
Identifying Targetable Vulnerabilities to Circumvent or Overcome Venetoclax Resistance in Diffuse Large B-Cell Lymphoma

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DLBCL Cell Lines
2.2. Generation of Venetoclax-Resistant Cell Lines
2.3. Inhibitors and Drug/Compound Screen
2.4. Cell Survival and Synergy Analyses
2.5. Western Blotting
2.6. Caspase-3/7 Apoptosis Analysis
2.7. Live/Dead Assay
2.8. RNA-Sequencing and Data Availability
2.9. Sequencing BCL2, BAX, and TP53
2.10. Flow Cytometry Intracellular Protein Analysis
2.11. Patient Samples
2.12. Statistics
3. Results
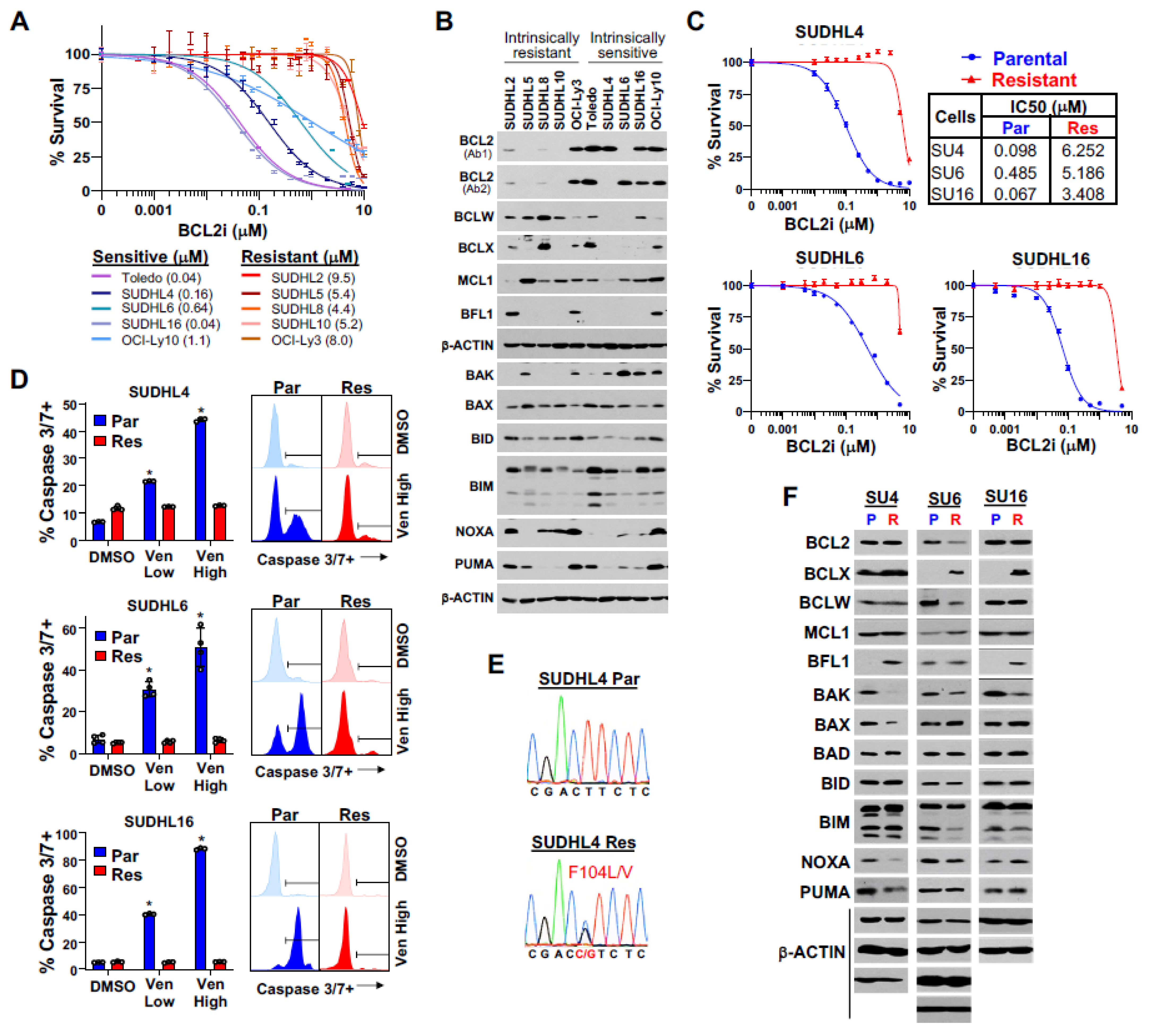
3.1. Differences in BCL2 Family Member Expression in Intrinsically Venetoclax-Sensitive and -Resistant DLBCL Cells
3.2. BCL2 Family Member Alterations in Acquired Venetoclax Resistance in DLBCL Cells
3.3. Overcoming Venetoclax Resistance in DLBCL Cells with Combination BCL2 Family Member Inhibition
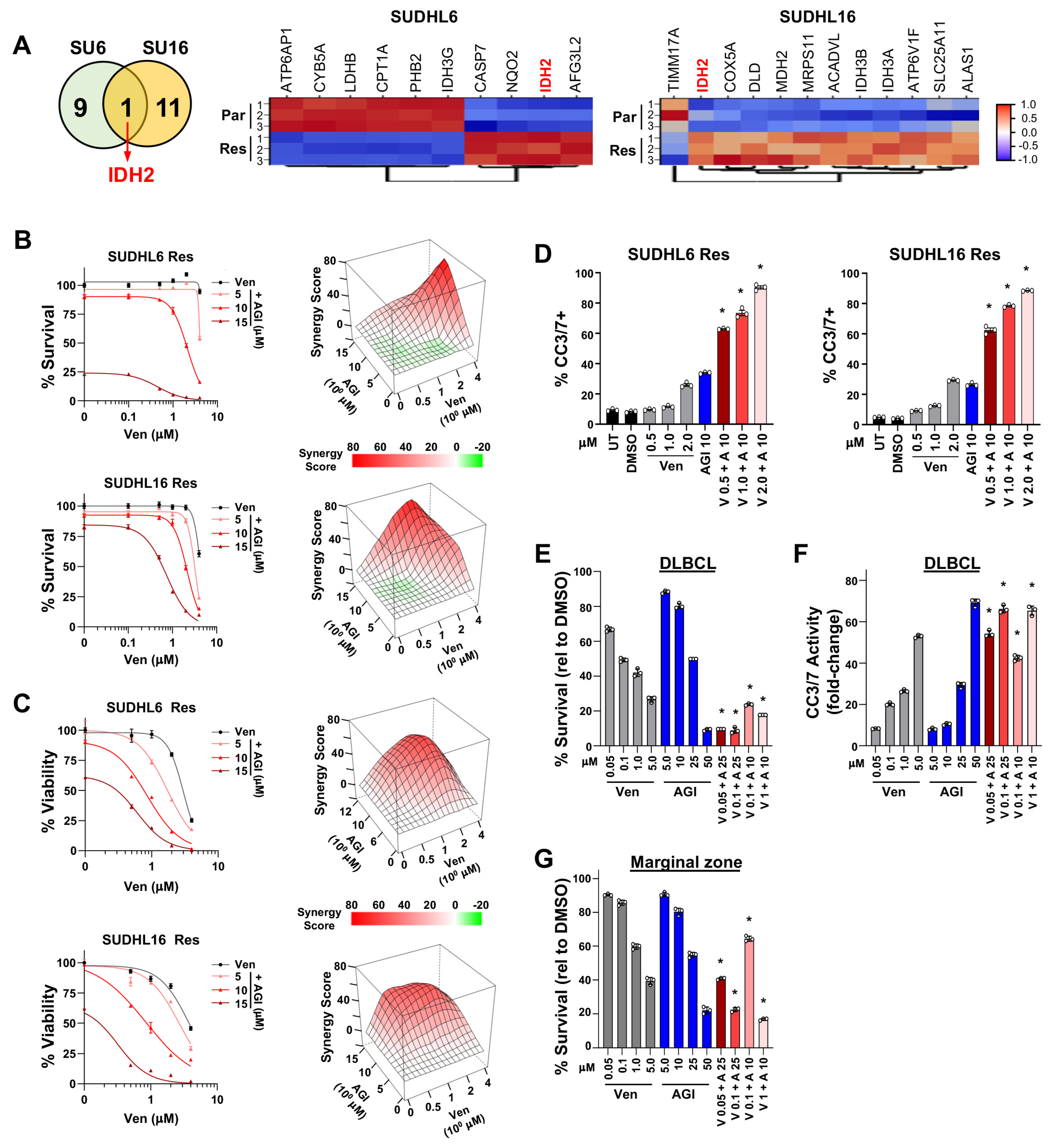
3.4. Combination Treatment of DLBCL, Follicular, and Marginal Zone Lymphoma Patient Samples with BCL2 Family Inhibitors
3.5. Transcriptomic and Drug Screen Analyses Identify Oxidative Phosphorylation as a Target-Able Vulnerability in Acquired Venetoclax-Resistant DLBCL
3.6. Synergistic Effects Co-Targeting BCL2 and IDH2 in Venetoclax-Resistant DLBCL
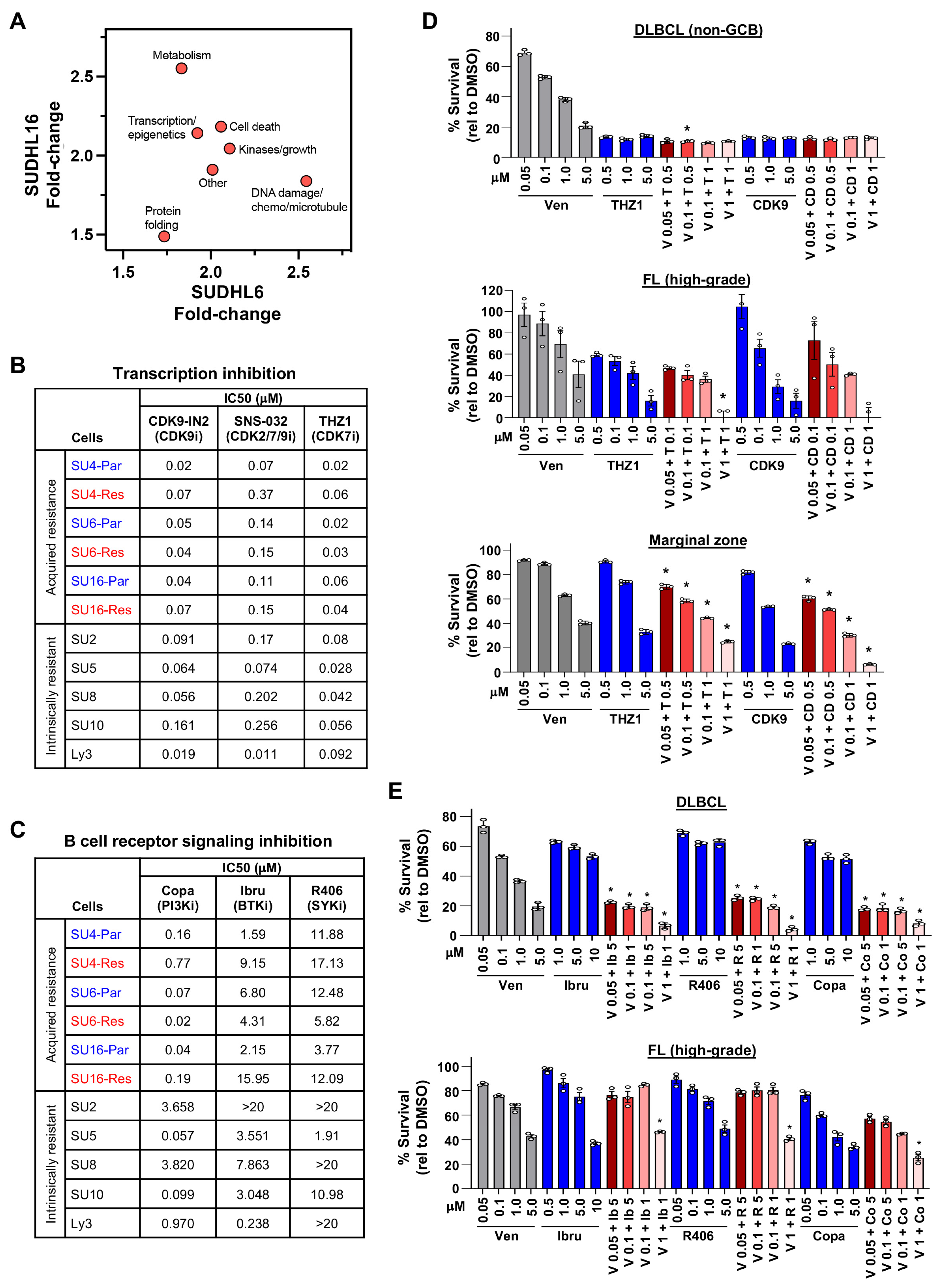
3.7. Drug/Compound Screens Identify Targetable Pathways to Circumvent DLBCL Venetoclax Resistance
3.8. DLBCL Patient Samples Were Sensitive to CDK7/9 and BCR Pathway Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Xu-Monette, Z.Y.; Pham, L.V.; Wang, X.; Tzankov, A.; Fang, X.; Zhu, F.; Visco, C.; Bhagat, G.; Dybkaer, K.; et al. Aggressive B-cell Lymphoma with MYC/TP53 Dual Alterations Displays Distinct Clinicopathobiological Features and Response to Novel Targeted Agents. Mol. Cancer Res. MCR 2021, 19, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 2018, 8, 636. [Google Scholar] [CrossRef]
- Vogler, M.; Walter, H.S.; Dyer, M.J.S. Targeting anti-apoptotic BCL2 family proteins in haematological malignancies—From pathogenesis to treatment. Br. J. Haematol. 2017, 178, 364–379. [Google Scholar] [CrossRef]
- Huang, J.Z.; Sanger, W.G.; Greiner, T.C.; Staudt, L.M.; Weisenburger, D.D.; Pickering, D.L.; Lynch, J.C.; Armitage, J.O.; Warnke, R.A.; Alizadeh, A.A.; et al. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood 2002, 99, 2285–2290. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef]
- Monni, O.; Joensuu, H.; Franssila, K.; Klefstrom, J.; Alitalo, K.; Knuutila, S. BCL2 overexpression associated with chromosomal amplification in diffuse large B-cell lymphoma. Blood 1997, 90, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Meyer, P.N.; Smith, L.M.; Johnson, N.A.; Vose, J.M.; Greiner, T.C.; Connors, J.M.; Staudt, L.M.; Rimsza, L.; Jaffe, E.; et al. BCL2 predicts survival in germinal center B-cell-like diffuse large B-cell lymphoma treated with CHOP-like therapy and rituximab. Clin. Cancer Res. 2011, 17, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Barrans, S.L.; Evans, P.A.; O’Connor, S.J.; Kendall, S.J.; Owen, R.G.; Haynes, A.P.; Morgan, G.J.; Jack, A.S. The t(14;18) is associated with germinal center-derived diffuse large B-cell lymphoma and is a strong predictor of outcome. Clin. Cancer Res. 2003, 9, 2133–2139. [Google Scholar] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Riedell, P.A.; Smith, S.M. Double hit and double expressors in lymphoma: Definition and treatment. Cancer 2018, 124, 4622–4632. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.; Greil, R.; Illes, A.; Johnson, N.A.; Larouche, J.F.; Lugtenburg, P.J.; Patti, C.; et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood 2021, 137, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Condoluci, A.; Rossi, D. Mechanisms of resistance to venetoclax. Blood 2022, 140, 2094–2096. [Google Scholar] [CrossRef] [PubMed]
- Thomalla, D.; Beckmann, L.; Grimm, C.; Oliverio, M.; Meder, L.; Herling, C.D.; Nieper, P.; Feldmann, T.; Merkel, O.; Lorsy, E.; et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood 2022, 140, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, R.; Tian, L.; Anderson, M.A.; Flensburg, C.; Jarratt, A.; Garnham, A.L.; Jabbari, J.S.; Peng, H.; Lew, T.E.; Teh, C.E.; et al. Single-cell multiomics reveal the scale of multilayered adaptations enabling CLL relapse during venetoclax therapy. Blood 2022, 140, 2127–2141. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genom. Proteom. Bioinform. 2022, 20, 587–596. [Google Scholar] [CrossRef]
- Adams, C.M.; Mitra, R.; Xiao, Y.; Michener, P.; Palazzo, J.; Chao, A.; Gour, J.; Cassel, J.; Salvino, J.M.; Eischen, C.M. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023, 13, 1210–1229. [Google Scholar] [CrossRef]
- Pham, L.V.; Huang, S.; Zhang, H.; Zhang, J.; Bell, T.; Zhou, S.; Pogue, E.; Ding, Z.; Lam, L.; Westin, J.; et al. Strategic Therapeutic Targeting to Overcome Venetoclax Resistance in Aggressive B-cell Lymphomas. Clin. Cancer Res. 2018, 24, 3967–3980. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.C.; Xiao, Y.; Lam, L.T.; Litvinovich, E.; Roberts-Rapp, L.; Souers, A.J.; Leverson, J.D. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. 2015, 5, e368. [Google Scholar] [CrossRef]
- Singh, G.; Guibao, C.D.; Seetharaman, J.; Aggarwal, A.; Grace, C.R.; McNamara, D.E.; Vaithiyalingam, S.; Waddell, M.B.; Moldoveanu, T. Structural basis of BAK activation in mitochondrial apoptosis initiation. Nat. Commun. 2022, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G.; Eberth, S.; Nagel, S.; MacLeod, R.A. Malignant hematopoietic cell lines: In vitro models for double-hit B-cell lymphomas. Leuk. Lymphoma 2016, 57, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef] [PubMed]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Brown, F.C.; Chua, C.C.; Dengler, M.A.; Pomilio, G.; Anstee, N.S.; Litalien, V.; Thompson, E.R.; Morley, T.D.; MacRaild, S.; et al. Acquired mutations in BAX confer resistance to BH3-mimetic therapy in Acute Myeloid Leukemia. Blood 2022, 141, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Lew, T.E.; Dengler, M.A.; Thompson, E.R.; Lin, V.S.; Chen, X.; Nguyen, T.; Panigrahi, A.; Handunnetti, S.M.; Carney, D.A.; et al. Clonal hematopoiesis, myeloid disorders and BAX-mutated myelopoiesis in patients receiving venetoclax for CLL. Blood 2022, 139, 1198–1207. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Thijssen, R.; Diepstraten, S.T.; Moujalled, D.M.; Chew, E.; Flensburg, C.; Shi, M.X.; Dengler, M.A.; Litalien, V.; MacRaild, S.; Chen, M.; et al. Intact TP53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 2021, 137, 2721–2735. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra240. [Google Scholar] [CrossRef]
- Szlavik, Z.; Ondi, L.; Csekei, M.; Paczal, A.; Szabo, Z.B.; Radics, G.; Murray, J.; Davidson, J.; Chen, I.; Davis, B.; et al. Structure-Guided Discovery of a Selective Mcl-1 Inhibitor with Cellular Activity. J. Med. Chem. 2019, 62, 6913–6924. [Google Scholar] [CrossRef]
- Merino, D.; Khaw, S.L.; Glaser, S.P.; Anderson, D.J.; Belmont, L.D.; Wong, C.; Yue, P.; Robati, M.; Phipson, B.; Fairlie, W.D.; et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 2012, 119, 5807–5816. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Mitra, R.; Gong, J.Z.; Eischen, C.M. Non-Hodgkin and Hodgkin Lymphomas Select for Overexpression of BCLW. Clin. Cancer Res. 2017, 23, 7119–7129. [Google Scholar] [CrossRef] [PubMed]
- Lossos, I.S.; Gascoyne, R.D. Transformation of follicular lymphoma. Best. Pract. Res. Clin. Haematol. 2011, 24, 147–163. [Google Scholar] [CrossRef]
- Casulo, C.; Friedberg, J. Transformation of marginal zone lymphoma (and association with other lymphomas). Best. Pract. Res. Clin. Haematol. 2017, 30, 131–138. [Google Scholar] [CrossRef]
- Nagasawa, J.; Mizokami, A.; Koshida, K.; Yoshida, S.; Naito, K.; Namiki, M. Novel HER2 selective tyrosine kinase inhibitor, TAK-165, inhibits bladder, kidney and androgen-independent prostate cancer in vitro and in vivo. Int. J. Urol. 2006, 13, 587–592. [Google Scholar] [CrossRef]
- Baccelli, I.; Gareau, Y.; Lehnertz, B.; Gingras, S.; Spinella, J.F.; Corneau, S.; Mayotte, N.; Girard, S.; Frechette, M.; Blouin-Chagnon, V.; et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 2019, 36, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, R.; Yang, Z.; Duan, Z.; Yin, D.; Zhou, Y. Biological Roles and Therapeutic Applications of IDH2 Mutations in Human Cancer. Front. Oncol. 2021, 11, 644857. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omede, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Tan, Z.; Lu, J.; Li, L.; Song, X.; Shi, F.; Xie, L.; You, S.; Luo, X.; et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1alpha in lung cancer. Theranostics 2018, 8, 4050–4061. [Google Scholar] [CrossRef]
- Zeng, P.; Lu, W.; Tian, J.; Qiao, S.; Li, J.; Glorieux, C.; Wen, S.; Zhang, H.; Li, Y.; Huang, P. Reductive TCA cycle catalyzed by wild-type IDH2 promotes acute myeloid leukemia and is a metabolic vulnerability for potential targeted therapy. J. Hematol. Oncol. 2022, 15, 30. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2019, 10, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef]
- Lasica, M.; Anderson, M.A. Review of Venetoclax in CLL, AML and Multiple Myeloma. J. Pers. Med. 2021, 11, 463. [Google Scholar] [CrossRef]
- Zhao, S.; Kanagal-Shamanna, R.; Navsaria, L.; Ok, C.Y.; Zhang, S.; Nomie, K.; Han, G.; Hao, D.; Hill, H.A.; Jiang, C.; et al. Efficacy of venetoclax in high risk relapsed mantle cell lymphoma (MCL)—Outcomes and mutation profile from venetoclax resistant MCL patients. Am. J. Hematol. 2020, 95, 623–629. [Google Scholar] [CrossRef]
- Adams, C.M.; Kim, A.S.; Mitra, R.; Choi, J.K.; Gong, J.Z.; Eischen, C.M. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J. Clin. Invest. 2017, 127, 635–650. [Google Scholar] [CrossRef]
- Thus, Y.J.; Eldering, E.; Kater, A.P.; Spaargaren, M. Tipping the balance: Toward rational combination therapies to overcome venetoclax resistance in mantle cell lymphoma. Leukemia 2022, 36, 2165–2176. [Google Scholar] [CrossRef]
- Yuda, J.; Will, C.; Phillips, D.C.; Abraham, L.; Alvey, C.; Avigdor, A.; Buck, W.; Besenhofer, L.; Boghaert, E.; Cheng, D.; et al. Selective MCL-1 inhibitor ABBV-467 is efficacious in tumor models but is associated with cardiac troponin increases in patients. Commun. Med. 2023, 3, 154. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Diepstraten, S.T.; Herold, M.J. Last but not least: BFL-1 as an emerging target for anti-cancer therapies. Biochem. Soc. Trans. 2022, 50, 1119–1128. [Google Scholar] [CrossRef]
- Boiko, S.; Proia, T.; San Martin, M.; Gregory, G.P.; Wu, M.M.; Aryal, N.; Hattersley, M.; Shao, W.; Saeh, J.C.; Fawell, S.E.; et al. Targeting Bfl-1 via acute CDK9 inhibition overcomes intrinsic BH3-mimetic resistance in lymphomas. Blood 2021, 137, 2947–2957. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.C.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 2019, 35, 752–766. [Google Scholar] [CrossRef]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Rava, M.; D’Andrea, A.; Nicoli, P.; Gritti, I.; Donati, G.; Doni, M.; Giorgio, M.; Olivero, D.; Amati, B. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci. Transl. Med. 2018, 10, eaan8723. [Google Scholar] [CrossRef]
- Guieze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384. [Google Scholar] [CrossRef]
- Esteve-Arenys, A.; Valero, J.G.; Chamorro-Jorganes, A.; Gonzalez, D.; Rodriguez, V.; Dlouhy, I.; Salaverria, I.; Campo, E.; Colomer, D.; Martinez, A.; et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 2018, 37, 1830–1844. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–1340. [Google Scholar] [CrossRef]
- Benjamini Y., Y. H. Controlling the False Discovery Rate-a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. BMethodological 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adams, C.M.; McBride, A.; Michener, P.; Shkundina, I.; Mitra, R.; An, H.H.; Porcu, P.; Eischen, C.M. Identifying Targetable Vulnerabilities to Circumvent or Overcome Venetoclax Resistance in Diffuse Large B-Cell Lymphoma. Cancers 2024, 16, 2130. https://doi.org/10.3390/cancers16112130
Adams CM, McBride A, Michener P, Shkundina I, Mitra R, An HH, Porcu P, Eischen CM. Identifying Targetable Vulnerabilities to Circumvent or Overcome Venetoclax Resistance in Diffuse Large B-Cell Lymphoma. Cancers. 2024; 16(11):2130. https://doi.org/10.3390/cancers16112130
Chicago/Turabian StyleAdams, Clare M., Amanda McBride, Peter Michener, Irina Shkundina, Ramkrishna Mitra, Hyun Hwan An, Pierluigi Porcu, and Christine M. Eischen. 2024. "Identifying Targetable Vulnerabilities to Circumvent or Overcome Venetoclax Resistance in Diffuse Large B-Cell Lymphoma" Cancers 16, no. 11: 2130. https://doi.org/10.3390/cancers16112130
APA StyleAdams, C. M., McBride, A., Michener, P., Shkundina, I., Mitra, R., An, H. H., Porcu, P., & Eischen, C. M. (2024). Identifying Targetable Vulnerabilities to Circumvent or Overcome Venetoclax Resistance in Diffuse Large B-Cell Lymphoma. Cancers, 16(11), 2130. https://doi.org/10.3390/cancers16112130