 40 hypoxia-responsive genes, including erythropoietin (EPO), insulin-like growth factor 2 (IGF2), vascular endothelial growth factor (VEGF), and angiopoietin (Ang)-1 and -2 [11]. HIF also upregulates platelet-derived growth factor (PDGF) proteins and activates oncogenic signaling pathways such as MAPK/RAS and PI3K/AKT [9]. HIF1α responds acutely to hypoxia, while HIF2α regulates tumor cell response to chronic hypoxia, making it a potential therapeutic target. BEV targeting VEGF-A shows promise in inhibiting HIF1α, especially in patients with chemoresistance [12,13]. However, the histologic and molecular heterogeneity of glioblastoma poses a challenge and requires research into novel multimodal therapies targeting hypoxia and HIF signaling pathways, including immunotherapy and nanoscale drug delivery [12,13]. Therefore, the aim of this systematic review is to investigate the role of hypoxia and HIFs in the development, progression, and therapeutic potential of glioblastoma.
2. Materials and Methods
2.1. Study Design and Registration
A systematic review of the literature was conducted to investigate the role of hypoxia and HIFs in glioblastoma. The methodology followed the established PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines [14]. This systematic review has been registered in the Open Science Framework (OSF) Register with the unique identifier OSF-REGISTRATIONS-8GD9K-V1 [15].
2.2. Search Strategy
On 15 January 2024, a search was conducted using the PICOS method to define the main search terms (Table 1). Three databases were searched: MEDLINE (PubMed), Web of Science (Clarivate Analytics, Philadelphia, PA, USA), and Scopus. The keywords “glioblastoma” and “hypoxia-inducible factors” were searched. A detailed search strategy can be found in Appendix A. Following the PRISMA guidelines, a checklist can be found in Appendix B.

Table 1.
PICOS strategy.
2.3. Study Selection
2.3.1. Inclusion and Exclusion Criteria
Strict inclusion and exclusion criteria were applied when conducting this systematic review to ensure the selection of relevant articles that contribute significantly to the understanding of the role of HIFs in glioblastoma. The inclusion criteria focused on articles that were written in English, directly related to the interaction between HIFs and glioblastoma, and contained data relevant to the objectives of the study. Conversely, the exclusion criteria were carefully defined to refine the selection process and exclude articles that may not align with the aims of the study. Excluded articles included book chapters, conference papers, reviews, non-English language literature, and articles that did not contain relevant data.
2.3.2. Included Studies
A total of 1318 entries were identified from PubMed (n = 558), Web of Science (n = 89), and Scopus (n = 671). Prior to the screening, 814 duplicate entries were removed using the EndNote software (21.3) for referencing. After automatic deduplication, all the remaining duplicate manuscripts were manually excluded. After this first step, 504 records were screened, and 36 records were excluded because they could not be found. Subsequently, 468 records were screened for eligibility, resulting in the exclusion of 84 book or book chapters, 57 conference papers, 49 reviews, 29 articles from non-English literature, and 145 articles without relevant data. Finally, 104 studies were deemed suitable and included in the systematic re-examination for analysis (Figure 1).
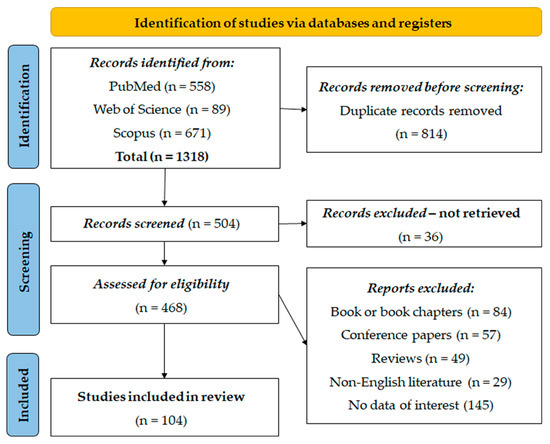
Figure 1.
PRISMA flow diagram.
2.4. Data Extraction
Data extraction from the studies was performed for didactic purposes, whereby the studies were divided into laboratory and clinical studies based on the tracking of different variables. The laboratory studies were further divided into genetic studies and drug-related studies as well as combined studies. For genetic studies, variables such as authors, country (year), study design, species, cell line(s), targeted HIF, related factors, role of HIF and related factors, gene modification, and the effect of gene modification were tracked. For drug studies, variables such as reference, country (year), study design, species, cell line(s), targeted HIF, related factors, role of HIF and related factors, target/system therapy, and pharmacologic effects were monitored. Combined laboratory studies tracked similar variables along with targeted therapy and pharmacologic effects. Clinical studies were reviewed for variables such as authors, country (year), study design, sample size (N), age, gender distribution, targeted HIF(s), and outcomes. The studies were first extracted into a single file using the EndNote software and then deduplicated. Data extraction was performed by eight researchers under the supervision of three senior researchers. Ambiguities in data extraction were resolved through online meetings and a final consensus.
2.5. Statistical Analysis
Descriptive statistics were used to present frequencies and absolute numbers, providing a quantitative summary of various key factors associated with the use of HIFs in glioblastoma. To improve the clarity and interpretation of results, graphical visualization was performed using Microsoft Excel (version 2021, Microsoft Corporation, Washington, DC, USA). BioRender (https://www.biorender.com/, accessed on 10 April 2024) license number RY26P9F0AG was used to design the scientific illustrations in the manuscript.
3. Results
3.1. Included Studies’ Characteristics
Among the 104 studies, contributions came from various countries. Studies from Canada, Egypt, Morocco, South Korea, Israel, the Netherlands, and Turkey together made up 1% of the total. Brazil, France, and Korea each contributed three studies (2.9%). The UK contributed four studies (3.8%), while Germany, India, and Japan each contributed five studies (4.8%). Italy contributed six studies (5.8%), and Taiwan contributed eight studies (7.7%). Significant contributions came from China with 24 studies (23.1%) and the United States with 28 studies (26.9%) (Figure 2).
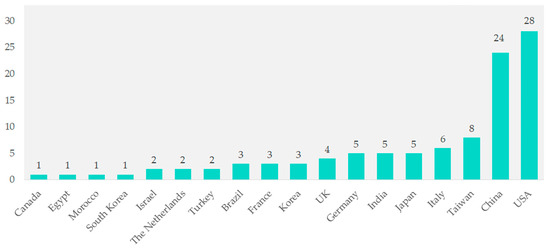
Figure 2.
Geographical distribution of included studies.
The years 2000, 2004, 2007, and 2009 each contributed 1%. In contrast, 2019 had 12 studies (11.5%), followed by 2018 with 11 studies (10.6%). From 2012 to 2022, annual contributions ranged from 8.7% to 9.6%. In 2021, the contribution was 7.7%, as shown in Figure 3.
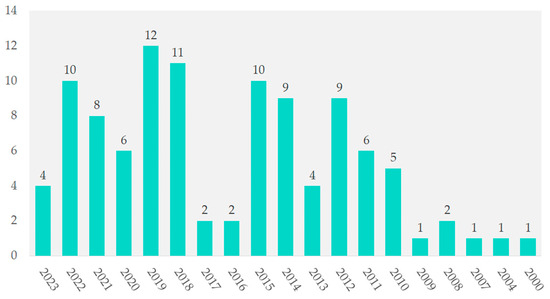
Figure 3.
Temporal distribution of included studies.
Most of the studies (90.4%) were laboratory-based, with 58.7% combining in vitro and in vivo methods. Pure in vitro studies made up 31.7%. Clinical research was less common, comprising 9.6% of the total, with prospective studies at 6.7% and retrospective studies at 2.9%, as shown in Figure 4.
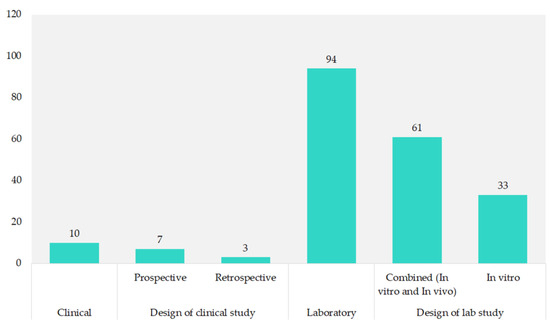
Figure 4.
Study design of included studies.
3.2. Role of HIF-Related Gene Modification in the Treatment of Glioblastoma
Table 2 shows 48 of the 94 laboratory studies (51%) that investigated the role of HIFs using different genetic methods in animal species and glioblastoma models. All the studies investigated HIF1α, while four studies also investigated the role of HIF2α in addition to HIF1α [14,15,16,17]. Among the included studies, deletion, overexpression, and transduction each accounted for 2.08% of the total (N = 1). Combined techniques and knockout methods each accounted for 6.25% (N = 3), while transfection was used in 22.92% of the studies (N = 11). Knockdown techniques in particular accounted for the majority of the research studies (58.33%, N = 28).
Table 2.
Laboratory studies with gene modification of HIFs in glioblastoma models and inoculated animals.
3.2.1. HIF’s Mechanisms Explored in Genetic Studies
The included studies have shed light on the complicated mechanisms in which HIF1α is involved. For example, Hashimoto et al. [16] have shown that AMPK boosts ATM expression via the transcription factor Sp1 under severe hypoxia, contributing to radioresistance. Conversely, Ho et al. [17] elucidated the role of MIR210HG in hypoxia-mediated glioma invasion and stemness formation, which is regulated by OCT1 and affects the expression of IGFBP2 and FGFR1. In addition, Ishikawa et al. [18] showed that HIF1α activates Ror1 transcription in glioblastoma and affects cancer progression by regulating cell proliferation and migration. Other related factors studied include miR-210-3p by Agrawal et al. [19]; CXCR7, CXCR4, and IDH1 by Bianco et al. [20]; C-Met and SF/HGF by Eckerich et al. [21]; S100A4/NMIIA axis by Inukai et al. [22]; NO and VEGF by Kimura et al. [23]; BAG3 by Li et al. [24]; and tryptophan 2,3-dioxygenase (TDO2) by Mohapatra et al. [27].
Studies have shown that HIF1α and HIF2α orchestrate several cellular processes that are crucial for the pathogenesis of glioblastoma. For example, HIF1α is involved in promoting radioresistance by modulating AMPK-mediated ATM expression [16], promoting glioma invasion and stem cell formation via regulating MIR210HG and OCT1 [16], and activating Ror1 transcription to influence cancer progression [18]. In addition, HIF1α is involved in the regulation of miR-210-3p, CXCR7, CXCR4, IDH1, C-Met, SF/HGF, the S100A4/NMIIA axis, NO, VEGF, BAG3, and TDO2, and influences various aspects of the glioblastoma biology such as angiogenesis, invasion, metabolism, and therapy resistance. Similarly, HIF2α contributes to glioblastoma progression by regulating genes such as GPx1, vasorin, beclin-1, and galectin-3, thereby influencing the response to oxidative stress, angiogenesis, autophagy, and cell survival.
3.2.2. Effect of Gene Modifications Related to HIFs
The collective results of various studies emphasize the multiple roles of HIFs and related factors in the pathogenesis of glioblastoma. For example, HIF1α was found to orchestrate regeneration resistance by upregulating AMPK-mediated ATM expression in severe hypoxia [16], while it promotes glioma invasion and stem cell formation by regulating MIR210HG and OCT1 [17]. In addition, HIF1α has been associated with the activation of Ror1 transcription to influence cancer progression [18], and it regulates miR-210-3p, CXCR7, CXCR4, IDH1, C-Met, SF/HGF, the S100A4/NMIIA axis, NO, VEGF, BAG3, and TDO2, and influences various aspects of the glioblastoma biology such as angiogenesis, invasion, metabolism and therapy resistance [19,20,21,22,23,24,25,26,27,28,30,31,32,33,34]. Similarly, HIF2α has been shown to regulate GPx1 to ensure resistance to oxidative stress and radiation [59], while contributing to glioblastoma progression via various mechanisms such as the DDX28-mediated regulation of eIF4E2-driven translation [49]. In addition, other factors such as miR-370-3p [61], NIX-mediated mitophagy [62], and p21 (CDKN1A)[63] have been identified as crucial players in glioblastoma pathogenesis, highlighting the complex interplay of genetic alterations in shaping the aggressive behavior of glioblastoma cells in the hypoxic tumor microenvironment.
3.3. Role of HIF-Related Targeted and Systematic Therapy of Glioblastoma
Table 3 shows a total of 26 laboratory studies addressing targeted and systemic therapies for glioblastoma in animal species with inoculated tumors or glioblastoma models. HIF1α was investigated in 25 of the 26 studies, while HIF2α was investigated in two studies.
Table 3.
Experimental investigations on hypoxia-inducible factors (HIFs) in glioblastoma models and animal subjects with induced tumors.
A plethora of studies clarify the different roles of HIF1α in the progression of glioblastoma and response to treatment. Nardinocchi et al. [64] demonstrated the zinc-induced degradation of HIF1α, which inhibits VEGF-mediated signaling pathways and improves cancer therapies. Maugeri et al. [65] emphasized the role of HIF1α in angiogenesis via the upregulation of VEGF, while Ma et al. [66] linked the overexpression of HIF1α to glucose metabolism, suggesting its involvement in metabolic adaptations. D’Amico et al. [67] showed that ADNP modulates the HIF signaling pathway and reduces VEGF secretion and migration. In addition, D’Alessio et al. [68] pointed to antiangiogenic therapy targeting HIF1α and related factors to inhibit neoangiogenic events in glioblastoma.
Several related factors influence HIF1α-mediated signaling pathways in glioblastoma. These include VEGF, which is influenced by zinc, as shown by Nardinocchi et al. [64], and PA-CAP, as shown by Maugeri et al. [65], suggesting its role in regulating angiogenesis. Ma et al. [64] highlighted the association of HIF1α with glucose metabolism through the upregulation of GLUT-1, GLUT-3, and HK2. D’Amico et al. [67] revealed the modulation of the HIF signaling pathway by ADNP, reducing VEGF secretion and migration. Other factors such as M2 receptors, CXCR4, POL5551, LonP1, CT-L, PPARα, and SUMO are involved in regulating various aspects of glioblastoma progression and response to therapy, as noted by Cristofaro et al. [69], Gagner et al. [89], Douglas et al. [72], Hofstetter et al. [76], and Bernstock et al. [83]. In addition, Lin et al. [71] highlighted the far-reaching influence of HIF1α on tumor cell behavior, while Lin et al. [88] investigated the regulation of pH-regulatory proteins in glioblastoma by hypoxia-induced HIF1α. In the field of glioblastoma therapy, various targeted and systematic approaches have emerged to target the complex signaling pathways mediated by HIF1α. As noted by Nardinocchi et al. [64], zinc induces the proteasomal degradation of HIF1α and could thus prevent tumor progression by suppressing VEGF, MDR1, and Bcl2 signaling pathways. PACAP, identified by Maugeri et al. [65], is promising as it inhibits the release of VEGF and thus prevents the formation of new vessels in the hypoxic microenvironment of glioblastoma. Ma et al. [64] showed that acriflavine in combination with PDT effectively suppresses HIF1α expression and increases the efficacy of PDT against glioblastoma. D’Amico et al. [67] showed that ADNP can modulate the HIF signaling pathway to decrease VEGF secretion and migration, which is a targeted therapy approach. Gagner et al. [89] demonstrated the potential of the combination of B20-4.1.1 and POL5551 in reducing glioma invasion and tumor spread. As noted by Arienti et al. [73], HBO shows promise in inhibiting proliferation, downregulating HIF1α expression, and reprogramming glucose metabolism, offering the potential for the systemic therapy of glioblastoma.
3.4. Role of Combined Gene and Targeted or Systematic Therapy of Glioblastoma
A total of 23 studies used a combined gene-modifying design and targeted or systematic therapy in the context of HIF in laboratory glioblastoma models (Table 4). All studies investigated HIF1α, while five studies also investigated the role of HIF2α in addition to HIF1α. Gene modification techniques included transduction (N = 1; 4.3%), combined techniques (N = 4; 17.4%), transfection (N = 8; 34.8%), and knockdown (N = 10; 43.5%).
Table 4.
Experimental investigations on hypoxia-inducible factors (HIFs) in glioblastoma models and animal subjects with induced tumors with combined genetic and targeted/systematic therapy.
Several studies have elucidated the multiple roles of HIFs in the pathogenesis and therapy of glioblastoma. Huang et al. [90] showed that the PI3K/Akt/mTOR/HIF1α signaling pathway enhances glioblastoma cell migration and invasion under hypoxia, with mTOR pathway siRNA suppressing these effects. Chhipa et al. [91] showed that the activation of the AMPK/CREB1 axis supports glioblastoma cell bioenergetics by increasing HIF1α transcription. Pang et al. [92] highlighted the role of HIF1α-regulated lysosomal protease LGMN in TAMs and showed that its blockade prolongs survival in glioblastoma models. Hu et al. [113] identified HIF1α and AMPK as the regulators of hypoxia-induced LC3 changes, BNIP3 expression, and p62 degradation, which affect autophagy and responsiveness to bevacizumab. Barliya et al. [95] linked Hsp90 to angiogenesis, migration, and invasion, and highlighted its mediation of the HIF1α-driven signaling pathways. Hsieh et al. [103] demonstrated Nox4-mediated ROS production under cyclic hypoxia, which affects HIF1α activity and tumor growth. Kannappan et al. [97] showed that NF-kB/HIF1α/HIF2α promotes EMT and metastasis. Joseph et al. [98] elucidated the HIF1α-ZEB1 axis in mesenchymal transition and invasion. These findings emphasize the complex involvement of HIFs in glioblastoma progression and point to potential therapeutic targets.
Several drugs targeting HIF1α and related signaling pathways have been evaluated for their effects on glioblastoma. Huang et al. [90] showed that inhibitors such as 2-mercaptoethanol, LY294002, rapamycin, and p70S6K siRNA inhibited the PI3K/Akt/mTOR signaling pathway and suppressed migration, invasion, and HIF1α expression in glioblastoma cells. Chhipa et al. [89] showed that AMPK inhibitor (bafilomycin) decreased the viability of glioblastoma stem cells (GSCs), while Pang et al. [100] found that anti-PD1 antibody synergistically blocked the HIF1α-LGMN axis with anti-PD1 therapy in glioblastoma. Hu et al. [113] showed that BEV and chloroquine reversed hypoxia-induced growth by increasing BNIP3 expression and blocking autophagy, respectively, while Kannappan et al. [95] showed that disulfiram selectively targeted hypoxia-induced GSCs and digoxin inhibited HIF1α mRNA translation. Other drugs such as chlorpromazine, echinomycin, fenofibrate, and R50922/R59949 inhibit glioblastoma growth via several mechanisms, including the blockade of dopamine signaling, interference with the HIF1α-PDGFD/PDGFRα-AKT pathway, and the induction of apoptosis [99,100,101,102]. Tempol and YC-1 inhibit tumor growth by blocking ROS production and the induction of ABCB1, respectively [92,101]. In addition, digitoxin reduces HIF1α protein accumulation, while AMD3100 increases radiosensitivity by inhibiting SDF-1/CXCR4 interactions [105,111].
3.5. Role of HIFs in Clinal Studies of Glioblastoma
Nine studies have investigated the expression and clinical significance of HIFs in glioblastoma (Table 5). Chen et al. [114] found that HIF1α expression correlated with high caveolin-1 (CAV1) expression, larger glioblastoma size, and shorter survival time. Bache et al. [115] observed higher expression of HIF2α, carbonic anhydrase 9 (CA9), vascular endothelial growth factor (VEGF), and other markers in glioblastoma compared to tumor-free brain tissue, with mRNA levels correlating with shorter survival. Erpolat et al. [116] reported that high levels of cytoplasmic and nuclear HIF1α and CA9 were associated with shorter survival, especially in patients with high hypoxia scores. Clara et al. [115] found that HIF1α expression correlated with increased vascular density, VEGF, and platelet-derived growth factor-C (PDGF-C) and survival. Other studies, such as those by Kaynar et al. [117] and Nobuyuki et al. [118], also emphasized the role of HIF1α in angiogenesis and radioresistance in glioblastoma. In addition, Ji et al. [119] showed that high HIF1α expression correlates with poorer outcomes and shorter survival, suggesting its potential as a prognostic marker. Sfifou et al. [120] found that negative HIF1α expression in conjunction with the positive expression of isocitrate dehydrogenase 1 (IDH1) was associated with a better prognosis. Potharaju et al. [121] observed the strong nuclear staining of HIF1α in a significant proportion of samples, which independently correlated with poor prognosis, especially in combination with the high expression of telomerase reverse transcriptase (TERT).
Table 5.
Included clinical studies.
3.6. Common HIF-Related Pathways in Glioblastoma
Glioblastoma involves a complex interplay of molecular signaling pathways, among which the PI3K/Akt/mTOR pathway stands out. This signaling pathway exerts a profound influence on the progression of glioblastoma and modulates important cellular processes such as migration, invasion, and the expression of HIF1α. The importance of this pathway is further emphasized by the fact that it can be modulated by PTEN-PI3K interactions, offering potential therapeutic opportunities (Figure 5). The intricate relationship between HIF1α and metabolic pathways adds another layer of complexity. HIF1α not only affects glucose metabolism by upregulating the glucose transporters GLUT-1 and GLUT-3 but also enhances glycolysis through the overexpression of hexokinase 2 (HK2). This metabolic switch contributes to the robustness of glioblastoma cells and allows them to thrive in the hypoxic tumor microenvironment. In addition, the therapeutic landscape in glioblastoma is evolving with the emergence of new strategies targeting HIF1α-related axes. The synergistic blockade of the HIF1α-LGMN axis, aided by AMPK inhibition and anti-PD1 antibody therapy [92], represents a promising approach to interrupting glioblastoma progression. Furthermore, interventions targeting VEGF [64,67,68,84,86], such as digoxin, offer potential opportunities to inhibit angiogenesis and overcome multidrug resistance (MDR) mediated by pathways involving Bcl2 [64]. Understanding and interfering with these pathways are key to developing more effective treatments for glioblastoma, a disease with poor prognosis and limited therapeutic options.
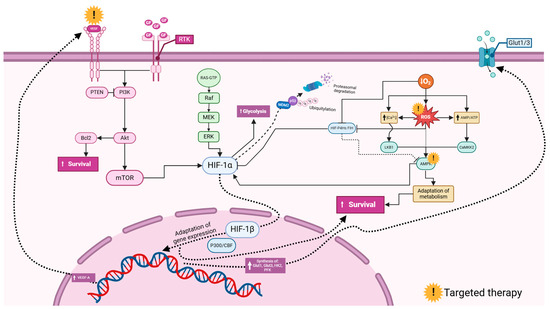
Figure 5.
Commonly investigated signaling pathways involving hypoxia-inducible factors (HIFs) in glioblastoma. Vascular endothelial growth factor (VEGF) and growth factors (GFs) activate Receptor Tyrosine Kinases (RTKs), triggering downstream signaling. The PI3K (Phosphoinositide 3-Kinase) pathway, inhibited by PTEN (Phosphatase and Tensin Homolog), activates Akt, leading to enhanced cell survival via the Bcl2 inhibition of apoptosis and mTOR (mechanistic target of rapamycin) promotion of growth. The RAS/MAPK (Mitogen-Activated Protein Kinase) pathway, through Raf, MEK, and ERK, also supports cell proliferation and survival. HIF-1α (hypoxia-inducible factor 1-alpha) under hypoxia increases glycolysis for energy production and gene expression for adaptation, regulated by HIF-1β (hypoxia-inducible factor 1-beta). HIF-1α stability is controlled by ubiquitination via MDM2 (Mouse Double Minute 2) and proteasomal degradation influenced by p53. Reactive oxygen species (ROS) and Calcium ion (Ca2+) signaling activate survival pathways, involving LKB1 (Liver Kinase B1), AMPK (AMP-activated Protein Kinase), and CaMK2 (Calcium/Calmodulin-Dependent Protein Kinase II). Glucose transporters Glut1/3 facilitate glucose uptake. This network highlights potential therapeutic targets, such as mTOR, PI3K, and HIF-1α, to disrupt glioblastoma cell survival and adaptation mechanisms. The figure was created using the BioRender online commercial platform.
4. Discussion
4.1. Research Trends
The high morbidity and mortality rate of glioblastoma has led to conventional treatments such as surgery, radiotherapy, and chemotherapy being re-evaluated due to their limited effectiveness. Researchers around the world, particularly in the United States and China, are exploring new treatments and incorporating molecular genetic features into diagnostics to better understand the pathogenesis of glioblastoma [2,124]. Despite an increase in in vitro and in vivo studies focusing on hypoxia-regulated genes, clinical trials remain limited, accounting for only 9.6% of the total. Advances in diagnostic methods, particularly next-generation sequencing, have led to significant growth in research [125,126]. However, the translation of promising laboratory results into clinical practice is challenging due to small sample sizes and geographic variation, making it difficult to develop standardized global diagnostic and treatment algorithms [66,116,121,123].
4.2. The Impact of HIF-Related Gene Modification on Glioblastoma Therapeutics
The importance of the knockdowns and knockouts of hypoxia-inducible factors lies in their ability to reveal the precise roles and functions of these factors in cellular processes and disease progression [127]. By elucidating the effects of manipulating hypoxia-inducible factors on glioblastoma progression, these techniques provide insights into potential therapeutic targets. Key findings include the functional importance of the interaction of N-cadherin and β-catenin on the radioresistance of glioblastoma stem cells. Elevated glucose-6-phosphatase (G6PC) levels contribute to resistance to glycolytic inhibition in glioblastoma cells [128]. AMPKα1 knockout affects glycolysis and tumorigenesis in a lymphoma mouse model. The overexpression of HHIF2α in AMPK knockdown GSCs possibly compensates for the loss of HIF1α. AMPKα knockdown decreases the expression of Sp1 and ATM under severe hypoxia and reduces radioresistance [91]. Moreover, the overexpression of MIR210HG enhances IGFBP2 and FGFR1 promoter activities under normoxia, which is inhibited by the suppression of OCT1, and decreases under hypoxia with MIR210HG or OCT1 knockdown. These findings emphasize the multifaceted role of hypoxia-inducible factors in glioblastoma, which includes radioresistance, migration, the regulation of gene expression, and metabolic processes [17].
Laboratory-based studies, such as those listed in Table 3, involve experimental manipulations and investigations performed on cells or animal models. This controlled environment allows researchers to isolate specific mechanisms, control variables, and collect preliminary data on the effects of hypoxia-inducible factors on glioblastoma. However, human clinical trials are challenging due to ethical considerations, difficulties in obtaining tumor samples, the heterogeneity of the patient population, and the complexity of studying hypoxia-inducible factors in the clinical setting [129]. Although laboratory-based studies provide valuable insights, they cannot fully reflect the complexity of human glioblastoma. Therefore, further research with human clinical trials is essential to validate the laboratory results and determine the clinical significance of hypoxia-inducible factors in glioblastoma.
4.3. Exploring HIF-Related Targeted and Systemic Therapies for Glioblastoma in Experimental Settings
Given the central role of HIF-1 in the pathophysiology of glioblastoma, the identification of a specific HIF-1 inhibitor holds promise for overcoming resistance to cytotoxic therapy and improving overall survival. Zinc is a potential candidate, as shown by Nardinocchi et al. [64], who observed its ability to induce the proteasomal degradation of HIF1α. While zinc showed similar effects in prostate cancer under hypoxic conditions, its efficacy was not present in the human RCC4 VHL-null cell line. Meanwhile, Maugeri et al. [65] found that PACAP inhibited the release of VEGF. D’Amico et al. [67] showed that this inhibition occurs through the activation of ADNP, a protein that is central to normal brain development and plays a dual role as an oncogene or tumor suppressor, depending on the tumor type. Although the involvement of PACAP in neurodegenerative diseases is well established, further investigation of the PACAP-ADNP axis in glioblastoma is warranted.
Another strategy for inhibiting VEGF is the use of BEV, an anti-VEGF monoclonal antibody that is frequently used in the treatment of glioblastomas. Preclinical and clinical studies have consistently shown that BEV is able to prolong progression-free and overall survival. However, a major challenge is to identify the patients who would benefit from this therapy, as many of them quickly develop resistance. This challenge is exacerbated by the lack of reliable biomarkers, as D’Alessio et al. [68] point out.
Despite BEV treatment, a significant proportion of glioblastoma cases (40–60%) continue to progress, as shown in the clinical studies by Hu et al. [113]. Ongoing randomized clinical trials are investigating the potential of combining chloroquine with the standard treatment of glioblastoma, but a significant benefit has not yet been demonstrated.
In a 2017 study, Gagner et al. [70] used glioma models with mice and administered the anti-VEGF antibody B20-4.1.1 and showed reduced tumor invasiveness in combination with POL5551, a CXCR4 antagonist previously shown to improve survival in immunodeficient mice when combined with other therapeutic modalities. Clinical trials with various CXCR4 antagonists are ongoing. For example, the study (NCT01339039) combines BEV with AMD3100 in patients with recurrent high-grade glioma, while another study (NCT01837095) is investigating POL6326 in combination with the chemotherapeutic agent eribulin in patients with metastatic breast cancer. Kioi et al. [111] investigated the SDF-1/CXCR4 inhibitor AMD3100 and reported its superior efficacy over VEGF blockade in reducing tumor tissue perfusion after radiotherapy.
Photodynamic therapy (PDT) has impressive complete remission rates of up to 90% for skin, head, and neck tumors as well as for early-stage lung and bladder cancer. However, the efficacy of PDT in the treatment of glioblastoma has been limited in the past. However, recent advances, such as the use of acriflavine (ACF) to inhibit HIF1α, as shown by Ma et al. [66], are promising. ACF, which is known for its safety profile, has extended median survival in patients with glioblastoma to 21 months after diagnosis. Since PDT usually upregulates HIF1α expression in most tumors, the integration of HIF inhibitors is crucial. Li et al. [108] have shown that PDT enhances the effect of TMZ by suppressing glycolytic metabolism. The role of immune cells and glycolysis-related enzymes should be further explored.
Hyperbaric oxygen therapy (HBO), which is used in the treatment of ischemic diseases, is also used in carcinoma therapy alongside radiotherapy [130]. Arienti et al. [73] demonstrated that HBO can inhibit the proliferation of glioma cells by increasing reactive oxygen species, which leads to DNA damage. However, preclinical studies often provide contradictory results. For example, Chen et al. [131] report the antitumor effects of HBOT, while there is evidence of tumor-promoting effects [132]. Although clinical studies support the use of HBOT as an adjunct to radiotherapy, a scientific rationale for this phenomenon remains elusive.
Cardiac glycosides that are effective in the treatment of malignancies have been identified as HIF1α inhibitors. The studies by Bar et al. [106], Joseph et al. [98], and Papale et al. [106] highlight the efficacy of digoxin, while Lee et al. [40] focused on digitoxin due to its liposolubility, suggesting the possible permeability of the blood–brain barrier.
Fenofibrate, known for the treatment of hyperlipidemia, has an anticancer effect that has been demonstrated in melanoma, medulloblastoma, and GBM. Trejo-Solis et al. [133] demonstrated its inhibition of glycolysis in GBM, while Lin et al. [71] elucidated the HIF1α inhibition of fenofibrate via multiple metabolic pathways. 2-Methoxyestradiol (2ME2) inhibits HIF1α, inhibits tumor growth, and is being tested in phase I and II in various cancers, including GBM, with promising efficacy and low toxicity. However, the development of resistance to 2ME2 remains enigmatic. Muh et al. [85] suggest PTEN analysis to predict patient response. Combination therapy with a PI3K inhibitor, such as LY294002, is suggested for improved efficacy.
In their effort to target glioma cell proliferation and improve the efficacy of TMZ, Douglas et al. [72] directed their research towards identifying a compound with the dual inhibition of LonP1 and CT-L. BT317 emerged as a promising candidate due to its ability to penetrate the blood–brain barrier, its low toxicity in animals, and its improved survival rates. However, in vivo tests with ritonavir led to the rapid development of resistance. In contrast, marizomib showed significant CNS toxicity in phase II studies and no improvement in survival was demonstrated in phase III trials. Hofstetter et al. [76] found that the inhibition of PP2A with LB1.2 enhanced the effect of TMZ on GBM and neuroblastoma in mouse studies, with no side effects observed during short-term monitoring.
Borneol, a terpene from traditional Chinese medicine, sensitizes cells to TMZ by promoting HIF1α degradation, as demonstrated by Lin et al. [88]. Previous studies have also shown that borneol enhances the efficacy of doxorubicin [134], curcumin [135], cisplatin [136], and radiotherapy [137]. Liu et al. [79] demonstrated in preclinical studies the usefulness of mannose as an adjunct to TMZ and to enhance radiotherapy, and achieved long-term survival in mice.
By combining methoxyamine and resveratrol with iododeoxyuridine, Khoei et al. [78] increased the sensitivity of GBM to radiotherapy. Ahmed et al. [104] noted that the sensitivity of GBM to cisplatin under hypoxic conditions may be independent of HIF and may be induced by the activation of CD133. Barliya et al. [95] investigated the effects of hypericin on the degradation of hsp90 and HIF1α in GBM and renal cell carcinoma cells, with modest results from phase I and phase II trials.
Hsieh et al. [103] reported the inhibition of HIF-1 activation and tumor growth by tempol, while Chou et al. [94] investigated the ability of YC-1 to enhance the efficacy of chemotherapy BCNU. Although not specific to HIF1, Chen et al. [114] demonstrated the synergistic effect of YC-1 with Bay 11-7082 by inhibiting Bcl-xL induction under hypoxia-induced TMZ resistance.
TAT-Lp15, a livin peptide inhibitor, sensitized GBM cells to radiotherapy and TMZ without affecting healthy tissues, as shown by Hsieh et al. [103]. In particular, the ability of TAT-Lp15 to cross the blood–brain barrier underscores its therapeutic potential and warrants further clinical validation.
Disulfiram, known for its ability to improve the efficacy of standard chemotherapies in various carcinomas while exhibiting low toxicity to healthy cells, is hampered by its short half-life in the bloodstream. To address this problem, Kannappan et al. [97] investigated DS-PLGA, an intravenously administered formulation that prolongs the residence time of disulfiram in the bloodstream and facilitates its penetration into GBM tissues without adverse effects on vital organs.
Sulfinosine (SF), known for its multiple anticancer effects via different metabolic pathways, has the potential to prevent cancer cells from developing resistance [138]. Dačević et al. [80] investigated the effect of SF in small-cell lung cancer and GBM and emphasized its ability to penetrate the CNS and its compatibility with other chemotherapeutic agents. Topotecan, which is approved for cervical, ovarian, and small-cell lung cancers, acts as both a DNA topoisomerase I inhibitor and a HIF1α inhibitor [139]. However, its efficacy in GBM remains limited, as noted by Bernstock et al. [83]. Nelfinavir and amprenavir, which have been shown to be effective in HIV therapy, inhibit both HIF1α and VEGF and could sensitize tumor cells to radiotherapy with minimal toxicity, as shown by Mait et al. [86].
Dominguez et al. [102] have identified DGKα as a promising therapeutic target for GBM and other carcinomas, with selective toxicity observed in malignant GBM cells when treated with the DGKα inhibitors R59022 and R59949. SGC707, a PRMT3 inhibitor, showed anticancer activity in GBM by inhibiting HIF1α and glycolysis while sparing normal brain cells, as found by Liao et al. [110].
Arecaidine propargyl ester (Ape) activates M2 muscarinic receptors, leading to cell cycle arrest in GBM stem cells, as reported by Cristofaro et al. [67]. WIN 55,212-2, a cannabinoid receptor agonist, induces GBM cell death, suggesting cannabinoids as potential anticancer agents according to Sugimoto et al. [87]. Paris saponin H, which is used in the treatment of lung cancer and malignant lymphoma, induces the apoptosis of gliomas, as shown by Bi et al. [77]. Although the insulin signaling pathway plays a crucial role in the progression of GBM, drugs targeting IGF1 await the successful completion of phase III trials as the molecular mechanisms involved are not yet fully understood, as noted by Lin et al. [71]. Echinomycin, a notable HIF1α inhibitor, induces apoptosis and inhibits GBM growth by targeting the HIF1α-PDGFD-PDGFRα axis, as found by Peng et al. [100].
4.4. Insights into HIF-Associated Discoveries from Clinical Investigations in GBM
Clinical studies consistently report the elevated expression of HIF1α in glioblastoma (GBM) tissues, suggesting its pivotal role in tumor progression. Chen et al. (2019) [114] observed significant HIF1α expression in both the nucleus and cytoplasm of GBM cells, correlating with tumor vasculature, indicating its involvement in angiogenesis. Similarly, the findings by Carlos Alfonsoe et al. [122] and Xiangjun et al. [119] linked HIF1α expression in GBM with increased vascular proliferation and poorer patient prognosis. Moreover, the research by Bache et al. [115] and El-Benhawy [123] described a diverse range of hypoxia-related factors, including HIF2α and OPN, contributing to the intricate tumor microenvironment, highlighting the multifaceted role of HIFs in tumor growth and survival under hypoxia.
Notably, the studies by Erpolat et al. [116] and Nobuyuki et al. [118] established a correlation between elevated HIF1α levels and reduced patient survival, indicating its potential as a prognostic marker. Conversely, the observations by Sfifou et al. [120] indicated longer survival in patients with negative HIF1α expression, reinforcing its prognostic value. High HIF expression levels correlate with aggressive GBM behavior, including rapid growth, enhanced invasiveness, and resistance to standard treatments, as demonstrated by Kaynar et al. [117] and Potharaju et al. [121], contributing to poorer patient outcomes.
These clinical findings underscore the importance of investigating hypoxia-induced tumor progression mechanisms in GBM. Developing targeted therapies to inhibit HIF activity, possibly in combination with existing treatments, holds promise for improving patient prognosis. Additionally, identifying novel biomarkers based on hypoxia-related factors could enhance early detection and treatment monitoring in GBM, ultimately improving patient outcomes. Future research efforts should focus on unraveling the complexities of the hypoxic tumor microenvironment to devise more effective interventions for managing GBM.
4.5. Advantages, Disadvantages, and Future Directions
Therapies targeting HIFs offer a promising avenue for combating GBM, a malignancy notorious for its resistance to conventional treatments. By specifically inhibiting HIF activity, these therapies hold potential for improving patient outcomes, particularly in cases where GBM displays elevated HIF expression levels [140]. Combining HIF-related therapies with established treatments like surgery, radiation, and chemotherapy may enhance their effectiveness, offering a more comprehensive approach to GBM management [141,142]. Research into HIFs in GBM provides crucial insights into tumor progression mechanisms, offering hope for the development of more potent therapeutic strategies. Moreover, exploring HIFs could lead to the identification of novel biomarkers for early diagnosis, prognosis assessment, and treatment response monitoring in patients with GBM [143].
However, challenges abound in the clinical application of HIF-related therapies. The lack of standardization in research methodologies impedes quantitative meta-analysis, while genetic mutations in GBM and therapy effects outside target sites present additional hurdles [13,144,145]. The complex and dynamic nature of the hypoxic tumor microenvironment may limit the efficacy of single-target HIF therapies, potentially leading to therapy resistance. Developing combination therapies or innovative treatment strategies may be necessary to address this issue. Despite encouraging preclinical results, limited clinical data exist on the efficacy of HIF-related therapies in patients with GBM, necessitating further extensive clinical trials for validation [137,146]. Safety concerns, including potential side effects and toxicity, especially when combined with other treatments, require thorough evaluation [147,148,149].
Moreover, the challenge lies in targeting HIFs without disrupting normal cellular responses to hypoxia, underscoring the need for precision in therapy development [150]. The absence of reliable biomarkers to identify patients who would benefit most from HIF-related therapies complicates treatment decisions and personalized care plans. Exploring combination therapies targeting multiple GBM progression pathways, conducting advanced clinical trials with diverse populations, and investigating the mechanisms of therapy resistance are crucial steps forward [151]. Additionally, advancing research to identify and validate biomarkers for early detection and treatment response monitoring is essential for the effective clinical implementation of HIF-related therapies in GBM management.
5. Conclusions
In conclusion, the evolving landscape of GBM research reflects a concerted effort to address the pressing challenges of poor patient outcomes associated with conventional treatments. While molecular genetic features have improved diagnostic capabilities, preclinical studies have highlighted the importance of HIFs as a therapeutic target, although clinical translation is limited. Overcoming challenges such as therapy resistance, safety concerns, and the absence of reliable biomarkers is crucial for the successful integration of HIF-related therapies into the treatment of GBM. By combining targeted approaches with conventional treatments, conducting large clinical trials, and testing combination therapies, researchers aim to optimize patient outcomes and pave the way for personalized treatment strategies in GBM. Ultimately, these multidisciplinary efforts promise to improve our understanding and treatment of GBM and provide hope for better patient care in the future.
Author Contributions
Conceptualization, E.B., H.B. and M.P.; methodology, E.B., H.B., A.D.-K. and S.K.V.; software, S.H.; validation, S.Š. and E.P.; formal analysis, E.B.; investigation, A.M.-A., S.Š., E.P., E.M.E., R.P. and T.K.; data curation, E.B., S.Š. and D.K.; writing—original draft preparation, A.N., A.D.-K., E.B., F.J.-B., S.Đ., S.Š., E.P. and R.P.; writing—review and editing, E.B., S.Š. and H.B.; visualization, S.H.; supervision, S.Đ. and M.P.; project administration, E.B.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
| Search | (Glioblastoma) AND (Hypoxia-Inducible Factors) |
| Base: MEDLINE (PubMed) | |
| Filter | None |
| Search query | (“glioblastoma”[MeSH Terms] OR “glioblastoma”[All Fields] OR “glioblastomas”[All Fields]) AND ((“hypoxia”[MeSH Terms] OR “hypoxia”[All Fields] OR “hypoxia s”[All Fields] OR “hypoxias”[All Fields]) AND (“induce”[All Fields] OR “induced”[All Fields] OR “inducer”[All Fields] OR “inducers”[All Fields] OR “induces”[All Fields] OR “inducibilities”[All Fields] OR “inducibility”[All Fields] OR “inducible”[All Fields] OR “inducing”[All Fields]) AND (“factor”[All Fields] OR “factor s”[All Fields] OR “factors”[All Fields])) |
| Results | 558 papers |
| Base: Web of Science | |
| Filter | None |
| Search query | TS = (“glioblastoma” OR “glioblastomas”) AND TS = (“hypoxia inducible factors” OR “hypoxia-inducible factors” OR “HIFs”) |
| Results | 89 papers |
| Base: Scopus | |
| Filter | None |
| Search query | TITLE-ABS-KEY(“glioblastoma”) AND TITLE-ABS-KEY(“hypoxia inducible factors” OR “HIFs”) |
| Results | 671 papers |
Appendix B
| Section/Topic | # | Checklist Item | Reported on Page # |
| Title | |||
| Title | 1 | Identify the report as a systematic review, meta-analysis, or both. | 1 |
| Abstract | |||
| Structured summary | 2 | Provide a structured summary including, as applicable: background; objectives; data sources; study eligibility criteria, participants, and interventions; study appraisal and synthesis methods; results; limitations; conclusions and implications of key findings; and systematic review registration number. | 1 |
| Introduction | |||
| Rationale | 3 | Describe the rationale for the review in the context of what is already known. | 2 |
| Objectives | 4 | Provide an explicit statement of the questions being addressed with reference to the participants, interventions, comparisons, outcomes, and study design (PICOS). | 2 |
| Methods | |||
| Protocol and registration | 5 | Indicate if a review protocol exists, if and where it can be accessed (e.g., Web address), and, if available, provide registration information including registration number. | 2 |
| Eligibility criteria | 6 | Specify study characteristics (e.g., PICOS and length of follow-up) and report characteristics (e.g., years considered, language and publication status) used as criteria for eligibility, giving rationale. | 3 |
| Information sources | 7 | Describe all information sources (e.g., databases with dates of coverage and contact with study authors to identify additional studies) in the search and date last searched. | 3 |
| Search | 8 | Present a full electronic search strategy for at least one database, including any limits used, such that it could be repeated. | Appendix A |
| Study selection | 9 | State the process for selecting studies (i.e., screening, eligibility, included in the systematic review, and, if applicable, included in the meta-analysis). | 3 |
| Data collection process | 10 | Describe the method of data extraction from the reports (e.g., piloted forms, independently and in duplicate) and any processes for obtaining and confirming data from the investigators. | 3 |
| Data items | 11 | List and define all variables for which data were sought (e.g., PICOS and funding sources) and any assumptions and simplifications made. | 3 |
| Risk of bias in individual studies | 12 | Describe the methods used for assessing the risk of bias in individual studies (including the specification of whether this was performed at the study or outcome level), and how this information is to be used in any data synthesis. | N/A |
| Summary measures | 13 | State the principal summary measures (e.g., risk ratio and the difference in means). | N/A |
| Synthesis of results | 14 | Describe the methods of handling data and combining the results of studies, if performed, including the measures of consistency (e.g., I2) for each meta-analysis. | N/A |
| Risk of bias across studies | 15 | Specify any assessment of the risk of bias that may affect the cumulative evidence (e.g., publication bias and selective reporting within studies). | N/A |
| Additional analyses | 16 | Describe the methods of additional analyses (e.g., sensitivity or subgroup analyses and meta-regression), if performed, indicating which were pre-specified. | N/A |
| Results | |||
| Study selection | 17 | Give the number of studies screened, assessed for eligibility, and included in the review, with reasons for exclusions at each stage, ideally with a flow diagram. | Figure 1 |
| Study characteristics | 18 | For each study, present characteristics for which data were extracted (e.g., study size, PICOS, and follow-up period) and provide the citations. | 3 |
| Risk of bias within studies | 19 | Present data on the risk of bias of each study and, if available, any outcome level assessment (see item 12). | N/A |
| Results of individual studies | 20 | For all outcomes considered (benefits or harms), present, for each study: (a) simple summary data for each intervention group and (b) effect estimates and confidence intervals, ideally with a forest plot. | Table 1, Table 2 and Table 3 |
| Synthesis of results | 21 | Present the results of each meta-analysis performed, including confidence intervals and the measures of consistency. | N/A |
| Risk of bias across studies | 22 | Present the results of any assessment of the risk of bias across studies (see item 15). | N/A |
| Additional analysis | 23 | Give the results of additional analyses, if performed (e.g., sensitivity or subgroup analyses and meta-regression [see item 16]). | N/A |
| Discussion | |||
| Summary of evidence | 24 | Summarize the main findings including the strength of evidence for each main outcome; consider their relevance to key groups (e.g., healthcare providers, users, and policy makers). | 4–8 |
| Limitations | 25 | Discuss limitations at the study and outcome level (e.g., risk of bias), and at the review level (e.g., the incomplete retrieval of identified research and reporting bias). | 10 |
| Conclusions | 26 | Provide a general interpretation of the results in the context of other evidence, and implications for future research. | 11 |
| Funding | |||
| Funding | 27 | Describe the sources of funding for the systematic review and other support (e.g., supply of data); and the role of funders for the systematic review. | 11 |
| N/A—Not Available | |||
References
- Shah, S. Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies. Med. Sci. 2024, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Begagić, E.; Bečulić, H.; Đuzić, N.; Džidić-Krivić, A.; Pugonja, R.; Muharemović, A.; Jaganjac, B.; Salković, N.; Sefo, H.; Pojskić, M. CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review. Biomedicines 2024, 12, 238. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Q.; Pierrevelcin, M.; Messe, M.; Lhermitte, B.; Blandin, A.F.; Papin, C.; Coca, A.; Dontenwill, M.; Entz-Werlé, N. Hypoxia Inducible Factors’ Signaling in Pediatric High-Grade Gliomas: Role, Modelization and Innovative Targeted Approaches. Cancers 2020, 12, 979. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Renfrow, J.J.; Soike, M.H.; Debinski, W.; Ramkissoon, S.H.; Mott, R.T.; Frenkel, M.B.; Sarkaria, J.N.; Lesser, G.J.; Strowd, R.E. Hypoxia-inducible factor 2α: A novel target in gliomas. Future Med. Chem. 2018, 10, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L.; Mumert, M.L.; Gillespie, D.L.; Kinney, A.Y.; Schabel, M.C.; Salzman, K.L. Preoperative dynamic contrast-enhanced MRI correlates with molecular markers of hypoxia and vascularity in specific areas of intratumoral microenvironment and is predictive of patient outcome. Neuro Oncol. 2014, 16, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Zhang, Z.; Fink, J.R.; Muzi, M.; Hanna, L.; Greco, E.; Prah, M.; Schmainda, K.M.; Mintz, A.; Kostakoglu, L.; et al. ACRIN 6684: Assessment of Tumor Hypoxia in Newly Diagnosed Glioblastoma Using 18F-FMISO PET and MRI. Clin. Cancer Res. 2016, 22, 5079–5086. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Anobile, D.P.; Montenovo, G.; Pecoraro, C.; Franczak, M.; Ait Iddouch, W.; Peters, G.J.; Riganti, C.; Giovannetti, E. Splicing deregulation, microRNA and notch aberrations: Fighting the three-headed dog to overcome drug resistance in malignant mesothelioma. Expert Rev. Clin. Pharmacol. 2022, 15, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, K.; Xiang, J.; Zhang, M.; Zhu, M.; Xi, Q. Hypoxia induces immunosuppression, metastasis and drug resistance in pancreatic cancers. Cancer Lett. 2023, 571, 216345. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Matthew, J.P.; Joanne, E.M.; Patrick, M.B.; Isabelle, B.; Tammy, C.H.; Cynthia, D.M.; Larissa, S.; Jennifer, M.T.; Elie, A.A.; Sue, E.B.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, O.R.; Peters, G.-J.Y.; Bakker, C.J.; Carlsson, R.; Coles, N.A.; Corker, K.S.; Feldman, G.; Moreau, D.; Nordström, T.; Pickering, J.S.; et al. Increasing the transparency of systematic reviews: Presenting a generalized registration form. Syst. Rev. 2023, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Urushihara, Y.; Murata, Y.; Fujishima, Y.; Hosoi, Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem. Biophys. Res. Commun. 2022, 590, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.H.; Shih, C.M.; Liu, A.J.; Chen, K.C. Hypoxia-inducible lncRNA MIR210HG interacting with OCT1 is involved in glioblastoma multiforme malignancy. Cancer Sci. 2022, 113, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ogura, Y.; Tanaka, K.; Nagashima, H.; Sasayama, T.; Endo, M.; Minami, Y. Ror1 is expressed inducibly by Notch and hypoxia signaling and regulates stem cell-like property of glioblastoma cells. Cancer Sci. 2023, 114, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Pandey, P.; Jha, P.; Dwivedi, V.; Sarkar, C.; Kulshreshtha, R. Hypoxic signature of microRNAs in glioblastoma: Insights from small RNA deep sequencing. BMC Genom. 2014, 15, 686. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.M.; Uno, M.; Oba-Shinjo, S.M.; Clara, C.A.; de Almeida Galatro, T.F.; Rosemberg, S.; Teixeira, M.J.; Nagahashi Marie, S.K. CXCR7 and CXCR4 Expressions in Infiltrative Astrocytomas and Their Interactions with HIF1α Expression and IDH1 Mutation. Pathol. Oncol. Res. 2015, 21, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Eckerich, C.; Zapf, S.; Fillbrandt, R.; Loges, S.; Westphal, M.; Lamszus, K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int. J. Cancer 2007, 121, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Yokoi, A.; Ishizuka, Y.; Hashimura, M.; Matsumoto, T.; Oguri, Y.; Nakagawa, M.; Ishibashi, Y.; Ito, T.; Kumabe, T.; et al. A functional role of S100A4/non-muscle myosin IIA axis for pro-tumorigenic vascular functions in glioblastoma. Cell Commun. Signal 2022, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Weisz, A.; Kurashima, Y.; Hashimoto, K.; Ogura, T.; D’Acquisto, F.; Addeo, R.; Makuuchi, M.; Esumi, H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: Control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 2000, 95, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Q.; Xu, S.; Wu, J.; Huang, Q.; Song, P.; Duan, F. Down-regulation of BAG3 inhibits proliferation and promotes apoptosis of glioblastoma multiforme through BAG3/HSP70/HIF-1α signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 4305–4318. [Google Scholar] [PubMed]
- Méndez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e224. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.R.; Sadik, A.; Tykocinski, L.O.; Dietze, J.; Poschet, G.; Heiland, I.; Opitz, C.A. Hypoxia Inducible Factor 1α Inhibits the Expression of Immunosuppressive Tryptophan-2,3-Dioxygenase in Glioblastoma. Front. Immunol. 2019, 10, 2762. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Savino, M.; Falchetti, M.L.; Illi, B.; Bozzo, F.; Valle, C.; Helmer-Citterich, M.; Ferrè, F.; Nasi, S.; Levi, A. c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget 2016, 7, 33257–33271. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.; Luo, X.; Lu, D.; Yuan, P.; Liu, B.; Xu, H.; Ye, M. Casein kinase 1α 1 is involved in the progression of glioblastoma through HIF-1α-mediated autophagy. J. Neurophysiol. 2022, 128, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.; Bookland, M.; Khalili, K. Astrocyte-elevated gene-1 (AEG-1) induction by hypoxia and glucose deprivation in glioblastoma. Cancer Biol. Ther. 2011, 11, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Rampazzo, E.; Abbadi, S.; Della Puppa, A.; Scienza, R.; D’Avella, D.; Denaro, L.; Te Kronnie, G.; Panchision, D.M.; Basso, G. Molecular mechanisms of HIF-1alpha modulation induced by oxygen tension and BMP2 in glioblastoma derived cells. PLoS ONE 2009, 4, e6206. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Polat, B.; Stein, S.; Guckenberger, M.; Hagemann, C.; Staab, A.; Katzer, A.; Anacker, J.; Flentje, M.; Vordermark, D. Inhibition of N-Myc down regulated gene 1 in in vitro cultured human glioblastoma cells. World J. Clin. Oncol. 2012, 3, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sesen, J.; Cammas, A.; Scotland, S.J.; Elefterion, B.; Lemarié, A.; Millevoi, S.; Mathew, L.K.; Seva, C.; Toulas, C.; Moyal, E.C.; et al. Int6/eIF3e is essential for proliferation and survival of human glioblastoma cells. Int. J. Mol. Sci. 2014, 15, 2172–2190. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Hu, F.; Huang, R.; Mackman, N.; Horowitz, J.M.; Jensen, R.L.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006, 66, 7067–7074. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Fan, J.; Yang, H.; Zhao, C.; Niu, W.; Fang, Z.; Chen, X. Heterogeneity of subsets in glioblastoma mediated by Smad3 palmitoylation. Oncogenesis 2021, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Voss, D.M.; Sloan, A.; Spina, R.; Ames, H.M.; Bar, E.E. The Alternative Splicing Factor, MBNL1, Inhibits Glioblastoma Tumor Initiation and Progression by Reducing Hypoxia-Induced Stemness. Cancer Res. 2020, 80, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yan, Q.; Liao, B.; Zhao, L.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Wu, L.; Deng, Y.; et al. The HIF1α/HIF2α-miR210-3p network regulates glioblastoma cell proliferation, dedifferentiation and chemoresistance through EGF under hypoxic conditions. Cell Death Dis. 2020, 11, 992. [Google Scholar] [CrossRef]
- Bae, W.Y.; Choi, J.S.; Nam, S.; Jeong, J.W. β-arrestin 2 stimulates degradation of HIF-1α and modulates tumor progression of glioblastoma. Cell Death Differ. 2021, 28, 3092–3104. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, Y.; She, X.; Sun, Y.; Fan, L.; Ren, X.; Fu, H.; Liu, C.; Li, P.; Zhao, C.; et al. Hypermethylated gene ANKDD1A is a candidate tumor suppressor that interacts with FIH1 and decreases HIF1α stability to inhibit cell autophagy in the glioblastoma multiforme hypoxia microenvironment. Oncogene 2019, 38, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Ozaki, S.; Kanemura, Y.; Suehiro, S.; Ohtsuka, Y.; Kohno, S.; Ohue, S.; et al. Hypoxia-induced phenotypic transition from highly invasive to less invasive tumors in glioma stem-like cells: Significance of CD44 and osteopontin as therapeutic targets in glioblastoma. Transl. Oncol. 2021, 14, 101137. [Google Scholar] [CrossRef] [PubMed]
- Choksi, S.; Lin, Y.; Pobezinskaya, Y.; Chen, L.; Park, C.; Morgan, M.; Li, T.; Jitkaew, S.; Cao, X.; Kim, Y.S.; et al. A HIF-1 target, ATIA, protects cells from apoptosis by modulating the mitochondrial thioredoxin, TRX2. Mol. Cell 2011, 42, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, E.; Chong, K.; Ryu, S.W.; Kim, C.; Choi, K.; Kim, J.H.; Choi, C. Atypical induction of HIF-1α expression by pericellular Notch1 signaling suffices for the malignancy of glioblastoma multiforme cells. Cell Mol. Life Sci. 2022, 79, 537. [Google Scholar] [CrossRef]
- Katakowski, M.; Charteris, N.; Chopp, M.; Khain, E. Density-Dependent Regulation of Glioma Cell Proliferation and Invasion Mediated by miR-9. Cancer Microenviron. 2016, 9, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Zhu, L.; Pan, H.; Li, W.; Zhou, Y.; Cong, Z.; Yan, F.; Chen, S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int. J. Cancer 2014, 135, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Saati, M.; Bensalah-Pigeon, H.; Ben M’Barek, K.; Gitton-Quent, O.; Bertrand, R.; Busso, D.; Mouthon, M.A.; Collura, A.; Junier, M.P.; et al. The HIF1α/JMY pathway promotes glioblastoma stem-like cell invasiveness after irradiation. Sci. Rep. 2020, 10, 18742. [Google Scholar] [CrossRef]
- Hu, Q.; Liu, F.; Yan, T.; Wu, M.; Ye, M.; Shi, G.; Lv, S.; Zhu, X. MicroRNA-576-3p inhibits the migration and proangiogenic abilities of hypoxia-treated glioma cells through hypoxia-inducible factor-1α. Int. J. Mol. Med. 2019, 43, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Paul, A.; Sen, E. Tumor necrosis factor α-induced hypoxia-inducible factor 1α-β-catenin axis regulates major histocompatibility complex class I gene activation through chromatin remodeling. Mol. Cell Biol. 2013, 33, 2718–2731. [Google Scholar] [CrossRef] [PubMed]
- Evagelou, S.L.; Bebenek, O.; Specker, E.J.; Uniacke, J. DEAD Box Protein Family Member DDX28 Is a Negative Regulator of Hypoxia-Inducible Factor 2α- and Eukaryotic Initiation Factor 4E2-Directed Hypoxic Translation. Mol. Cell Biol. 2020, 40, e00610-19. [Google Scholar] [CrossRef] [PubMed]
- Ikemori, R.Y.; Machado, C.M.; Furuzawa, K.M.; Nonogaki, S.; Osinaga, E.; Umezawa, K.; de Carvalho, M.A.; Verinaud, L.; Chammas, R. Galectin-3 up-regulation in hypoxic and nutrient deprived microenvironments promotes cell survival. PLoS ONE 2014, 9, e111592. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Huang, H.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem Cell 2018, 22, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Bordji, K.; Grandval, A.; Cuhna-Alves, L.; Lechapt-Zalcman, E.; Bernaudin, M. Hypoxia-inducible factor-2α (HIF-2α), but not HIF-1α, is essential for hypoxic induction of class III β-tubulin expression in human glioblastoma cells. FEBS J. 2014, 281, 5220–5236. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Heller, S.; Wanka, C.; Rieger, J.; Steinbach, J.P. Knockdown of the TP53-Induced Glycolysis and Apoptosis Regulator (TIGAR) Sensitizes Glioma Cells to Hypoxia, Irradiation and Temozolomide. Int. J. Mol. Sci. 2019, 20, 1061. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Potdar, A.A.; Gong, Y.; Eswarappa, S.M.; Donnola, S.; Lathia, J.D.; Hambardzumyan, D.; Rich, J.N.; Fox, P.L. Profilin-1 phosphorylation directs angiocrine expression and glioblastoma progression through HIF-1α accumulation. Nat. Cell Biol. 2014, 16, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhu, K.; Yang, Z.; Zhou, Y.; Xia, Z.; Ren, J.; Zhao, Y.; Wu, G.; Liu, C. Hypoxia-Induced Autophagy Is Involved in Radioresistance via HIF1A-Associated Beclin-1 in Glioblastoma Multiforme. Heliyon 2023, 9, e12820. [Google Scholar] [CrossRef] [PubMed]
- Coma, S.; Shimizu, A.; Klagsbrun, M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adhes. Migr. 2011, 5, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.J.; Chiang, J.Y.; Chang, H.J.; Chen, D.C.; Wang, H.L.; Yang, H.A.; Wei, K.Y.; Huang, Y.C.; Wang, C.C.; Wei, S.T.; et al. Cellular and exosomal GPx1 are essential for controlling hydrogen peroxide balance and alleviating oxidative stress in hypoxic glioblastoma. Redox Biol. 2023, 65, 102831. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, A.R.; Durden, D.L. MDM2 regulates hypoxic hypoxia-inducible factor 1α stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J. Biol. Chem. 2014, 289, 22785–22797. [Google Scholar] [CrossRef] [PubMed]
- Lulli, V.; Buccarelli, M.; Ilari, R.; Castellani, G.; De Dominicis, C.; Di Giamberardino, A.; QG, D.A.; Giannetti, S.; Martini, M.; Stumpo, V.; et al. Mir-370-3p Impairs Glioblastoma Stem-Like Cell Malignancy Regulating a Complex Interplay between HMGA2/HIF1A and the Oncogenic Long Non-Coding RNA (lncRNA) NEAT1. Int. J. Mol. Sci. 2020, 21, 3610. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zhang, Y.; Celiku, O.; Zhang, W.; Song, H.; Williams, B.J.; Giles, A.J.; Rich, J.N.; Abounader, R.; Gilbert, M.R.; et al. Mitochondrial NIX Promotes Tumor Survival in the Hypoxic Niche of Glioblastoma. Cancer Res. 2019, 79, 5218–5232. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; et al. A positive feedback circuit comprising p21 and HIF-1α aggravates hypoxia-induced radioresistance of glioblastoma by promoting Glut1/LDHA-mediated glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Pantisano, V.; Puca, R.; Porru, M.; Aiello, A.; Grasselli, A.; Leonetti, C.; Safran, M.; Rechavi, G.; Givol, D.; et al. Zinc downregulates HIF-1α and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE 2010, 5, e15048. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Saccone, S.; Federico, C.; Rasà, D.M.; Caltabiano, R.; Broggi, G.; Giunta, S.; Musumeci, G.; D’Agata, V. Effect of PACAP on Hypoxia-Induced Angiogenesis and Epithelial-Mesenchymal Transition in Glioblastoma. Biomedicines 2021, 9, 965. [Google Scholar] [CrossRef]
- Ma, S.; Wang, F.; Dong, J.; Wang, N.; Tao, S.; Du, J.; Hu, S. Inhibition of hypoxia-inducible factor 1 by acriflavine renders glioblastoma sensitive for photodynamic therapy. J. Photochem. Photobiol. B 2022, 234, 112537. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Maugeri, G.; Magrì, B.; Giunta, S.; Saccone, S.; Federico, C.; Pricoco, E.; Broggi, G.; Caltabiano, R.; Musumeci, G.; et al. Modulatory activity of ADNP on the hypoxia-induced angiogenic process in glioblastoma. Int. J. Oncol. 2023, 62, 14. [Google Scholar] [CrossRef]
- D’Alessio, A.; Proietti, G.; Lama, G.; Biamonte, F.; Lauriola, L.; Moscato, U.; Vescovi, A.; Mangiola, A.; Angelucci, C.; Sica, G. Analysis of angiogenesis related factors in glioblastoma, peritumoral tissue and their derived cancer stem cells. Oncotarget 2016, 7, 78541–78556. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, I.; Limongi, C.; Piscopo, P.; Crestini, A.; Guerriero, C.; Fiore, M.; Conti, L.; Confaloni, A.; Tata, A.M. M2 Receptor Activation Counteracts the Glioblastoma Cancer Stem Cell Response to Hypoxia Condition. Int. J. Mol. Sci. 2020, 21, 1700. [Google Scholar] [CrossRef] [PubMed]
- Gagner, J.P.; Sarfraz, Y.; Ortenzi, V.; Alotaibi, F.M.; Chiriboga, L.A.; Tayyib, A.T.; Douglas, G.J.; Chevalier, E.; Romagnoli, B.; Tuffin, G.; et al. Multifaceted C-X-C Chemokine Receptor 4 (CXCR4) Inhibition Interferes with Anti-Vascular Endothelial Growth Factor Therapy-Induced Glioma Dissemination. Am. J. Pathol. 2017, 187, 2080–2094. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Luo, J.; Wang, Z.; Cai, X. Borneol promotes autophagic degradation of HIF-1α and enhances chemotherapy sensitivity in malignant glioma. PeerJ 2024, 12, e16691. [Google Scholar] [CrossRef] [PubMed]
- Douglas, C.; Lomeli, N.; Lepe, J.; Di, K.; Nandwana, N.K.; Vu, T.; Pham, J.; Kenney, M.C.; Das, B.; Bota, D.A. Discovery and Validation of Novel LonP1 and Proteasome Inhibitor in IDH1-R132H Malignant Astrocytoma Models. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Arienti, C.; Pignatta, S.; Zanoni, M.; Zamagni, A.; Cortesi, M.; Sarnelli, A.; Romeo, A.; Arpa, D.; Longobardi, P.; Bartolini, D.; et al. High-pressure oxygen rewires glucose metabolism of patient-derived glioblastoma cells and fuels inflammasome response. Cancer Lett. 2021, 506, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.W.; Liao, A.; Qutub, A.A. Simulation predicts IGFBP2-HIF1α interaction drives glioblastoma growth. PLoS Comput. Biol. 2015, 11, e1004169. [Google Scholar] [CrossRef]
- Lund, E.L.; Høg, A.; Olsen, M.W.; Hansen, L.T.; Engelholm, S.A.; Kristjansen, P.E. Differential regulation of VEGF, HIF1alpha and angiopoietin-1, -2 and -4 by hypoxia and ionizing radiation in human glioblastoma. Int. J. Cancer 2004, 108, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, C.P.; Burkhardt, J.K.; Shin, B.J.; Gürsel, D.B.; Mubita, L.; Gorrepati, R.; Brennan, C.; Holland, E.C.; Boockvar, J.A. Protein phosphatase 2A mediates dormancy of glioblastoma multiforme-derived tumor stem-like cells during hypoxia. PLoS ONE 2012, 7, e30059. [Google Scholar] [CrossRef]
- Bi, L.; Liu, Y.; Yang, Q.; Zhou, X.; Li, H.; Liu, Y.; Li, J.; Lu, Y.; Tang, H. Paris saponin H inhibits the proliferation of glioma cells through the A1 and A3 adenosine receptor-mediated pathway. Int. J. Mol. Med. 2021, 47, 30. [Google Scholar] [CrossRef] [PubMed]
- Khoei, S.; Shoja, M.; Mostaar, A.; Faeghi, F. Effects of resveratrol and methoxyamine on the radiosensitivity of iododeoxyuridine in U87MG glioblastoma cell line. Exp. Biol. Med. 2016, 241, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xu, X.; Li, C.; Li, C.; Li, Y.; Yin, S.; Yu, S.; Chen, X.Q. Mannose synergizes with chemoradiotherapy to cure cancer via metabolically targeting HIF-1 in a novel triple-negative glioblastoma mouse model. Clin. Transl. Med. 2020, 10, e226. [Google Scholar] [CrossRef] [PubMed]
- Dačević, M.; Isaković, A.; Podolski-Renić, A.; Isaković, A.M.; Stanković, T.; Milošević, Z.; Rakić, L.; Ruždijić, S.; Pešić, M. Purine nucleoside analog--sulfinosine modulates diverse mechanisms of cancer progression in multi-drug resistant cancer cell lines. PLoS ONE 2013, 8, e54044. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Kimura, T.; Sadahiro, H.; Kawano, H.; Takubo, K.; Suzuki, M.; Ikeda, E. Histological Characterization of the Tumorigenic “Peri-Necrotic Niche” Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma. PLoS ONE 2016, 11, e0147366. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, D.; Zhang, Z.; Wang, Y.; Zhang, Z.; Liang, Z.; Liu, F.; Chen, L. Photodynamic therapy enhances the cytotoxicity of temozolomide against glioblastoma via reprogramming anaerobic glycolysis. Photodiagn. Photodyn. Ther. 2023, 42, 103342. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kögel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci. Rep. 2017, 7, 7425. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Di Vito, M.; Frati, A.; Pellegrini, L.; De Santis, E.; Sette, G.; Eramo, A.; Sale, P.; Mari, E.; Santoro, A.; et al. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflamm. 2011, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Muh, C.R.; Joshi, S.; Singh, A.R.; Kesari, S.; Durden, D.L.; Makale, M.T. PTEN status mediates 2ME2 anti-tumor efficacy in preclinical glioblastoma models: Role of HIF1α suppression. J. Neurooncol. 2014, 116, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Ishibashi, H.; Nakamura, H.; Yachie, A.; Ohno-Shosaku, T. Hypoxia-induced inhibition of the endocannabinoid system in glioblastoma cells. Oncol. Rep. 2017, 38, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lai, S.W.; Shen, C.K.; Chen, C.W.; Tsai, C.F.; Liu, Y.S.; Lu, D.Y.; Huang, B.R. Fenofibrate inhibits hypoxia-inducible factor-1 alpha and carbonic anhydrase expression through activation of AMP-activated protein kinase/HO-1/Sirt1 pathway in glioblastoma cells. Environ. Toxicol. 2021, 36, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Gagner, J.P.; Lechpammer, M.; Zagzag, D. Induction and Assessment of Hypoxia in Glioblastoma Cells In Vitro. Methods Mol. Biol. 2018, 1741, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ding, X.; Ye, H.; Wang, J.; Shao, J.; Huang, T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. Neuroreport 2018, 29, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Guo, S.; Khan, F.; Dunterman, M.; Ali, H.; Liu, Y.; Huang, Y.; Chen, P. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep. Med. 2023, 4, 101238. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Wang, C.C.; Wu, C.P.; Lin, Y.J.; Lee, Y.C.; Cheng, Y.W.; Hsieh, C.H. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro Oncol. 2012, 14, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Barliya, T.; Mandel, M.; Livnat, T.; Weinberger, D.; Lavie, G. Degradation of HIF-1alpha under hypoxia combined with induction of Hsp90 polyubiquitination in cancer cells by hypericin: A unique cancer therapy. PLoS ONE 2011, 6, e22849. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shyu, W.C.; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011, 6, e23945. [Google Scholar] [CrossRef] [PubMed]
- Kannappan, V.; Liu, Y.; Wang, Z.; Azar, K.; Kurusamy, S.; Kilari, R.S.; Armesilla, A.L.; Morris, M.R.; Najlah, M.; Liu, P.; et al. PLGA-Nano-Encapsulated Disulfiram Inhibits Hypoxia-Induced NF-κB, Cancer Stem Cells, and Targets Glioblastoma In Vitro and In Vivo. Mol. Cancer Ther. 2022, 21, 1273–1284. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.P.; Shireman, J.M.; Huang, M.; Miska, J.; Atashi, F.; Baisiwala, S.; Hong Park, C.; Saathoff, M.R.; Warnke, L.; Xiao, T.; et al. Activation of Dopamine Receptor 2 Prompts Transcriptomic and Metabolic Plasticity in Glioblastoma. J. Neurosci. 2019, 39, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, Y.; Ge, P.; Bailey, C.; Zhang, P.; Zhang, D.; Meng, Z.; Qi, C.; Chen, Q.; Chen, J.; et al. The HIF1α-PDGFD-PDGFRα axis controls glioblastoma growth at normoxia/mild-hypoxia and confers sensitivity to targeted therapy by echinomycin. J. Exp. Clin. Cancer Res. 2021, 40, 278. [Google Scholar] [CrossRef]
- Han, D.; Wei, W.; Chen, X.; Zhang, Y.; Wang, Y.; Zhang, J.; Wang, X.; Yu, T.; Hu, Q.; Liu, N.; et al. NF-κB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget 2015, 6, 26119–26128. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Lin, Y.J.; Wu, C.P.; Lee, H.T.; Shyu, W.C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-independent, CD133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell. Oncol. 2018, 41, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Cheul Oh, S.; Giles, A.J.; Jung, J.; Gilbert, M.R.; Park, D.M. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1α in human glioma stem cells. Oncotarget 2017, 8, 40233–40245. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Wang, C.C.; Lin, Y.J.; Wu, C.P.; Hsieh, C.H. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J. Transl. Med. 2015, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Zhang, M.; Feng, Y.K.; Wang, X.J. IDH1-R132H Suppresses Glioblastoma Malignancy through FAT1-ROS-HIF-1α Signaling. Neurol. India 2020, 68, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Pan, M.H.; Wang, L.; Li, W.; Jiang, C.; He, J.; Abouzid, K.; Liu, L.Z.; Shi, Z.; Jiang, B.H. Hypoxia-mediated mitochondria apoptosis inhibition induces temozolomide treatment resistance through miR-26a/Bad/Bax axis. Cell Death Dis. 2018, 9, 1128. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Luo, Z.; Lin, Y.; Chen, H.; Chen, T.; Xu, L.; Orgurek, S.; Berry, K.; Dzieciatkowska, M.; Reisz, J.A.; et al. PRMT3 drives glioblastoma progression by enhancing HIF1A and glycolytic metabolism. Cell Death Dis. 2022, 13, 943. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Boso, D.; Rampazzo, E.; Zanon, C.; Bresolin, S.; Maule, F.; Porcù, E.; Cani, A.; Della Puppa, A.; Trentin, L.; Basso, G.; et al. HIF-1α/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics 2019, 9, 4860–4877. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, X.F. HIF-miR-215-KDM1B promotes glioma-initiating cell adaptation to hypoxia. Cell Cycle 2016, 15, 1939–1940. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cheng, X.; Wang, X.; Wang, J.; Wen, X.; Xie, C.; Liao, C. Clinical implications of hypoxia-inducible factor-1α and caveolin-1 overexpression in isocitrate dehydrogenase-wild type glioblastoma multiforme. Oncol. Lett. 2019, 17, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Bache, M.; Rot, S.; Keßler, J.; Güttler, A.; Wichmann, H.; Greither, T.; Wach, S.; Taubert, H.; Söling, A.; Bilkenroth, U.; et al. mRNA expression levels of hypoxia-induced and stem cell-associated genes in human glioblastoma. Oncol. Rep. 2015, 33, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Erpolat, O.P.; Gocun, P.U.; Akmansu, M.; Ozgun, G.; Akyol, G. Hypoxia-related molecules HIF-1α, CA9, and osteopontin: Predictors of survival in patients with high-grade glioma. Strahlenther. Onkol. 2013, 189, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kaynar, M.Y.; Sanus, G.Z.; Hnimoglu, H.; Kacira, T.; Kemerdere, R.; Atukeren, P.; Gumustas, K.; Canbaz, B.; Tanriverdi, T. Expression of hypoxia inducible factor-1alpha in tumors of patients with glioblastoma multiforme and transitional meningioma. J. Clin. Neurosci. 2008, 15, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Matsuo, T.; Nagata, I. Protocol of radiotherapy for glioblastoma according to the expression of HIF-1. Brain Tumor Pathol. 2004, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Tang, Y.; Zhou, Y.; Zhu, L.; Gao, C.; Li, W.; You, W.; Yu, B.; et al. Correlation of Nrf2 and HIF-1α in glioblastoma and their relationships to clinicopathologic features and survival. Neurol. Res. 2013, 35, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Sfifou, F.; Hakkou, E.M.; Bouaiti, E.A.; Slaoui, M.; Errihani, H.; Al Bouzidi, A.; Abouqal, R.; El Ouahabi, A.; Cherradi, N. Correlation of immunohistochemical expression of HIF-1alpha and IDH1 with clinicopathological and therapeutic data of moroccan glioblastoma and survival analysis. Ann. Med. Surg. 2021, 69, 102731. [Google Scholar] [CrossRef] [PubMed]
- Potharaju, M.; Mathavan, A.; Mangaleswaran, B.; Patil, S.; John, R.; Ghosh, S.; Kalavakonda, C.; Ghosh, M.; Verma, R.S. Clinicopathological Analysis of HIF-1alpha and TERT on Survival Outcome in Glioblastoma Patients: A Prospective, Single Institution Study. J. Cancer 2019, 10, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Clara, C.A.; Marie, S.K.; de Almeida, J.R.; Wakamatsu, A.; Oba-Shinjo, S.M.; Uno, M.; Neville, M.; Rosemberg, S. Angiogenesis and expression of PDGF-C, VEGF, CD105 and HIF-1α in human glioblastoma. Neuropathology 2014, 34, 343–352. [Google Scholar] [CrossRef] [PubMed]
- El-Benhawy, S.A.; Sakr, O.A.; Fahmy, E.I.; Ali, R.A.; Hussein, M.S.; Nassar, E.M.; Salem, S.M.; Abu-Samra, N.; Elzawawy, S. Assessment of Serum Hypoxia Biomarkers Pre- and Post-radiotherapy in Patients with Brain Tumors. J. Mol. Neurosci. 2022, 72, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Begagić, E.; Pugonja, R.; Bečulić, H.; Čeliković, A.; Tandir Lihić, L.; Kadić Vukas, S.; Čejvan, L.; Skomorac, R.; Selimović, E.; Jaganjac, B.; et al. Molecular Targeted Therapies in Glioblastoma Multiforme: A Systematic Overview of Global Trends and Findings. Brain Sci. 2023, 13, 1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Yu, H.Y.; Zhao, G.X.; Jiang, X.Z.; Gao, G.; Wei, B.J. Global research trends in immunotherapy for glioma: A comprehensive visualization and bibliometric analysis. Front. Endocrinol. 2023, 14, 1273634. [Google Scholar] [CrossRef] [PubMed]
- Łaba, A.E.; Ziółkowski, P. Trends in glioblastoma treatment research: An analysis of clinical trials and literature. Neurol. Neurochir. Pol. 2021, 55, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013, 15, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C. Use of laboratory studies for the design, explanation, and validation of human micronutrient intervention studies. J. Nutr. 2012, 142, 157s–160s. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martinez, O.; García-Montero, C.; Callejón-Peláez, E.; Sáez, M.A.; Álvarez-Mon, M.A.; García-Honduvilla, N.; Monserrat, J.; Álvarez-Mon, M.; Bujan, J.; et al. A General Overview on the Hyperbaric Oxygen Therapy: Applications, Mechanisms and Translational Opportunities. Medicina 2021, 57, 864. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Tsuneyama, K.; Yen, M.-H.; Lee, J.-T.; Chen, J.-L.; Huang, S.-M. Hyperbaric oxygen suppressed tumor progression through the improvement of tumor hypoxia and induction of tumor apoptosis in A549-cell-transferred lung cancer. Sci. Rep. 2021, 11, 12033. [Google Scholar] [CrossRef]
- Herrera-Campos, A.B.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Fernández-Cortés, M.; Montuenga, L.M.; Oliver, F.J.; Garcia-Diaz, A. Implications of Hyperoxia over the Tumor Microenvironment: An Overview Highlighting the Importance of the Immune System. Cancers 2022, 14, 2740. [Google Scholar] [CrossRef]
- Trejo-Solis, C.; Silva-Adaya, D.; Serrano-García, N.; Magaña-Maldonado, R.; Jimenez-Farfan, D.; Ferreira-Guerrero, E.; Cruz-Salgado, A.; Castillo-Rodriguez, R.A. Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. Int. J. Mol. Sci. 2023, 24, 17633. [Google Scholar] [CrossRef] [PubMed]
- Beylerli, O.; Beilerli, A.; Shumadalova, A.; Wang, X.; Yang, M.; Sun, H.; Teng, L. Therapeutic effect of natural polyphenols against glioblastoma. Front. Cell Dev. Biol. 2022, 10, 1036809. [Google Scholar] [CrossRef] [PubMed]
- Luís, Â.; Amaral, L.; Domingues, F.; Pereira, L.; Cascalheira, J.F. Action of Curcumin on Glioblastoma Growth: A Systematic Review with Meta-Analysis of Animal Model Studies. Biomedicines 2024, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, F.; Omata, D.; Munakata, L.; Kageyama, S.; Maruyama, K.; Kudo, N.; Suzuki, R. Brain Delivery of Cisplatin Using Microbubbles in Combination with Ultrasound as an Effective Therapy for Glioblastoma. Pharmaceuticals 2023, 16, 1599. [Google Scholar] [CrossRef] [PubMed]
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Muniraj, N.; Siddharth, S.; Sharma, D. Bioactive Compounds: Multi-Targeting Silver Bullets for Preventing and Treating Breast Cancer. Cancers 2019, 11, 1563. [Google Scholar] [CrossRef] [PubMed]
- Di Nunzio, M.R.; Douhal, A. Robust Inclusion Complex of Topotecan Comprised within a Rhodamine-Labeled β-Cyclodextrin: Competing Proton and Energy Transfer Processes. Pharmaceutics 2023, 15, 1620. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T. Current Status and Future Perspective in Glioma Invasion Research. Brain Sci. 2024, 14, 309. [Google Scholar] [CrossRef] [PubMed]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewska, S.; Sławski, J.; Collawn, J.F.; Bartoszewski, R. HIF-1-Induced hsa-miR-429: Understanding Its Direct Targets as the Key to Developing Cancer Diagnostics and Therapies. Cancers 2023, 15, 2903. [Google Scholar] [CrossRef] [PubMed]
- Englert-Golon, M.; Tokłowicz, M.; Żbikowska, A.; Sajdak, S.; Kotwicka, M.; Andrusiewicz, M. Differential Expression of HIF1A, EPAS1, and VEGF Genes in Benign and Malignant Ovarian Neoplasia. Cancers 2022, 14, 4899. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Ramar, V.; Guo, S.; Hudson, B.; Liu, M. Progress in Glioma Stem Cell Research. Cancers 2024, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Oishi, T.; Koizumi, S.; Kurozumi, K. Molecular Mechanisms and Clinical Challenges of Glioma Invasion. Brain Sci. 2022, 12, 291. [Google Scholar] [CrossRef] [PubMed]
- Qannita, R.A.; Alalami, A.I.; Harb, A.A.; Aleidi, S.M.; Taneera, J.; Abu-Gharbieh, E.; El-Huneidi, W.; Saleh, M.A.; Alzoubi, K.H.; Semreen, M.H.; et al. Targeting Hypoxia-Inducible Factor-1 (HIF-1) in Cancer: Emerging Therapeutic Strategies and Pathway Regulation. Pharmaceuticals 2024, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Pantazopoulou, V.; Jeannot, P.; Rosberg, R.; Berg, T.J.; Pietras, A. Hypoxia-Induced Reactivity of Tumor-Associated Astrocytes Affects Glioma Cell Properties. Cells 2021, 10, 613. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Moreno-Murciano, P.; Oriol-Caballo, M.; López-Blanch, R.; Pineda, B.; Gutiérrez-Arroyo, J.L.; Loras, A.; Gonzalez-Bonet, L.G.; Martinez-Cadenas, C.; Estrela, J.M.; et al. Glioblastoma Therapy: Past, Present and Future. Int. J. Mol. Sci. 2024, 25, 2529. [Google Scholar] [PubMed]
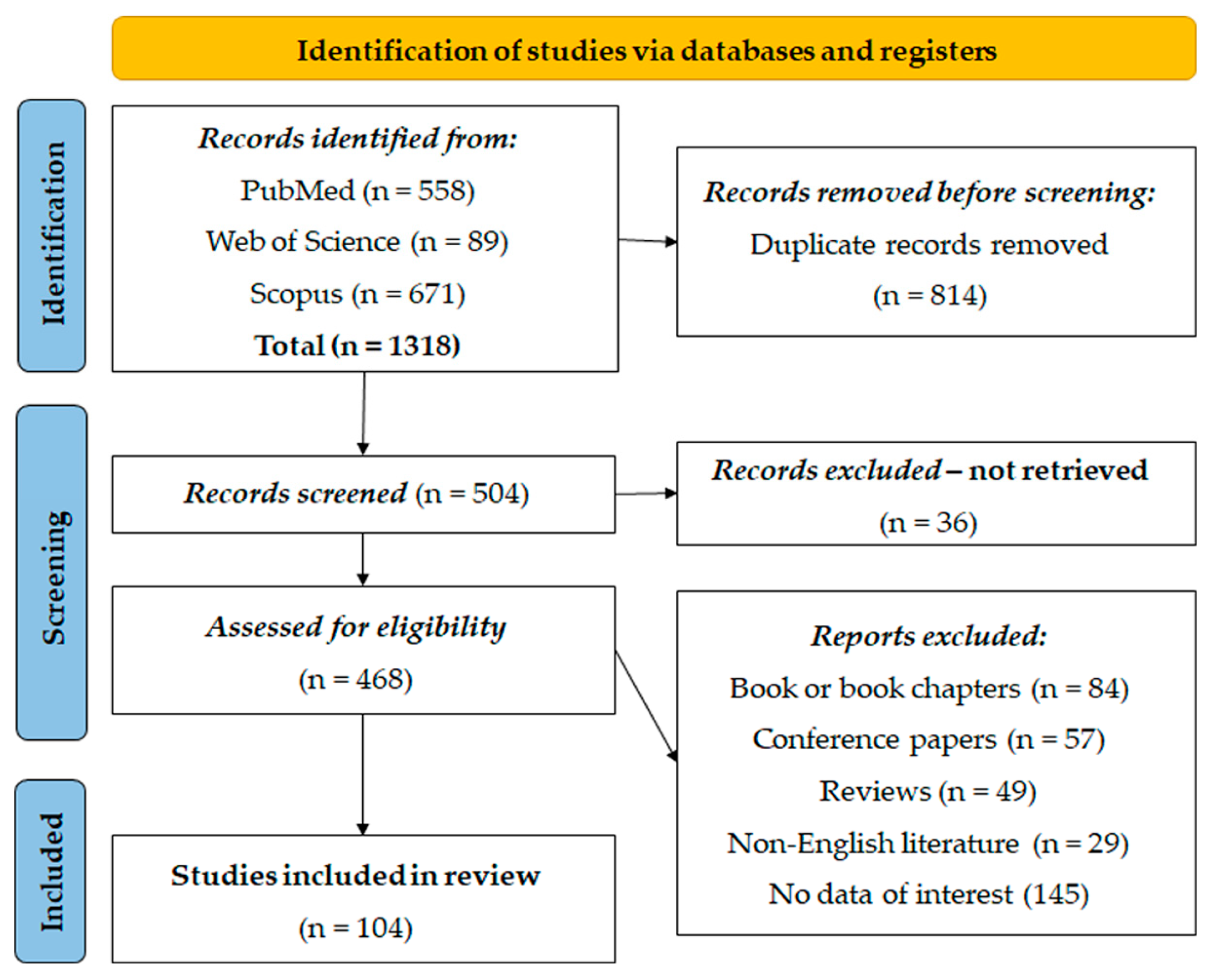
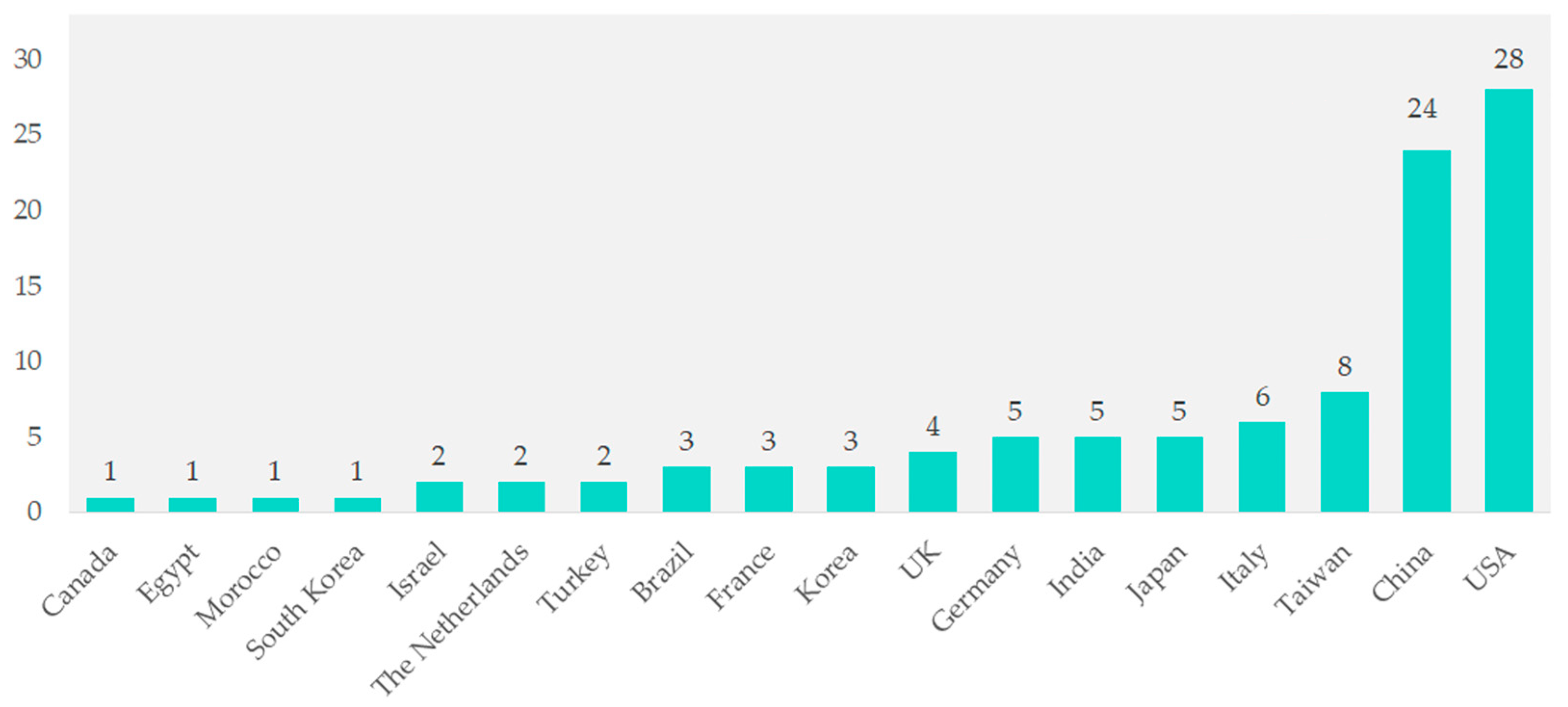
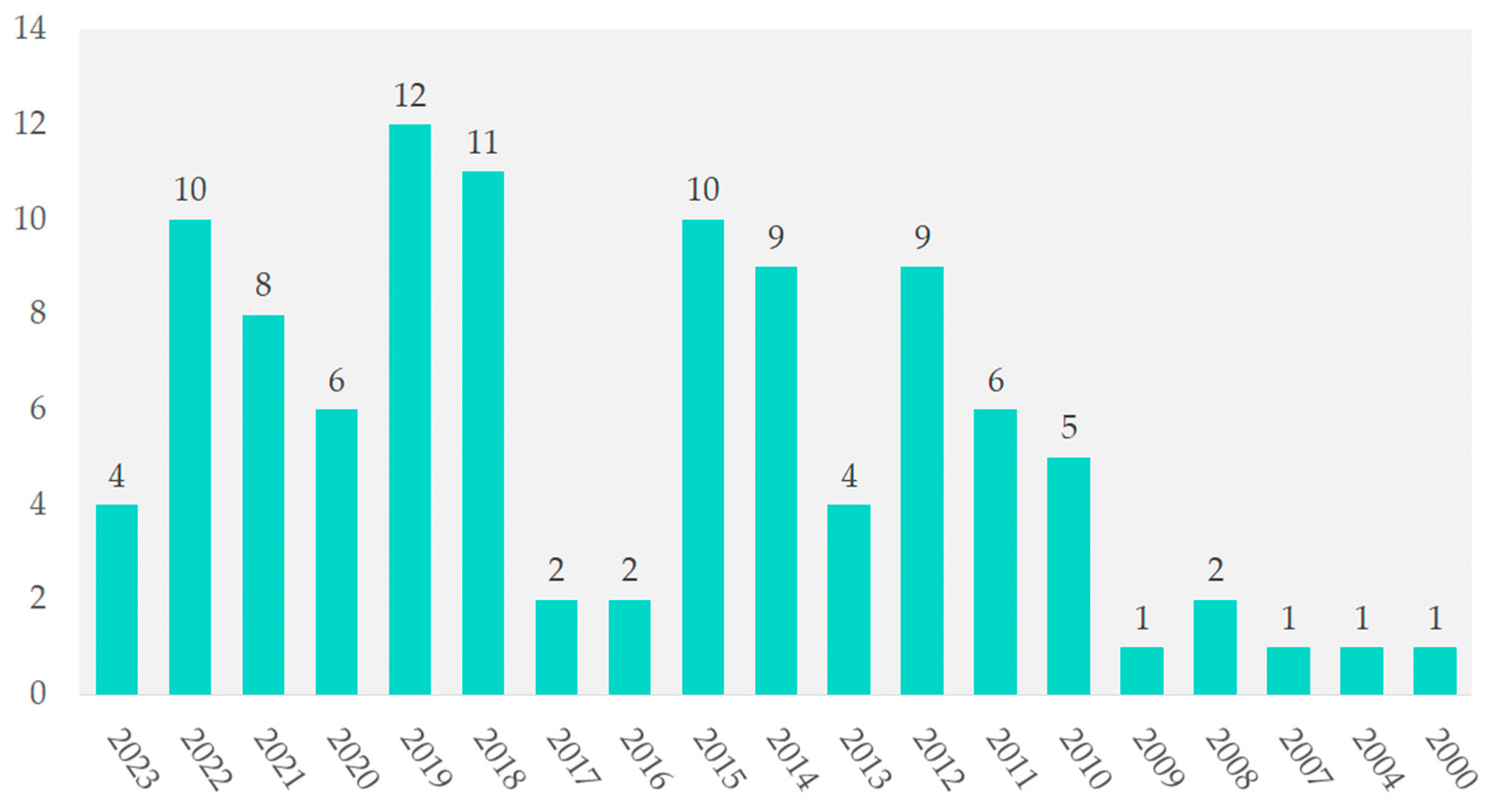
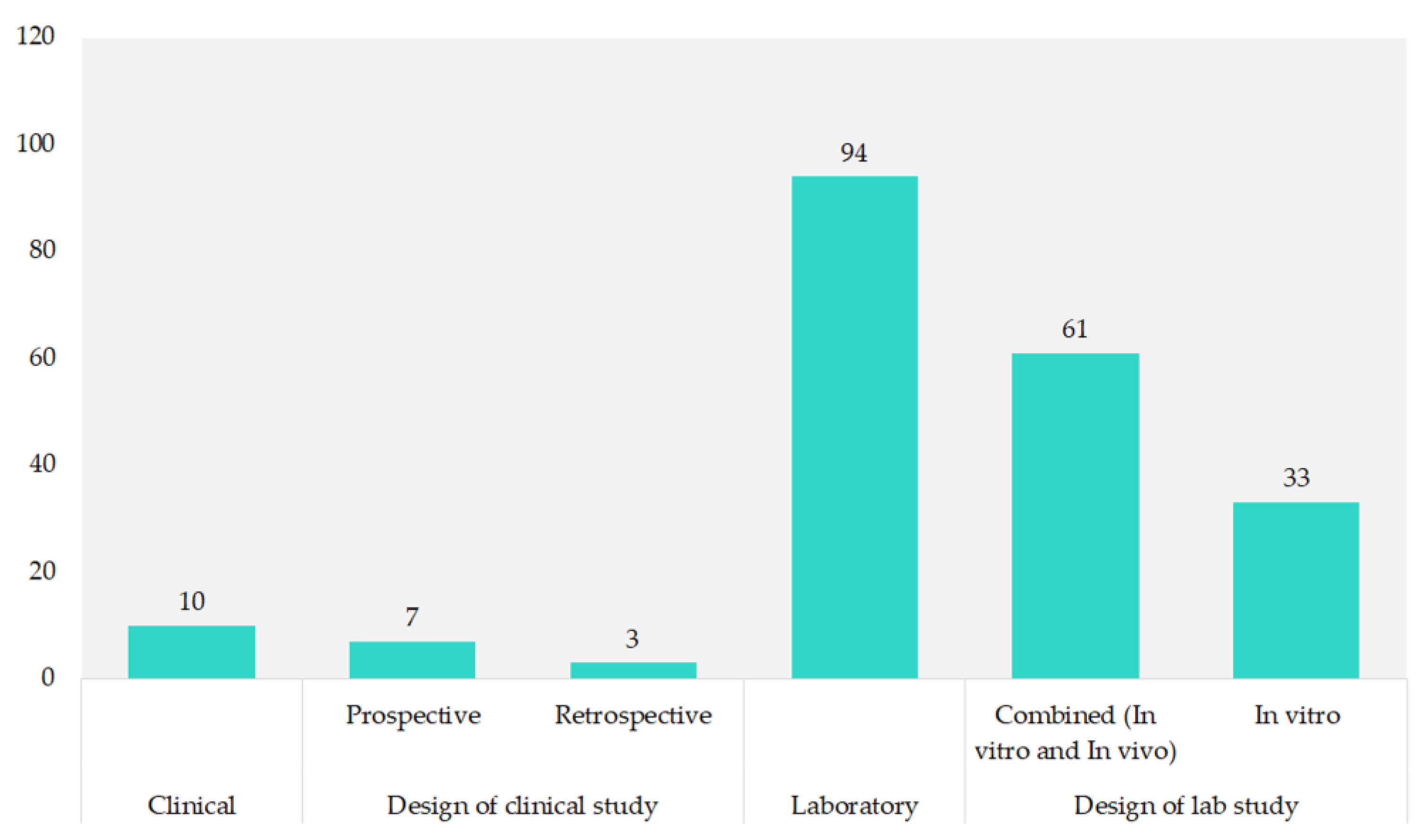
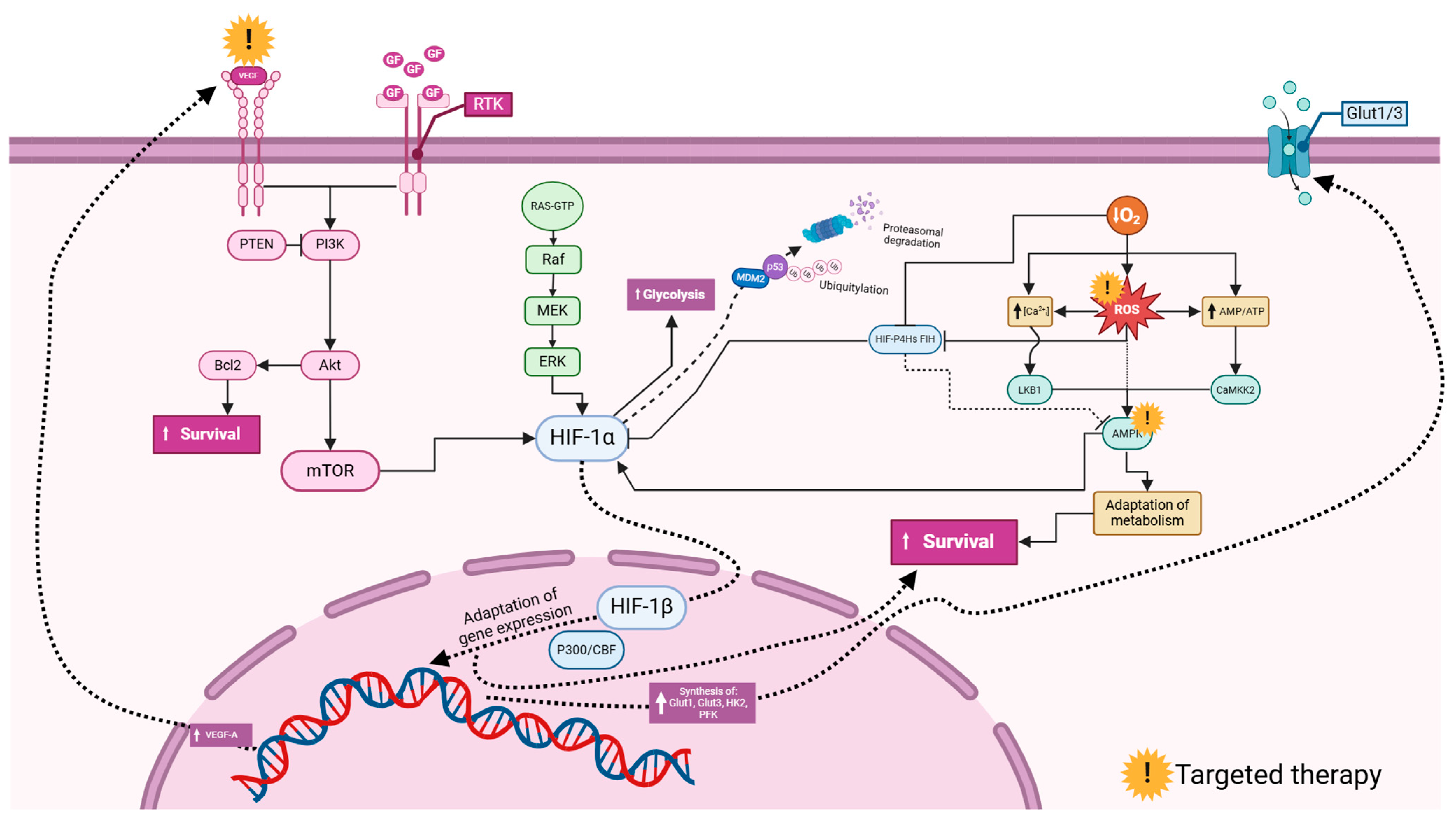
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).