
Importance of Michaelis Constants for Cancer Cell Redox Balance and Lactate Secretion—Revisiting the Warburg Effect

Abstract
:Simple Summary
Abstract
1. Introduction
2. Glucose and Lactate Metabolism in Normal Body Tissues

{kind=link}
{kind=link}
{kind=link}
| Sample | Metabolite | Concentration (mM) | Ratio | References | |||
|---|---|---|---|---|---|---|---|
| Means | Range 1 | SD | n 2 | ||||
| Blood | lactate (resting) | 1.597 | 0.800–3.500 | 0.738 | 10 | [lac]/[pyr] 22.0 | [42,43,44,45,46,47,48,49,50] |
| pyruvate (resting) | 0.073 | 0.032–0.120 | 0.027 | 7 | [46,47,48,49,50,51,52] | ||
| lactate (exercising) | 6.200 | 5.500–7.500 | 0.748 | 5 | - | [45,47,49,50,52] | |
| RBC | lactate | 1.021 | 0.200–1.870 | 0.454 | 7 | [lac]/[pyr] 16.7 | [43,44,46,53,54,55] |
| pyruvate | 0.061 | 0.043–0.083 | 0.013 | 5 | [46,53,54,55] | ||
| NAD+ | 0.051 | 0.040–0.062 | 0.011 | 2 | [NAD+]/[NADH] 1.9 | [46,55] | |
| NADH | 0.027 | 0.027 | 0.000 | 1 | [55] | ||
| Tumor cell line | lactate | 13.033 | 2.000–35.525 | 10.905 | 12 | [lac]/[pyr] 17.8 | [39,56,57] |
| pyruvate | 0.733 | 0.130–5.880 | 1.326 | 19 | [56,57,58,59,60] | ||
| NAD+ | 0.486 | 0.470–0.502 | 0.016 | 2 | [NAD+]/[NADH] 6.9 | [58,59] | |
| NADH | 0.070 | 0.065–0.075 | 0.005 | 2 | [58,59] | ||
3. Key Enzymes and Metabolites at the Pyruvate Junction
4. Switching to the Tumor Metabolic Phenotype
5. Maintaining the Redox Balance under the Forced Growth Conditions in Cancer Cells
6. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Warburg, O. Tests on surviving carcinoma cultures. Biochem. Z. 1923, 142, 317–333. [Google Scholar]
- Warburg, O. On the formation of lactic acid with growth. Biochem. Z. 1925, 160, 307–311. [Google Scholar]
- Cori, C.F.; Cori, G.T. The carbohydrate metabolism of tumors: I. The free sugar, lactic acid, and glycogen content of malignant tumors. J. Biol. Chem. 1925, 64, 11–22. [Google Scholar] [CrossRef]
- Cori, C.F.; Cori, G.T. The carbohydrate metabolism of tumors: II. Changes in the sugar, lactic acid, and CO2-combining power of blood passing through a tumor. J. Biol. Chem. 1925, 65, 397–405. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Otto, A.M. Warburg effect(s)-a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016, 4, 5. [Google Scholar] [CrossRef]
- Kariagina, A.; Lunt, S.Y.; McCormick, J.J. Genomic and metabolomic analysis of step-wise malignant transformation in human skin fibroblasts. Carcinogenesis 2020, 41, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Sheraj, I.; Guray, N.T.; Banerjee, S. A pan-cancer transcriptomic study showing tumor specific alterations in central metabolism. Sci. Rep. 2021, 11, 13637. [Google Scholar] [CrossRef]
- Creighton, C.J. Gene Expression Profiles in Cancers and Their Therapeutic Implications. Cancer J. 2023, 29, 9–14. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Tannock, I.F. Oxygen diffusion and the distribution of cellular radiosensitivity in tumours. Br. J. Radiol. 1972, 45, 515–524. [Google Scholar] [CrossRef]
- Grimes, D.R.; Kelly, C.; Bloch, K.; Partridge, M. A method for estimating the oxygen consumption rate in multicellular tumour spheroids. J. R. Soc. Interface 2014, 11, 20131124. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Teoh, S.T.; Lunt, S.Y. Metabolism in cancer metastasis: Bioenergetics, biosynthesis, and beyond. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 10, e1406. [Google Scholar] [CrossRef]
- Bonnay, F.; Veloso, A.; Steinmann, V.; Kocher, T.; Abdusselamoglu, M.D.; Bajaj, S.; Rivelles, E.; Landskron, L.; Esterbauer, H.; Zinzen, R.P.; et al. Oxidative Metabolism Drives Immortalization of Neural Stem Cells during Tumorigenesis. Cell 2020, 182, 1490–1507.e19. [Google Scholar] [CrossRef]
- Fan, T.W.; Lane, A.N.; Higashi, R.M.; Farag, M.A.; Gao, H.; Bousamra, M.; Miller, D.M. Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 2009, 8, 41. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Kallinowski, F.; Tyler, G.; Mueller-Klieser, W.; Vaupel, P. Growth-related changes of oxygen consumption rates of tumor cells grown in vitro and in vivo. J. Cell Physiol. 1989, 138, 183–191. [Google Scholar] [CrossRef]
- Vaupel, P. Physiological properties of malignant tumours. NMR Biomed. 1992, 5, 220–225. [Google Scholar] [CrossRef]
- Chandel, N.S. Glycolysis. Cold Spring Harb. Perspect. Biol. 2021, 13, a040535. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria. Cold Spring Harb. Perspect. Biol. 2021, 13, a040543. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Zeng, X.; Cowan, A.J.; Wang, L.; Chen, L.; Morscher, R.J.; Reyes, J.; Frezza, C.; Hwang, H.Y.; et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. 2019, 30, 594–606.e3. [Google Scholar] [CrossRef]
- Chandel, N.S. Carbohydrate Metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040568. [Google Scholar] [CrossRef] [PubMed]
- Everse, J.; Kaplan, N.O. Lactate dehydrogenases: Structure and function. Adv. Enzymol. Relat. Areas Mol. Biol. 1973, 37, 61–133. [Google Scholar]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; W.H. Freeman & Company: New York, NY, USA, 2008; p. 1158. [Google Scholar]
- Goto, T.; Sugawara, K.; Nakamura, S.; Kidokoro, S.I.; Wakui, H.; Nunomura, W. Enzymatic and thermodynamic profiles of a heterotetramer lactate dehydrogenase isozyme in swine. Biochem. Biophys. Res. Commun. 2016, 479, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Heinova, D.; Rosival, I.; Avidar, Y.; Bogin, E. Lactate dehydrogenase isoenzyme distribution and patterns in chicken organs. Res. Vet. Sci. 1999, 67, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, J.; Tateyama, K.; Syuto, B.; Too, K. Lactate dehydrogenase and creatine phosphokinase isoenzymes in tissues of laboratory animals. Jpn. J. Vet. Res. 1990, 38, 19–29. [Google Scholar] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Perez-Escuredo, J.; Van Hee, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Cucchi, D.; Mauro, C. Lactate transporters as therapeutic targets in cancer and inflammatory diseases. Expert Opin. Ther. Targets 2018, 22, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. [Google Scholar] [CrossRef]
- Mathew, M.; Nguyen, N.T.; Bhutia, Y.D.; Sivaprakasam, S.; Ganapathy, V. Metabolic Signature of Warburg Effect in Cancer: An Effective and Obligatory Interplay between Nutrient Transporters and Catabolic/Anabolic Pathways to Promote Tumor Growth. Cancers 2024, 16, 504. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Harris, J.E.; Hernandez, A.; Gladden, L.B. Blood lactate measurements and analysis during exercise: A guide for clinicians. J. Diabetes Sci. Technol. 2007, 1, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Rost, F.; Seelig, J. Simultaneous separation of intracellular and extracellular lactate NMR signals of human erythrocytes. Magn. Reson. Med. 2007, 58, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Lormes, W.; Emmert, J.; Liu, Y.; Lehmann, M.; Steinacker, J.M. Lactate concentration in plasma and red blood cells during incremental exercise. Int. J. Sports Med. 2000, 21, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Hurley, B.F.; Hagberg, J.M.; Allen, W.K.; Seals, D.R.; Young, J.C.; Cuddihee, R.W.; Holloszy, J.O. Effect of training on blood lactate levels during submaximal exercise. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Minakami, S.; Suzuki, C.; Saito, T.; Yoshikawa, H. Studies on erythrocyte glycolysis. I. Determination of the glycolytic intermediates in human erythrocytes. J. Biochem. 1965, 58, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.; Golam, K.; Kumar, P.; Patil, N.; Sadacharan, K. Perioperative blood lactate levels, pyruvate levels, and lactate-pyruvate ratio in children undergoing cardiopulmonary bypass for congenital heart disease. Indian J. Crit. Care Med. 2005, 9, 145–150. [Google Scholar]
- Ceperuelo-Mallafre, V.; Reverte, L.; Peraire, J.; Madeira, A.; Maymo-Masip, E.; Lopez-Dupla, M.; Gutierrez-Valencia, A.; Ruiz-Mateos, E.; Buzon, M.J.; Jorba, R.; et al. Circulating pyruvate is a potent prognostic marker for critical COVID-19 outcomes. Front. Immunol. 2022, 13, 912579. [Google Scholar] [CrossRef]
- Carter, H.; Jones, A.M.; Doust, J.H. Changes in blood lactate and pyruvate concentrations and the lactate-to-pyruvate ratio during the lactate minimum speed test. J. Sports Sci. 2000, 18, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Pianosi, P.; Seargeant, L.; Haworth, J.C. Blood lactate and pyruvate concentrations, and their ratio during exercise in healthy children: Developmental perspective. Eur. J. Appl. Physiol. Occup. Physiol. 1995, 71, 518–522. [Google Scholar] [CrossRef]
- Landon, J.; Fawcett, J.K.; Wynn, V. Blood pyruvate concentration measured by a specific method in control subjects. J. Clin. Pathol. 1962, 15, 579–584. [Google Scholar] [CrossRef]
- Freund, H.; Marbach, J.; Ott, C.; Lonsdorfer, J.; Heitz, A.; Zouloumian, P.; Kehayoff, P. Blood pyruvate recovery curves after short heavy submaximal exercise in man. Eur. J. Appl. Physiol. Occup. Physiol. 1980, 43, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; Meo, F.; Scapigliati, A.; Sciarra, M.; Schiavello, R. Red blood cell energy metabolism during cardiopulmonary bypass. J. Cardiovasc. Surg. 1999, 40, 653–657. [Google Scholar]
- Niessner, H.; Beutler, E. Fluorometric analysts of glycolytic intermediates in human red blood cells. Biochem. Med. 1973, 8, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.E.; Omachi, A. Measured and calculated NAD+-NADH ratios in human erythrocytes. Biochim. Biophys. Acta 1974, 354, 1–10. [Google Scholar] [CrossRef]
- Rost, L.M.; Brekke Thorfinnsdottir, L.; Kumar, K.; Fuchino, K.; Eide Langorgen, I.; Bartosova, Z.; Kristiansen, K.A.; Bruheim, P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites 2020, 10, 74. [Google Scholar] [CrossRef]
- Xie, J.; Dai, C.; Hu, X. Evidence That Does Not Support Pyruvate Kinase M2 (PKM2)-catalyzed Reaction as a Rate-limiting Step in Cancer Cell Glycolysis. J. Biol. Chem. 2016, 291, 8987–8999. [Google Scholar] [CrossRef] [PubMed]
- Park, J.O.; Rubin, S.A.; Xu, Y.F.; Amador-Noguez, D.; Fan, J.; Shlomi, T.; Rabinowitz, J.D. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat. Chem. Biol. 2016, 12, 482–489. [Google Scholar] [CrossRef]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Hu, W.; Wang, Y.; Wu, H.; Zeng, S.; Ying, M.; Hu, X. The thermodynamic state of aerobic glycolytic flux plays a critical role in stabilizing aerobic glycolytic flux of cancer cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hui, S.; Cowan, A.J.; Zeng, X.; Yang, L.; TeSlaa, T.; Li, X.; Bartman, C.; Zhang, Z.; Jang, C.; Wang, L.; et al. Quantitative Fluxomics of Circulating Metabolites. Cell Metab. 2020, 32, 676–688.e4. [Google Scholar] [CrossRef]
- Opie, L.H.; Evans, J.R.; Shipp, J.C. Effect of Fasting on Glucose and Palmitate Metabolism of Perfused Rat Heart. Am. J. Physiol. 1963, 205, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C. Myocardial lactate metabolism during exercise. Med. Sci. Sports Exerc. 1991, 23, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, A.; Nemati, N.; Lamberti, G.; Trefny, M.; Krogsdam, A.; Siller, A.; Hortnagl, P.; Schumacher, P.; Sopper, S.; Sandbichler, A.; et al. (13)C tracer analysis reveals the landscape of metabolic checkpoints in human CD8(+) T cell differentiation and exhaustion. Front. Immunol. 2023, 14, 1267816. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Bartman, C.R.; Jankowski, C.S.R.; Zhang, Z.; Xu, X.; Xing, X.; Wang, L.; Lu, W.; Hui, S.; Rabinowitz, J.D. The Source of Glycolytic Intermediates in Mammalian Tissues. Cell Metab. 2021, 33, 367–378.e5. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Guo, C.; Hu, X. The quantitative relationship between isotopic and net contributions of lactate and glucose to the tricarboxylic acid (TCA) cycle. J. Biol. Chem. 2019, 294, 9615–9630. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Ng, C.P.; Jones, O.; Fung, T.S.; Ryu, K.W.; Li, D.; Thompson, C.B. Lactate activates the mitochondrial electron transport chain independently of its metabolism. Mol. Cell 2023, 83, 3904–3920.e7. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Waste Not, Want Not: Lactate Oxidation Fuels the TCA Cycle. Cell Metab. 2017, 26, 803–804. [Google Scholar] [CrossRef]
- Schormann, N.; Hayden, K.L.; Lee, P.; Banerjee, S.; Chattopadhyay, D. An overview of structure, function, and regulation of pyruvate kinases. Protein Sci. 2019, 28, 1771–1784. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Gassaway, B.M.; Cardone, R.L.; Padyana, A.K.; Petersen, M.C.; Judd, E.T.; Hayes, S.; Tong, S.; Barber, K.W.; Apostolidi, M.; Abulizi, A.; et al. Distinct Hepatic PKA and CDK Signaling Pathways Control Activity-Independent Pyruvate Kinase Phosphorylation and Hepatic Glucose Production. Cell Rep. 2019, 29, 3394–3404.e9. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, D.R.; Cardenas, J.M. Kinetic properties of pyruvate kinase hybrids formed with native type L and inactivated type M subunits. J. Biol. Chem. 1975, 250, 4931–4936. [Google Scholar] [CrossRef] [PubMed]
- Reynard, A.M.; Hass, L.F.; Jacobsen, D.D.; Boyer, P.D. The correlation of reaction kinetics and substrate binding with the mechanism of pyruvate kinase. J. Biol. Chem. 1961, 236, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- van Berkel, T.J.; Koster, J.F.; Hulsmann, W.C. Some kinetic properties of the allosteric M-type pyruvate kinase from rat liver; influence of pH and the nature of amino acid inhibition. Biochim. Biophys. Acta 1973, 321, 171–180. [Google Scholar] [CrossRef] [PubMed]
- van Berkel, T.J.C. Pyruvate Kinase Isoenzymes. Medical Dissertation, Erasmus Universiteit te Rotterdam, Rotterdam, The Netherlands, 1974. [Google Scholar]
- Eigenbrodt, E.; Schoner, W. Purification and properties of the pyruvate kinase isoenzymes type L and M2 from chicken liver. Hoppe Seylers. Z. Physiol. Chem. 1977, 358, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, B.; Baranowski, T. Kinetic properties of human muscle pyruvate kinase. Mol. Cell. Biochem. 1982, 45, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.R.; Cottam, G.L. Isozymes of pyruvate kinase in vertebrates: Their physical, chemical, kinetic and immunological properties. Int. J. Biochem. 1978, 9, 785–793. [Google Scholar] [CrossRef]
- Consler, T.G.; Woodard, S.H.; Lee, J.C. Effects of primary sequence differences on the global structure and function of an enzyme: A study of pyruvate kinase isozymes. Biochemistry 1989, 28, 8756–8764. [Google Scholar] [CrossRef]
- van Berkel, J.C. Some kinetic properties of M2-type pyruvate kinase from rat liver at physiological Mg2+ concentration. Biochim. Biophys. Acta 1974, 370, 140–152. [Google Scholar] [CrossRef] [PubMed]
- van Berkel, T.J.; de Jonge, H.R.; Koster, J.F.; Hulsmann, W.C. Kinetic evidence for the presence of two forms of M2-type pyruvate kinase in rat small intestine. Biochem. Biophys. Res. Commun. 1974, 60, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.P.; O’Reilly, F.J.; Wear, M.A.; O’Neill, J.R.; Fothergill-Gilmore, L.A.; Hupp, T.; Walkinshaw, M.D. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc. Natl. Acad. Sci. USA 2013, 110, 5881–5886. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.M.; Howell, A.J.; Hosios, A.M.; Li, Z.; Israelsen, W.J.; Vander Heiden, M.G. Cancer-associated mutations in human pyruvate kinase M2 impair enzyme activity. FEBS Lett. 2020, 594, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Harano, Y.; Sue, F.; Morimura, H. Crystallization, characterization and metabolic regulation of two types of pyruvate kinase isolated from rat tissues. J. Biochem. 1967, 62, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.F.; Slee, R.G.; Staal, G.E.; van Berkel, T.J. The influence of glucose I,6-diphosphate on the enzymatic activity of pyruvate kinase. Biochim. Biophys. Acta 1972, 258, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P.; Lewis, C.A. Kinetic properties of the partially purified pyruvate dehydrogenase complex of ox brain. Biochem. J. 1973, 131, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kiselevsky, Y.V.; Ostrovtsova, S.A.; Strumilo, S.A. Kinetic characterization of the pyruvate and oxoglutarate dehydrogenase complexes from human heart. Acta Biochim. Pol. 1990, 37, 135–139. [Google Scholar]
- Czygier, M.; Strumilo, S.A. Basic properties of the pyruvate dehydrogenase complex isolated from aurochs heart. Acta Biochim. Pol. 1994, 41, 453–457. [Google Scholar] [CrossRef]
- Fang, R.; Nixon, P.F.; Duggleby, R.G. Identification of the catalytic glutamate in the E1 component of human pyruvate dehydrogenase. FEBS Lett. 1998, 437, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gong, X.; Yan, X.; Peng, T.; Baker, J.C.; Li, L.; Robben, P.M.; Ravindran, S.; Andersson, L.A.; Cole, A.B.; et al. Reaction mechanism for mammalian pyruvate dehydrogenase using natural lipoyl domain substrates. Arch. Biochem. Biophys. 2001, 386, 123–135. [Google Scholar] [CrossRef]
- Wu, Y.G.; Chen, W.Y.; Zhang, Z.W.; Yang, G.Z.; Li, W.; Duggleby, R.G. Biochemical characterization of two mutants of human pyruvate dehydrogenase, F205L and T231A of the E1alpha subunit. J. Inherit. Metab. Dis. 2003, 26, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.; Golbik, R.; Brauer, J.; Lilie, H.; Schroder-Tittmann, K.; Hinze, E.; Korotchkina, L.G.; Patel, M.S.; Tittmann, K. Direct kinetic evidence for half-of-the-sites reactivity in the E1 component of the human pyruvate dehydrogenase multienzyme complex through alternating sites cofactor activation. Biochemistry 2006, 45, 12775–12785. [Google Scholar] [CrossRef]
- Pavlu-Pereira, H.; Lousa, D.; Tome, C.S.; Florindo, C.; Silva, M.J.; de Almeida, I.T.; Leandro, P.; Rivera, I.; Vicente, J.B. Structural and functional impact of clinically relevant E1alpha variants causing pyruvate dehydrogenase complex deficiency. Biochimie 2021, 183, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Jitrapakdee, S.; Walker, M.E.; Wallace, J.C. Functional expression, purification, and characterization of recombinant human pyruvate carboxylase. Biochem. Biophys. Res. Commun. 1999, 266, 512–517. [Google Scholar] [CrossRef]
- Barash, V.; Khassis, S.; Granat, M.; Gutman, A. Pyruvate carboxylase activity in chorionic villi: Possibility of application to prenatal diagnosis. Enzyme 1988, 39, 54–59. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Jenkins, W.T. Alanine aminotransferase. I. Purification and properties. J. Biol. Chem. 1967, 242, 91–100. [Google Scholar] [CrossRef]
- Hopper, S.; Segal, H.L. Kinetic studies of rat liver glutamicalanine transaminase. J. Biol. Chem. 1962, 237, 3189–3195. [Google Scholar] [CrossRef]
- Bulos, B.; Handler, P. Kinetics of Beef Heart Glutamic-Alanine Transaminase. J. Biol. Chem. 1965, 240, 3283–3294. [Google Scholar] [CrossRef]
- McAllister, C.H.; Facette, M.; Holt, A.; Good, A.G. Analysis of the enzymatic properties of a broad family of alanine aminotransferases. PLoS ONE 2013, 8, e55032. [Google Scholar] [CrossRef] [PubMed]
- Peracchi, A.; Polverini, E. Using Steady-State Kinetics to Quantitate Substrate Selectivity and Specificity: A Case Study with Two Human Transaminases. Molecules 2022, 27, 1398. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Jenkins, W.T. Alanine aminotransferase. II. The basis for substrate specificity. J. Biol. Chem. 1967, 242, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bergmeyer, H.U.; Scheibe, P.; Wahlefeld, A.W. Optimization of methods for aspartate aminotransferase and alanine aminotransferase. Clin. Chem. 1978, 24, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Qi, L.; Qiao, J.; Zhang, H.; Ma, H. Study on alanine aminotransferase kinetics by microchip electrophoresis. Anal. Biochem. 2012, 421, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Hakala, M.T.; Glaid, A.J.; Schwert, G.W. Lactic dehydrogenase. II. Variation of kinetic and equilibrium constants with temperature. J. Biol. Chem. 1956, 221, 191–209. [Google Scholar] [CrossRef]
- Nisselbaum, J.S.; Packer, D.E.; Bodansky, O. Comparison of the Actions of Human Brain, Liver, and Heart Lactic Dehydrogenase Variants on Nucleotide Analogues and on Substrate Analogues in the Absence and in the Presence of Oxalate and Oxamate. J. Biol. Chem. 1964, 239, 2830–2834. [Google Scholar] [CrossRef]
- Boland, M.J.; Gutfreund, H. Pig heart lactate dehydrogenase. Binding of pyruvate and the interconversion of pyruvate-containing ternary complexes. Biochem. J. 1975, 151, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Nitisewojo, P.; Hultin, H.O. A comparison of some kinetic properties of soluble and bound lactate dehydrogenase isoenzymes at different temperatures. Eur. J. Biochem. 1976, 67, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Aragon, J.J.; Feliu, J.E.; Frenkel, R.A.; Sols, A. Permeabilization of animal cells for kinetic studies of intracellular enzymes: In situ behavior of the glycolytic enzymes of erythrocytes. Proc. Natl. Acad. Sci. USA 1980, 77, 6324–6328. [Google Scholar] [CrossRef]
- Fernandez-Velasco, D.A.; Garza-Ramos, G.; Ramirez, L.; Shoshani, L.; Darszon, A.; Tuena de Gomez-Puyou, M.; Gomez-Puyou, A. Activity of heart and muscle lactate dehydrogenases in all-aqueous systems and in organic solvents with low amounts of water. Effect of guanidine chloride. Eur. J. Biochem. 1992, 205, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.S.; Moran, J.; Murtaza, B.N.; Muhammad, K.; Ahmad, H. Cloning, E. coli expression, and characterization of heart lactate dehydrogenase B from river buffalo (Bubalus bubalis). Anim. Biotechnol. 2014, 25, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, L.; Wei, D.; Li, X.; Xu, L.; Wei, L. Enzymatic Kinetic Properties of the Lactate Dehydrogenase Isoenzyme C(4) of the Plateau Pika (Ochotona curzoniae). Int. J. Mol. Sci. 2016, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Mali, A.V.; Bhise, S.S.; Katyare, S.S.; Hegde, M.V. Altered Kinetics Properties of Erythrocyte Lactate Dehydrogenase in Type II Diabetic Patients and Its Implications for Lactic Acidosis. Indian J. Clin. Biochem. 2018, 33, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Gammon, S.T.; Sutton, M.N.; Zacharias, N.M.; Bhattacharya, P.; Piwnica-Worms, D. Excess exogenous pyruvate inhibits lactate dehydrogenase activity in live cells in an MCT1-dependent manner. J. Biol. Chem. 2021, 297, 100775. [Google Scholar] [CrossRef] [PubMed]
- Pasti, A.P.; Rossi, V.; Di Stefano, G.; Brigotti, M.; Hochkoeppler, A. Human lactate dehydrogenase A undergoes allosteric transitions under pH conditions inducing the dissociation of the tetrameric enzyme. Biosci. Rep. 2022, 42, BSR20212654. [Google Scholar] [CrossRef] [PubMed]
- Al-Kasimy, L.N.; Al Jebur, L.A.; Maher, F.T. A Kinetic and Thermodynamic Study of The Enzyme Lactate Dehydrogenase Purified from Cardiac Patients’ Blood. HIV Nurs. 2023, 23, 1257–1266. [Google Scholar]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Buhl, S.N.; Jackson, K.Y.; Vanderlinde, R.E. The effect of temperature on the kinetic constants of human lactate dehydrogenase 1 and 5. Clin. Chim. Acta 1977, 80, 265–270. [Google Scholar] [CrossRef]
- Nakae, Y.; Stoward, P.J. The diverse Michaelis constants and maximum velocities of lactate dehydrogenase in situ in various types of cell. Histochem. J. 1994, 26, 292–297. [Google Scholar] [CrossRef]
- Pettit, S.M.; Nealon, D.A.; Henderson, A.R. Purification of lactate dehydrogenase isoenzyme-5 from human liver. Clin. Chem. 1981, 27, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Talaiezadeh, A.; Shahriari, A.; Tabandeh, M.R.; Fathizadeh, P.; Mansouri, S. Kinetic characterization of lactate dehydrogenase in normal and malignant human breast tissues. Cancer Cell Int. 2015, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Nakae, Y.; Stoward, P.J. Kinetic parameters of lactate dehydrogenase in liver and gastrocnemius determined by three quantitative histochemical methods. J. Histochem. Cytochem. 1997, 45, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Maeda, T.; Suzuki, A.; Baba, Y. Cancer metabolism: New insights into classic characteristics. Jpn. Dent. Sci. Rev. 2018, 54, 8–21. [Google Scholar] [CrossRef]
- Marbaniang, C.; Kma, L. Dysregulation of Glucose Metabolism by Oncogenes and Tumor Suppressors in Cancer Cells. Asian Pac. J. Cancer Prev. 2018, 19, 2377–2390. [Google Scholar]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Ancey, P.B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [PubMed]
- Bentaib, A.; De Tullio, P.; Chneiweiss, H.; Hermans, E.; Junier, M.P.; Leprince, P. Metabolic reprogramming in transformed mouse cortical astrocytes: A proteomic study. J. Proteom. 2015, 113, 292–314. [Google Scholar] [CrossRef]
- Wettersten, H.I. Reprogramming of Metabolism in Kidney Cancer. Semin. Nephrol. 2020, 40, 2–13. [Google Scholar] [CrossRef]
- Sugiyama, K.; Ebinuma, H.; Nakamoto, N.; Sakasegawa, N.; Murakami, Y.; Chu, P.S.; Usui, S.; Ishibashi, Y.; Wakayama, Y.; Taniki, N.; et al. Prominent steatosis with hypermetabolism of the cell line permissive for years of infection with hepatitis C virus. PLoS ONE 2014, 9, e94460. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. Oncogenic virus-induced aerobic glycolysis and tumorigenesis. J. Cancer 2018, 9, 3699–3706. [Google Scholar] [CrossRef] [PubMed]
- Gerresheim, G.K.; Bathke, J.; Michel, A.M.; Andreev, D.E.; Shalamova, L.A.; Rossbach, O.; Hu, P.; Glebe, D.; Fricke, M.; Marz, M.; et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. Int. J. Mol. Sci. 2019, 20, 1321. [Google Scholar] [CrossRef]
- Gerresheim, G.K.; Roeb, E.; Michel, A.M.; Niepmann, M. Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells. Cells. 2019, 8, 1410. [Google Scholar] [CrossRef]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8(+) T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663.e8. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Fenton, A.W. A critical review of the role of M(2)PYK in the Warburg effect. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Cros, M.; Hemmerlin, C.; Ferretti, S.; Zhang, J.; Gounarides, J.S.; Yin, H.; Muller, A.; Haberkorn, A.; Chene, P.; Sellers, W.R.; et al. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc. Natl. Acad. Sci. USA 2013, 110, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Bentaib, A.; De Tullio, P.; Chneiweiss, H.; Hermans, E.; Junier, M.P.; Leprince, P. Data in support of metabolic reprogramming in transformed mouse cortical astrocytes: A proteomic study. Data Brief 2015, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bluemlein, K.; Gruning, N.M.; Feichtinger, R.G.; Lehrach, H.; Kofler, B.; Ralser, M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011, 2, 393–400. [Google Scholar] [CrossRef]
- Meoni, G.; Lorini, S.; Monti, M.; Madia, F.; Corti, G.; Luchinat, C.; Zignego, A.L.; Tenori, L.; Gragnani, L. The metabolic fingerprints of HCV and HBV infections studied by Nuclear Magnetic Resonance Spectroscopy. Sci. Rep. 2019, 9, 4128. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. ‘Why do tumour cells glycolyse?’: From glycolysis through citrate to lipogenesis. Mol. Cell Biochem. 2005, 280, 1–8. [Google Scholar] [CrossRef]
- Chandel, N.S. Lipid Metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040576. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Amino Acid Metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040584. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Hata, K.; Hirose, K.; Okui, T.; Toyosawa, S.; Uzawa, N.; Nishimura, R.; Yoneda, T. The lactate sensor GPR81 regulates glycolysis and tumor growth of breast cancer. Sci. Rep. 2022, 12, 6261. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, E.; Badisa, R.; Mack, N.; Cassim, S.; Zdralevic, M.; Pouyssegur, J.; Soliman, K.F.A. Whole-transcriptome Analysis of Fully Viable Energy Efficient Glycolytic-null Cancer Cells Established by Double Genetic Knockout of Lactate Dehydrogenase A/B or Glucose-6-Phosphate Isomerase. Cancer Genom. Proteom. 2020, 17, 469–497. [Google Scholar] [CrossRef]
- Zdralevic, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Mahar, R.; Donabedian, P.L.; Merritt, M.E. HDO production from [2H7]glucose Quantitatively Identifies Warburg Metabolism. Sci. Rep. 2020, 10, 8885. [Google Scholar] [CrossRef]
- Chen, P.H.; Cai, L.; Huffman, K.; Yang, C.; Kim, J.; Faubert, B.; Boroughs, L.; Ko, B.; Sudderth, J.; McMillan, E.A.; et al. Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Mol. Cell 2019, 76, 838–851.e5. [Google Scholar] [CrossRef] [PubMed]
- Damasio, A. Feeling and Knowing Making Minds Conscious; Pantheon Books: New York, NY, USA, 2021; ISBN 13-978-1524747558. [Google Scholar]
- Matschinsky, F.M.; Wilson, D.F. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years after Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front. Physiol. 2019, 10, 148. [Google Scholar] [CrossRef]
- Wang, Z.H.; Peng, W.B.; Zhang, P.; Yang, X.P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cui, J. Targeting the lactic acid metabolic pathway for antitumor therapy. Mol. Ther. Oncolytics 2023, 31, 100740. [Google Scholar] [CrossRef] [PubMed]
- Dorneburg, C.; Fischer, M.; Barth, T.F.E.; Mueller-Klieser, W.; Hero, B.; Gecht, J.; Carter, D.R.; de Preter, K.; Mayer, B.; Christner, L.; et al. LDHA in Neuroblastoma Is Associated with Poor Outcome and Its Depletion Decreases Neuroblastoma Growth Independent of Aerobic Glycolysis. Clin. Cancer Res. 2018, 24, 5772–5783. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Di Matteo, M.; Serneels, J.; Pal, R.; Cafarello, S.T.; Lanza, M.; Sanchez-Martin, C.; Evert, M.; Castegna, A.; Calvisi, D.F.; et al. Deletion of Lactate Dehydrogenase-A Impairs Oncogene-Induced Mouse Hepatocellular Carcinoma Development. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 609–624. [Google Scholar] [CrossRef] [PubMed]
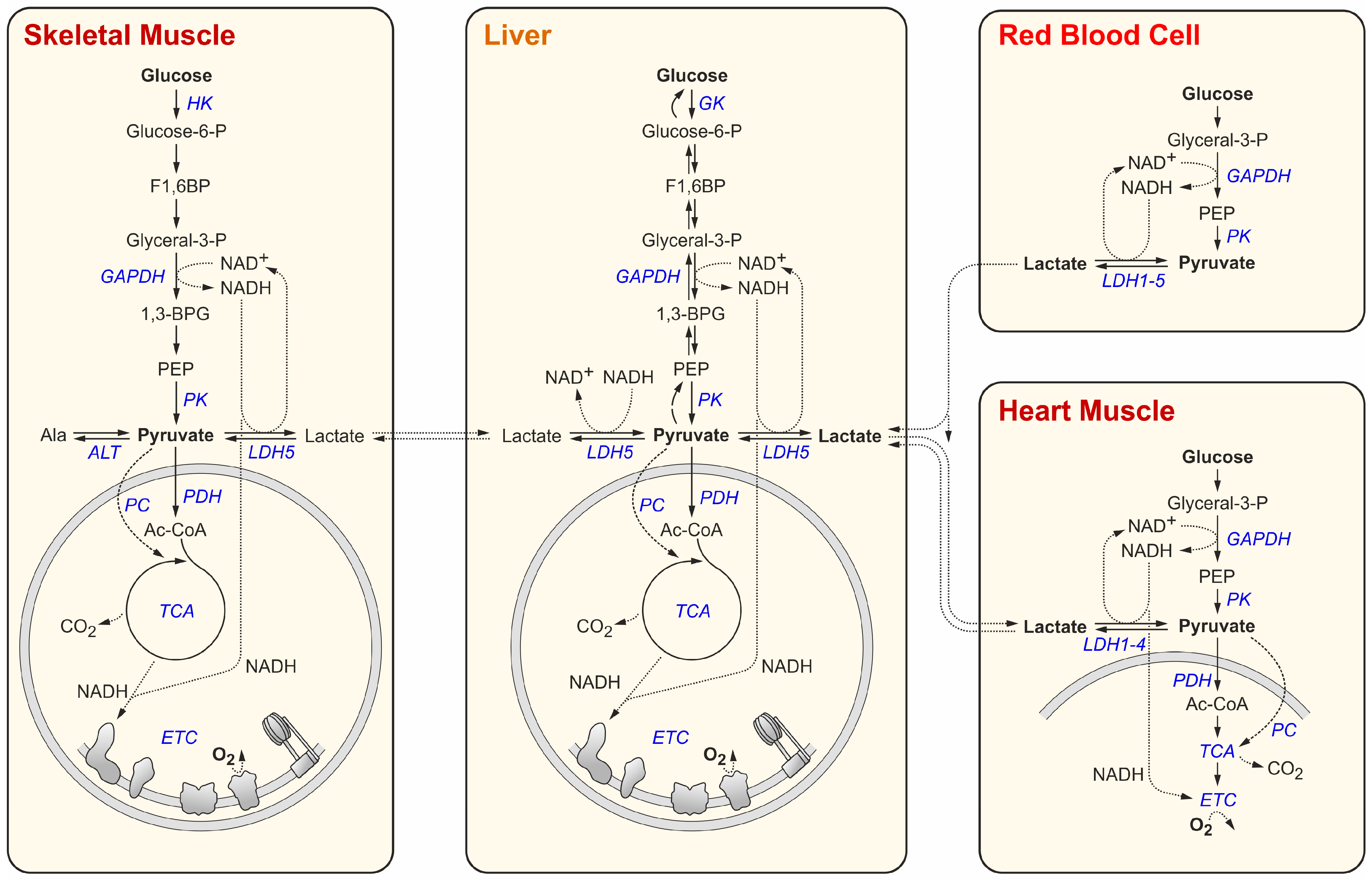
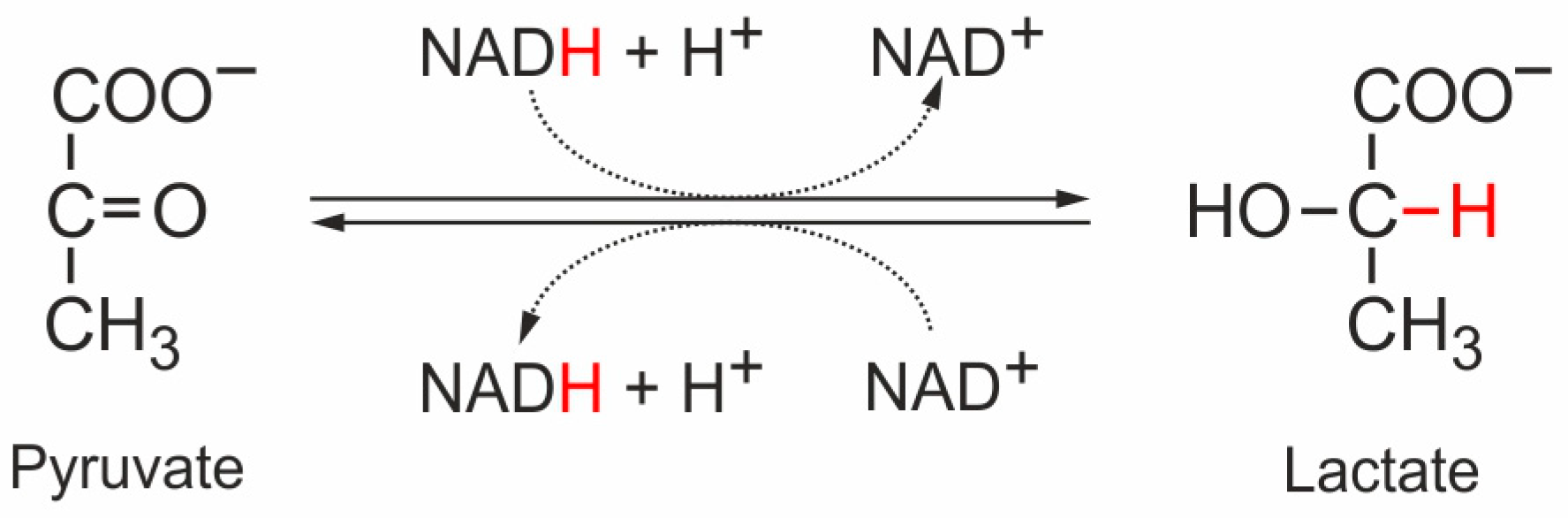
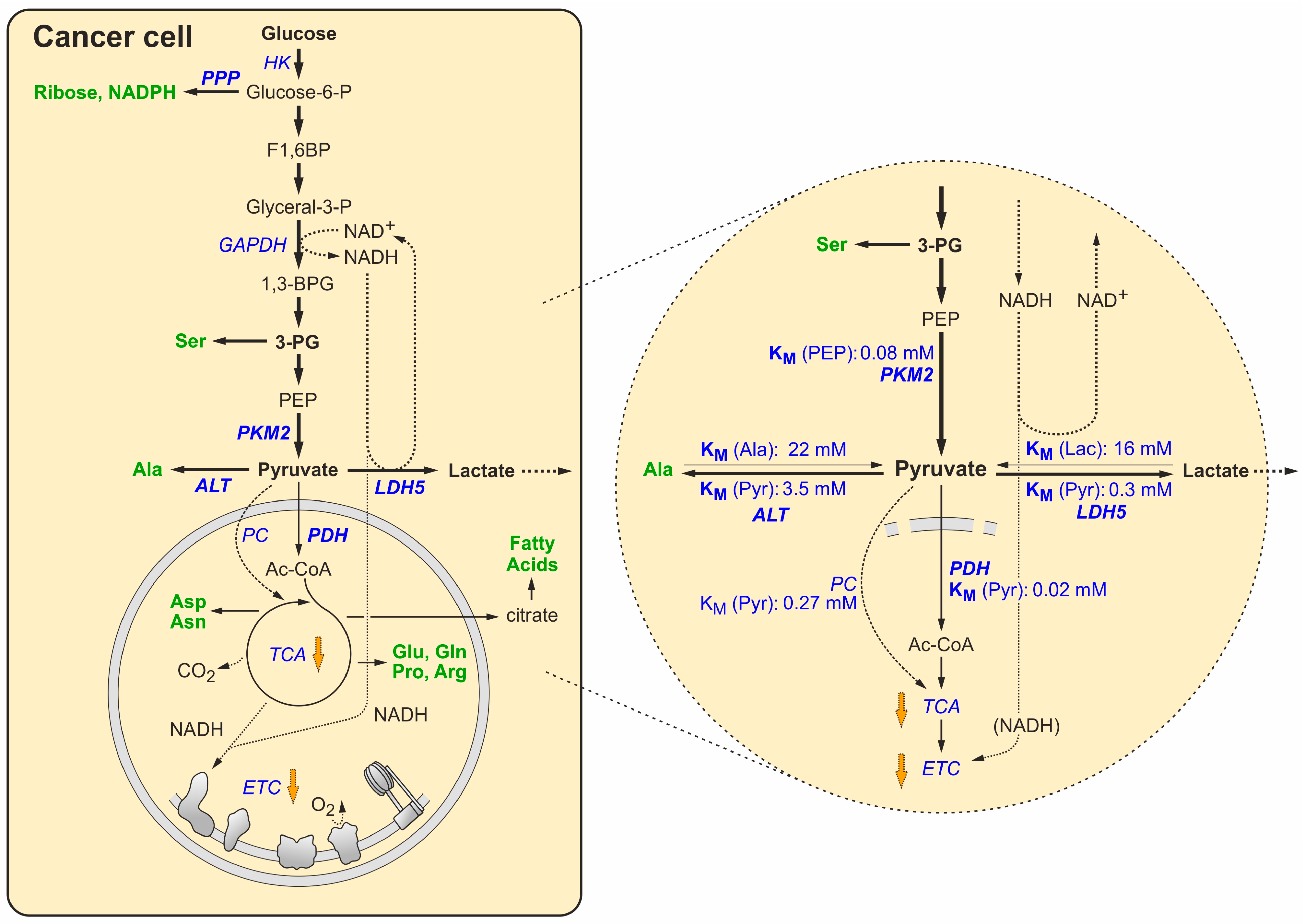
| Enzyme | Subtype | Cell Type | Normal/ Tumor | Substrate | Allosteric Effector 1 | KM (mM) Means | KM (mM) Range 2 | KM (mM)SD | n 3 | References |
|---|---|---|---|---|---|---|---|---|---|---|
| PK | M1 | muscle | normal | PEP | - | 0.057 | 0.032–0.085 | 0.019 | 7 | [76,77,78,79,80,81,82] |
| M2 | many | normal | PEP | - | 0.421 | 0.140–0.860 | 0.223 | 8 | [77,78,79,81,82,83,84,85] | |
| tumor | PEP | - | 0.648 | 0.130–2.100 | 0.542 | 9 | [57,86,87] | |||
| normal | PEP | F1,6BP | 0.064 | 0.030–0.100 | 0.026 | 4 | [77,78,85,88] | |||
| tumor | PEP | F1,6BP | 0.103 | 0.030–0.170 | 0.057 | 3 | [57,86,87] | |||
| L, R | liver, RBC | normal | PEP | - | 0.736 | 0.500–1.100 | 0.178 | 8 | [75,78,79,81,88,89] | |
| normal | PEP | F1,6BP | 0.073 | 0.060–0.090 | 0.013 | 3 | [75,81] | |||
| PDH | normal | pyruvate | - | 0.020 | 0.005–0.043 | 0.011 | 9 | [90,91,92,93,94,95,96,97] | ||
| PC | normal | pyruvate | - | 0.265 | 0.230–0.300 | 0.035 | 2 | [98,99] | ||
| GPT/ ALT | normal | pyruvate | - | 2.800 | 0.070–12.50 | 4.858 | 5 | [100,101,102,103,104] | ||
| normal | Ala | - | 22.003 | 10.12–34.00 | 7.735 | 8 | [100,101,102,103,104,105,106,107] | |||
| normal | Glu | - | 9.830 | 3.22–15.00 | 4.568 | 4 | [101,102,103,107] | |||
| normal | 2-OG | - | 0.523 | 0.100–1.100 | 0.356 | 6 | [100,101,102,103,105,106] | |||
| LDH | 1 (B, H) | heart, (RBC) | normal | pyruvate | - | 0.100 | 0.034–0.243 | 0.063 | 17 | [30,32,106,108,109,110,111,112,113,114,115,116,117,118,119,120] |
| normal | lactate | - | 5.916 | 2.000–9.690 | 2.911 | 8 | [30,32,108,109,115,121,122] | |||
| normal | NADH | - | 0.038 | 0.014–0.069 | 0.023 | 3 | [32,112,116] | |||
| normal | NAD+ | - | 0.123 | 0.075–0.170 | 0.039 | 3 | [30,109,121] | |||
| 5 (A, M) | liver, muscle, (heart) (RBC) | normal | pyruvate | - | 0.288 | 0.095–0.630 | 0.173 | 12 | [30,32,109,111,113,115,117,118,120,123,124] | |
| normal 4 | pyruvate | - | 0.630 | - | - | 1 4 | [124] | |||
| tumor 4 | pyruvate | - | 0.780 | - | - | 1 4 | [124] | |||
| normal | lactate | - | 15.940 | 6.880–40.000 | 7.759 | 15 | [30,32,109,115,121,122,124,125] | |||
| normal 4 | lactate | - | 10.730 | - | - | 1 4 | [124] | |||
| tumor 4 | lactate | - | 21.780 | - | - | 1 4 | [124] | |||
| normal | NADH | - | 0.173 | 0.016–0.330 | 0.157 | 2 | [32,124] | |||
| normal 4 | NADH | - | 0.300 | - | - | 1 4 | [124] | |||
| tumor 4 | NADH | - | 0.330 | - | - | 1 4 | [124] | |||
| normal | NAD+ | - | 0.337 | 0.220–0.500 | 0.119 | 3 | [109,121,124] | |||
| normal | NAD+ | - | 0.500 | 0.220–0.500 | 0.119 | 3 | [124] | |||
| tumor | NAD+ | 0.990 | - | - | 1 | [124] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niepmann, M. Importance of Michaelis Constants for Cancer Cell Redox Balance and Lactate Secretion—Revisiting the Warburg Effect. Cancers 2024, 16, 2290. https://doi.org/10.3390/cancers16132290
Niepmann M. Importance of Michaelis Constants for Cancer Cell Redox Balance and Lactate Secretion—Revisiting the Warburg Effect. Cancers. 2024; 16(13):2290. https://doi.org/10.3390/cancers16132290
Chicago/Turabian StyleNiepmann, Michael. 2024. "Importance of Michaelis Constants for Cancer Cell Redox Balance and Lactate Secretion—Revisiting the Warburg Effect" Cancers 16, no. 13: 2290. https://doi.org/10.3390/cancers16132290
APA StyleNiepmann, M. (2024). Importance of Michaelis Constants for Cancer Cell Redox Balance and Lactate Secretion—Revisiting the Warburg Effect. Cancers, 16(13), 2290. https://doi.org/10.3390/cancers16132290