
Medullary Thyroid Cancer: Molecular Drivers and Immune Cellular Milieu of the Tumour Microenvironment—Implications for Systemic Treatment

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
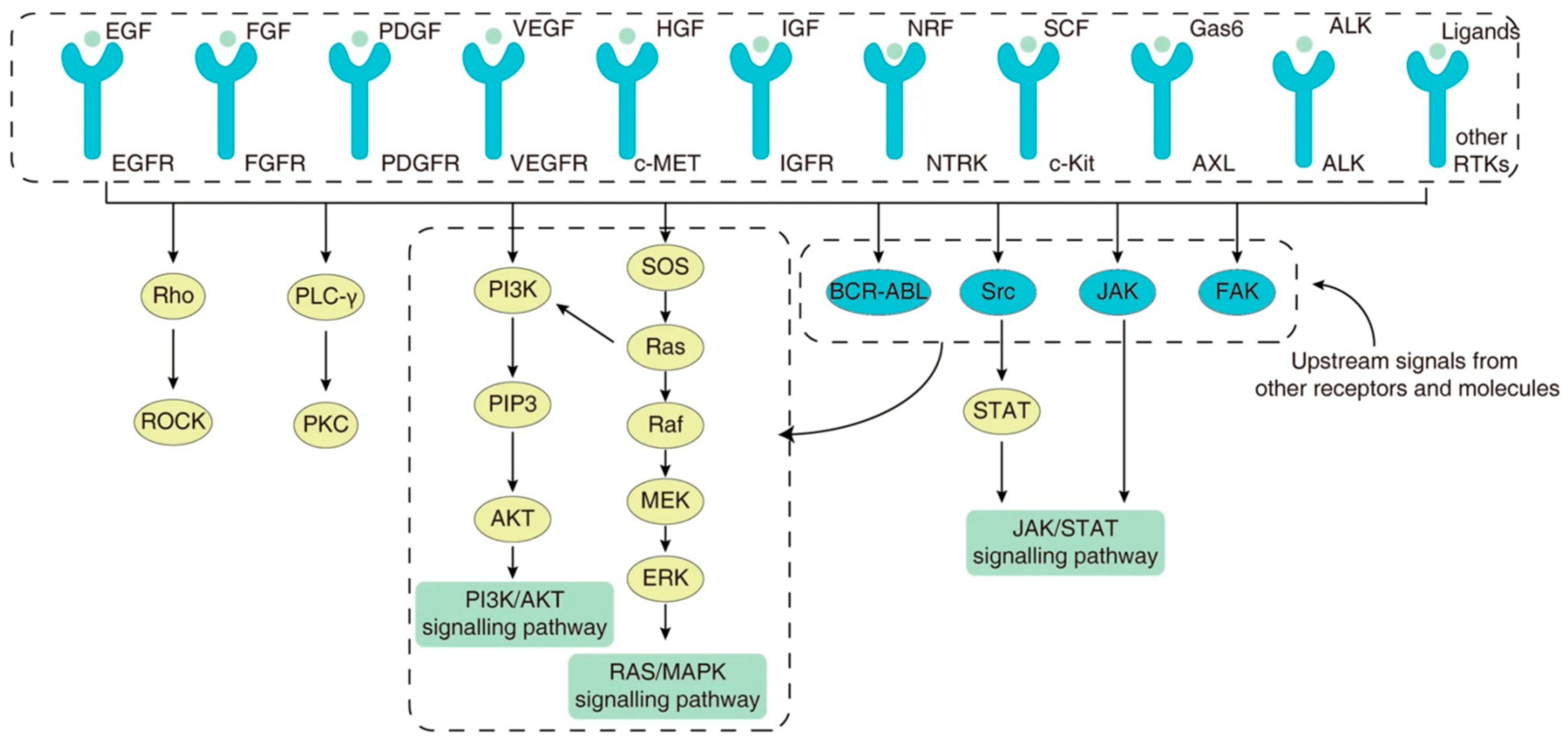
3. Genetic and Molecular Drivers of Disease
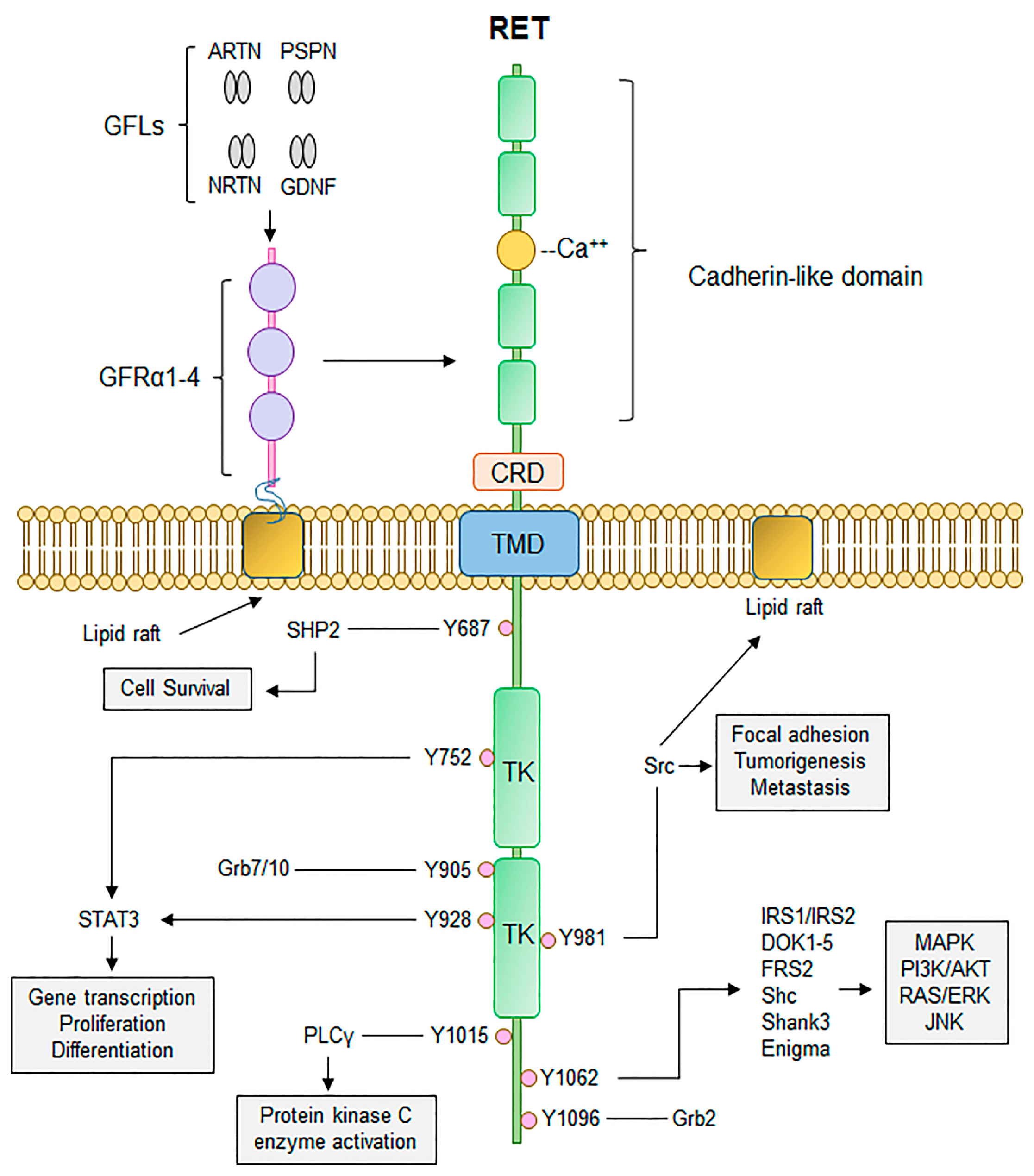
4. The RET Protooncogene and Hereditary Disease
5. Sporadic Disease
6. Molecular Subtyping
7. Interplay between the Immune System and Medullary Thyroid Cancer
An Overview of the Immune System and Carcinogenesis
8. Mechanisms of Immune Evasion
8.1. Immune Suppression Mediated by the Tumour Microenvironment
8.2. Immune Suppression through Surface Receptor Co-Stimulatory Inhibition
8.2.1. Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA-4)
8.2.2. PD-1
9. Emerging Co-Inhibitory Receptors
Tumour-Infiltrating Lymphocytes (TILs)
10. Efficacy of Immunotherapy
11. Current Systemic Treatment Options in MTC
11.1. Targeted Therapy with Pathway Inhibition
11.2. Escape of Pathway Inhibition
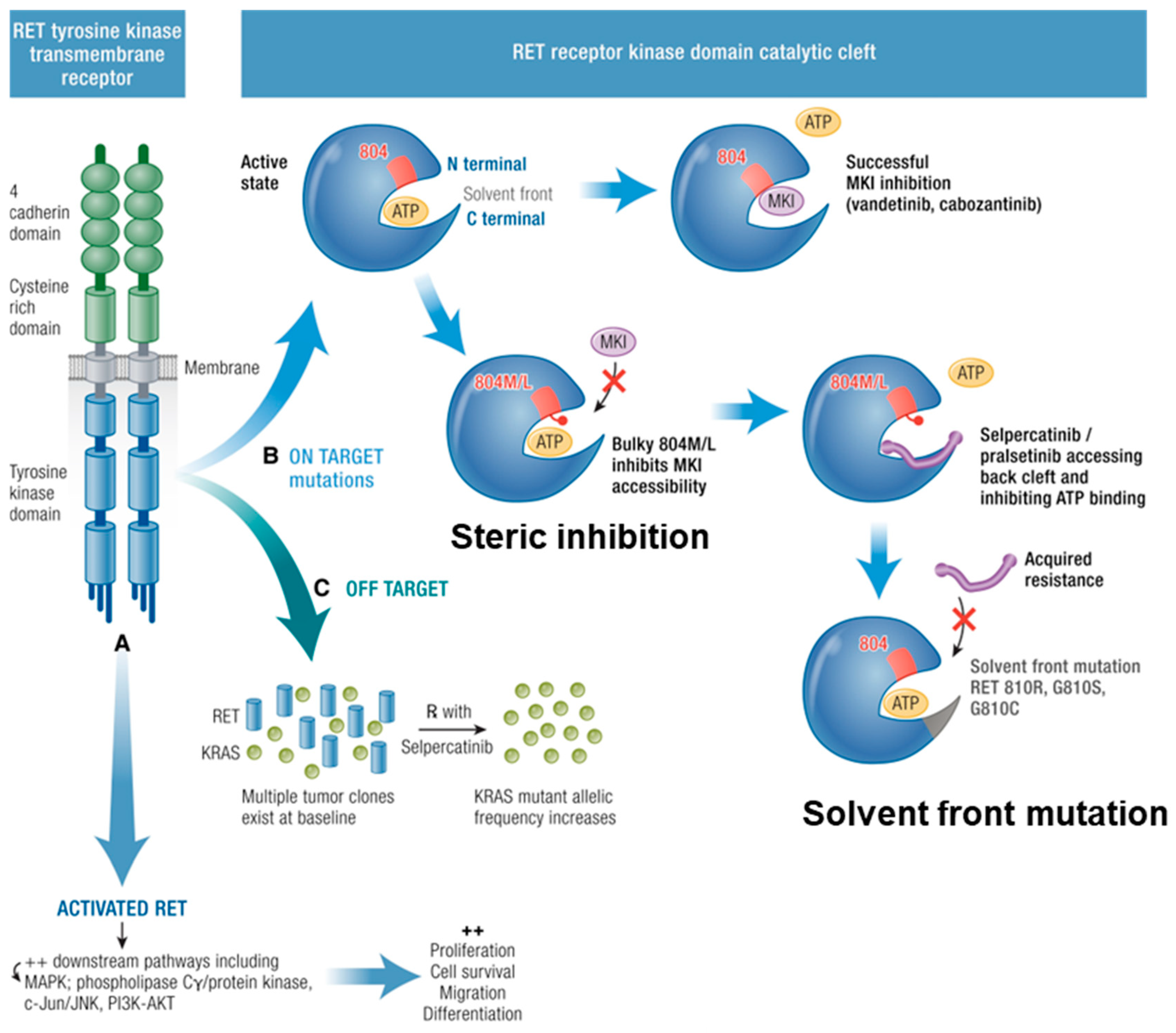
12. On-Target Resistance
13. Off-Target Resistance
14. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma: The American Thyroid Association Guidelines Task Force on medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Lin, R.; Sosa, J.A. Prognosis of medullary thyroid carcinoma: Demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 107, 2134–2142. [Google Scholar] [CrossRef]
- Papachristos, A.J.; Nicholls, L.E.; Mechera, R.; Aniss, A.M.; Robinson, B.; Clifton-Bligh, R.; Gill, A.J.; Learoyd, D.; Sidhu, S.B.; Glover, A. Management of medullary thyroid cancer: Patterns of recurrence and outcomes of reoperative surgery. Oncologist 2023, 28, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Dadu, R.; Hu, M.I. Medullary thyroid carcinoma. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 540–546. [Google Scholar] [CrossRef]
- Elisei, R.; Romei, C.; Cosci, B.; Agate, L.; Bottici, V.; Molinaro, E.; Sculli, M.; Miccoli, P.; Basolo, F.; Grasso, L. RET genetic screening in patients with medullary thyroid cancer and their relatives: Experience with 807 individuals at one center. J. Clin. Endocrinol. Metab. 2007, 92, 4725–4729. [Google Scholar] [CrossRef]
- Sippel, R.S.; Kunnimalaiyaan, M.; Chen, H. Current management of medullary thyroid cancer. Oncologist 2008, 13, 539–547. [Google Scholar] [CrossRef]
- Hincza-Nowak, K.; Kowalik, A.; Walczyk, A.; Pałyga, I.; Gąsior-Perczak, D.; Płusa, A.; Kopczyński, J.; Chrapek, M.; Góźdź, S.; Kowalska, A. Immune Profiling of Medullary Thyroid Cancer—An Opportunity for Immunotherapy. Genes 2021, 12, 1534. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.G.; Inabnet, W.B.; Lo, C.Y. Endocrine Surgery; Springer: London, UK, 2009. [Google Scholar]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Pützer, B.M. Mechanisms of disease: Cancer targeting and the impact of oncogenic RET for medullary thyroid carcinoma therapy. Nat. Clin. Pract. Oncol. 2006, 3, 564–574. [Google Scholar] [CrossRef]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Regua, A.T.; Najjar, M.; Lo, H.-W. RET signaling pathway and RET inhibitors in human cancer. Front. Oncol. 2022, 12, 932353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Oczko-Wojciechowska, M.; Swierniak, M.; Krajewska, J.; Kowalska, M.; Kowal, M.; Stokowy, T.; Wojtas, B.; Rusinek, D.; Pawlaczek, A.; Czarniecka, A. Differences in the transcriptome of medullary thyroid cancer regarding the status and type of RET gene mutations. Sci. Rep. 2017, 7, 42074. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Shi, X.; Zhao, J.-J.; Guan, H.; Zhang, T.-T.; Wen, S.-S.; Liao, T.; Hu, J.-Q.; Liu, W.-Y.; Wang, Y.-L. Genomic and transcriptomic characterization of sporadic medullary thyroid carcinoma. Thyroid 2020, 30, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Minna, E.; Romeo, P.; Dugo, M.; De Cecco, L.; Aiello, A.; Pistore, F.; Carenzo, A.; Greco, A.; Borrello, M.G. Medullary thyroid carcinoma mutational spectrum update and signaling-type inference by transcriptional profiles: Literature meta-analysis and study of tumor samples. Cancers 2022, 14, 1951. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Cosci, B.; Renzini, G.; Bottici, V.; Molinaro, E.; Agate, L.; Passannanti, P.; Viola, D.; Biagini, A.; Basolo, F. RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin. Endocrinol. 2011, 74, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; Van Amstel, H.K.P.; Lips, C.J.; Nishisho, I.; Takai, S.-I. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET Mutation Consortium analysis. JAMA 1996, 276, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- McGregor, L.M.; McCune, B.K.; Graff, J.R.; McDowell, P.R.; Romans, K.E.; Yancopoulos, G.D.; Ball, D.W.; Baylin, S.B.; Nelkin, B.D. Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc. Natl. Acad. Sci. USA 1999, 96, 4540–4545. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, D.; Koczan, D.; Ricken, P.; Rimpler, U.; Pahnke, J.; Li, Z.; Pützer, B.M. Transcriptome analysis in mouse tumors induced by Ret-MEN2/FMTC mutations reveals subtype-specific role in survival and interference with immune surveillance. Endocr.-Relat. Cancer 2009, 16, 211–224. [Google Scholar] [CrossRef]
- Ameur, N.; Lacroix, L.; Roucan, S.; Roux, V.; Broutin, S.; Talbot, M.; Dupuy, C.; Caillou, B.; Schlumberger, M.; Bidart, J.-M. Aggressive inherited and sporadic medullary thyroid carcinomas display similar oncogenic pathways. Endocr.-Relat. Cancer 2009, 16, 1261–1272. [Google Scholar] [CrossRef]
- Smith-Hicks, C.L.; Sizer, K.C.; Powers, J.F.; Tischler, A.S.; Costantini, F. C-cell hyperplasia, pheochromocytoma and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2B. EMBO J. 2000, 19, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Hereditary cancer: Two hits revisited. J. Cancer Res. Clin. Oncol. 1996, 122, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Torres-Cruz, J.; Pack, S.D.; Koch, C.A.; Vortmeyer, A.O.; Mannan, P.; Lubensky, I.A.; Gagel, R.F.; Zhuang, Z. Amplification and overexpression of mutant RET in multiple endocrine neoplasia type 2-associated medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Travagli, J.; Caillou, B.; Laplanche, A.; Bidart, J.; Schlumberger, M.; Baudin, E. Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome: Influence of the stage on the clinical course. Cancer 2002, 94, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N. Comprehensive genetic characterization of human thyroid cancer cell lines: A validated panel for preclinical studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Barletta, J.A.; Nosé, V.; Sadow, P.M. Genomics and epigenomics of medullary thyroid carcinoma: From sporadic disease to familial manifestations. Endocr. Pathol. 2021, 32, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, Y.; Shen, C.; Zhang, Y.; Shi, R.; Zhang, F.; Liao, T.; Lv, G.; Zhu, Z.; Jiao, L. Integrated proteogenomic characterization of medullary thyroid carcinoma. Cell Discov. 2022, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Viswanathan, K.; Ahadi, M.S.; Ahmadi, S.; Alzumaili, B.; Bani, M.-A.; Baudin, E.; Behrman, D.B.; Capelletti, M.; Chau, N.G. Association of the genomic profile of medullary thyroid carcinoma with tumor characteristics and clinical outcomes in an international multicenter study. Thyroid 2024, 34, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Erickson, T.A.; Zhang, L.; Ellison, K.; Rivard, C.J.; Sams, S.; Hirsch, F.R.; Haugen, B.R.; French, J.D. Comprehensive Immune Profiling of Medullary Thyroid Cancer. Thyroid 2020, 30, 1263–1279. [Google Scholar] [CrossRef]
- Abraham, D.; Jackson, N.; Gundara, J.S.; Zhao, J.; Gill, A.J.; Delbridge, L.; Robinson, B.G.; Sidhu, S.B. MicroRNA profiling of sporadic and hereditary medullary thyroid cancer identifies predictors of nodal metastasis, prognosis, and potential therapeutic targets. Clin. Cancer Res. 2011, 17, 4772–4781. [Google Scholar] [CrossRef]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F. The concept of immunological surveillance. Immunol. Asp. Neoplasia 1970, 13, 1–27. [Google Scholar]
- Birkeland, S.A.; Storm, H.H.; Lamm, L.U.; Barlow, L.; Blohmé, I.; Forsberg, B.; Eklund, B.; Fjeldborg, O.; Friedberg, M.; Frödin, L. Cancer risk after renal transplantation in the Nordic countries, 1964–1986. Int. J. Cancer 1995, 60, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.W.; Vaidya, S.A.; Cheng, G. The art of war: Innate and adaptive immune responses. Cell. Mol. Life Sci. 2003, 60, 2604–2621. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The concept of immune surveillance against tumors. The first theories. Oncotarget 2017, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nature Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Galdiero, M.R.; Ruffilli, I.; Elia, G.; Ragusa, F.; Paparo, S.R.; Patrizio, A.; Mazzi, V.; Varricchi, G. Immune and inflammatory cells in thyroid cancer microenvironment. Int. J. Mol. Sci. 2019, 20, 4413. [Google Scholar] [CrossRef]
- Rao, D.; Verburg, F.; Renner, K.; Peeper, D.S.; Lacroix, R.; Blank, C.U. Metabolic profiles of regulatory T cells in the tumour microenvironment. Cancer Immunol. Immunother. 2021, 70, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef]
- Modica, R.; Minotta, R.; Liccardi, A.; Cannavale, G.; Benevento, E.; Colao, A. Evaluation of Neutrophil-to-Lymphocyte Ratio (NLR), Platelet-to-Lymphocyte Ratio (PLR) and Systemic Immune-Inflammation Index (SII) as Potential Biomarkers in Patients with Sporadic Medullary Thyroid Cancer (MTC). J. Pers. Med. 2023, 13, 953. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Shang, Z.; Wang, W.; Gu, C.; Wei, Y.; Zhu, Y.; Yang, C.; Zhang, T.; Zhu, Y.; Zhu, Y.; et al. Immune Co-inhibitory Receptors CTLA-4, PD-1, TIGIT, LAG-3, and TIM-3 in Upper Tract Urothelial Carcinomas: A Large Cohort Study. J. Immunother. 2023, 46, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Kern, R.; Panis, C. CTLA-4 expression and its clinical significance in breast cancer. Arch. Immunol. Ther. Exp. 2021, 69, 16. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, C.-W.; Tan, L.-C.; Wen, S.-S.; Liao, T.; Zhang, Y.; Chen, T.-Z.; Ma, B.; Yu, P.-C.; Lu, Z.-W. Immune co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: A large cohort study. J. Clin. Endocrinol. Metab. 2021, 106, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Saltiki, K.; Simeakis, G.; Anagnostou, E.; Zapanti, E.; Anastasiou, E.; Alevizaki, M. Different outcomes in sporadic versus familial medullary thyroid cancer. Head Neck 2019, 41, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar]
- Chowdhury, S.; Veyhl, J.; Jessa, F.; Polyakova, O.; Alenzi, A.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016, 7, 32318. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, M.; Rebecchini, C.; Saglietti, C.; Bulliard, J.-L.; Marino, L.; de Leval, L.; Sykiotis, G.P. Very low expression of PD-L1 in medullary thyroid carcinoma. Endocr.-Relat. Cancer 2017, 24, L35–L38. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yu, P.-C.; Lei, B.-W.; Li, C.-W.; Zhang, Y.; Tan, L.-C.; Shi, R.-L.; Wang, J.; Ma, B.; Xu, W.-B. Association between programmed death-ligand 1 expression and clinicopathological characteristics, structural recurrence, and biochemical recurrence/persistent disease in medullary thyroid carcinoma. Thyroid 2019, 29, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Bi, Y.; Ren, X.; Bai, X.; Meng, Y.; Luo, Y.; Cao, J.; Zhang, Y.; Liang, Z. PD-1/PD-L1 expressions in medullary thyroid carcinoma: Clinicopathologic and prognostic analysis of Chinese population. Eur. J. Surg. Oncol. 2019, 45, 353–358. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, rv324–rv328. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Lorch, J.H.; Barletta, J.A.; Nehs, M.; Uppaluri, R.; Alexander, E.K.; Haddad, R.I.; Hanna, G.J.; Margalit, D.N.; Tishler, R.B.; Schoenfeld, J.D. A phase II study of nivolumab (N) plus ipilimumab (I) in radioidine refractory differentiated thyroid cancer (RAIR DTC) with exploratory cohorts in anaplastic (ATC) and medullary thyroid cancer (MTC). J. Clin. Oncol. 2020, 38, 6513. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K. Tim-3 inhibits T helper type 1–mediated auto-and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yuan, L.; Gao, Q.; Yuan, P.; Zhao, P.; Yuan, H.; Fan, H.; Li, T.; Qin, P.; Han, L. Circulating and tumor-infiltrating Tim-3 in patients with colorectal cancer. Oncotarget 2015, 6, 20592. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, P.; Liang, T.; Wang, L.; Hu, L. TIM-3 is a potential prognostic marker for patients with solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 31705–31713. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhang, X.; Xia, X.; Zhang, C.; Liang, X.; Gao, L.; Zhang, X.; Ma, C. Ectopic expression of TIM-3 in lung cancers: A potential independent prognostic factor for patients with NSCLC. Am. J. Clin. Pathol. 2012, 137, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Gallel, P.; Pallares, J.; Dolcet, X.; Llobet, D.; Eritja, N.; Santacana, M.; Yeramian, A.; Palomar-Asenjo, V.; Lagarda, H.; Mauricio, D.; et al. Nuclear factor-kappaB activation is associated with somatic and germ line RET mutations in medullary thyroid carcinoma. Hum. Pathol. 2008, 39, 994–1001. [Google Scholar] [CrossRef]
- Matias-Guiu, X.; De Lellis, R. Medullary thyroid carcinoma: A 25-year perspective. Endocr. Pathol. 2014, 25, 21–29. [Google Scholar] [CrossRef]
- Hincza-Nowak, K.; Kowalik, A.; Walczyk, A.; Palyga, I.; Gasior-Perczak, D.; Plusa, A.; Kopczynski, J.; Chrapek, M.; Gozdz, S.; Kowalska, A. CD276 as a Candidate Target for Immunotherapy in Medullary Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 10019. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; Van De Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I. Assessing tumor infiltrating lymphocytes in solid tumors: A practical review for pathologists and proposal for a standardized method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv. Anat. Pathol. 2017, 24, 311. [Google Scholar] [PubMed]
- Fuchs, T.L.; Sioson, L.; Sheen, A.; Jafari-Nejad, K.; Renaud, C.J.; Andrici, J.; Ahadi, M.; Chou, A.; Gill, A.J. Assessment of tumor-infiltrating lymphocytes using International TILs Working Group (ITWG) system is a strong predictor of overall survival in colorectal carcinoma: A study of 1034 patients. Am. J. Surg. Pathol. 2020, 44, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Pénault-Llorca, F. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Rago, T.; Di Coscio, G.; Ugolini, C.; Scutari, M.; Basolo, F.; Latrofa, F.; Romani, R.; Berti, P.; Grasso, L.; Braverman, L. Clinical features of thyroid autoimmunity are associated with thyroiditis on histology and are not predictive of malignancy in 570 patients with indeterminate nodules on cytology who had a thyroidectomy. Clin. Endocrinol. 2007, 67, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, C.I.; Hall, P.; Dickman, P.W.; Zedenius, J. Clinically significant prognostic factors for differentiated thyroid carcinoma: A population-based, nested case–control study. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 106, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, S.; Kawai, K.; Matsumoto, Y.; Mukuta, T.; Morita, T.; Hirai, K.; Matsuzuka, F.; Kakudoh, K.; Kuma, K.; Tamai, H. The correlation between papillary thyroid carcinoma and lymphocytic infiltration in the thyroid gland. J. Clin. Endocrinol. Metab. 1995, 80, 3421–3424. [Google Scholar]
- Scopsi, L.; Collini, P.; Sampietro, G.; Boracchi, P.; Pilotti, S. Prognostic impact of thyroid lymphocytic infiltration in patients with medullary thyroid carcinoma. Thyroid 1996, 6, 613–617. [Google Scholar] [CrossRef] [PubMed]
- French, J.D.; Weber, Z.J.; Fretwell, D.L.; Said, S.; Klopper, J.P.; Haugen, B.R. Tumor-associated lymphocytes and increased FoxP3+ regulatory T cell frequency correlate with more aggressive papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2010, 95, 2325–2333. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Menicali, E.; Guzzetti, M.; Morelli, S.; Moretti, S.; Puxeddu, E. Immune landscape of thyroid cancers: New insights. Front. Endocrinol. 2021, 11, 637826. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.-j.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Poehnert, D.; Müller, J.; Scheumann, G.; Koch, M.; Lück, R. Regulatory T cells in peripheral blood, lymph node, and thyroid tissue in patients with medullary thyroid carcinoma. World J. Surg. 2010, 34, 1481–1487. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M. First-line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 2019, 37, 992. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013, 98, E364–E369. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.; Carey, F.; Pratt, N.; Steele, R. The colorectal adenoma–carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019, 36, 385–401.e388. [Google Scholar] [CrossRef]
- Ruan, X.; Yi, J.; Hu, L.; Zhi, J.; Zeng, Y.; Hou, X.; Huang, J.; Gu, P.; Hao, W.; Gao, M. Reduced MHC class II expression in medullary thyroid cancer identifies patients with poor prognosis. Endocr.-Relat. Cancer 2022, 29, 87–98. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.; Gaweł, D.; Godlewska, M. Novel inhibitor-based therapies for thyroid cancer—An update. Int. J. Mol. Sci. 2021, 22, 11829. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on fundamental mechanisms of thyroid cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S. Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Jozaghi, Y.; Zafereo, M.; Williams, M.D.; Gule-Monroe, M.K.; Wang, J.; Grubbs, E.G.; Vaporciyan, A.; Hu, M.I.; Busaidy, N.; Dadu, R.; et al. Neoadjuvant selpercatinib for advanced medullary thyroid cancer. Head Neck 2021, 43, E7–E12. [Google Scholar] [CrossRef]
- Gild, M.L.; Clifton-Bligh, R.J.; Wirth, L.J.; Robinson, B.G. Medullary Thyroid Cancer: Updates and Challenges. Endocr. Rev. 2023, 44, 934–946. [Google Scholar] [CrossRef]
- Hadoux, J.; Elisei, R.; Brose, M.S.; Hoff, A.O.; Robinson, B.G.; Gao, M.; Jarzab, B.; Isaev, P.; Kopeckova, K.; Wadsley, J. Phase 3 trial of selpercatinib in advanced RET-mutant medullary thyroid cancer. N. Engl. J. Med. 2023, 389, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Bullock, M.; Tsang, V.; Clifton-Bligh, R.J.; Robinson, B.G.; Wirth, L.J. Challenges and Strategies to Combat Resistance Mechanisms in Thyroid Cancer Therapeutics. Thyroid 2023, 33, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, T.; Mooers, B.H.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer. Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef] [PubMed]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [PubMed]
- Terzyan, S.S.; Shen, T.; Liu, X.; Huang, Q.; Teng, P.; Zhou, M.; Hilberg, F.; Cai, J.; Mooers, B.H.; Wu, J. Structural basis of resistance of mutant RET protein-tyrosine kinase to its inhibitors nintedanib and vandetanib. J. Biol. Chem. 2019, 294, 10428–10437. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef]
- Drilon, A.E.; Zhai, D.; Rogers, E.; Deng, W.; Zhang, X.; Ung, J.; Lee, D.; Rodon, L.; Graber, A.; Zimmerman, Z.F. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J. Clin. Oncol. 2020, 38, 3616. [Google Scholar] [CrossRef]
- Nakaoku, T.; Kohno, T.; Araki, M.; Niho, S.; Chauhan, R.; Knowles, P.P.; Tsuchihara, K.; Matsumoto, S.; Shimada, Y.; Mimaki, S. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat. Commun. 2018, 9, 625. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Vaquero, J.; Lobe, C.; Tahraoui, S.; Clapéron, A.; Mergey, M.; Merabtene, F.; Wendum, D.; Coulouarn, C.; Housset, C.; Desbois-Mouthon, C. The IGF2/IR/IGF1R pathway in tumor cells and myofibroblasts mediates resistance to EGFR inhibition in cholangiocarcinoma. Clin. Cancer Res. 2018, 24, 4282–4296. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Chiang, C.-L.; Hung, J.-Y.; Lee, M.-H.; Su, W.-C.; Wu, S.-Y.; Wei, Y.-F.; Lee, K.-Y.; Tseng, Y.-H.; Su, J. Resistance profiles of anaplastic lymphoma kinase tyrosine kinase inhibitors in advanced non–small-cell lung cancer: A multicenter study using targeted next-generation sequencing. Eur. J. Cancer 2021, 156, 1–11. [Google Scholar] [CrossRef]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 2016, 150, 1646–1658.e1617. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gong, K.; Ali, S.; Ali, N.; Shallwani, S.; Hatanpaa, K.J.; Pan, E.; Mickey, B.; Burma, S.; Wang, D.H. A TNF–JNK–Axl–ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat. Neurosci. 2017, 20, 1074–1084. [Google Scholar] [CrossRef]
- Ou, B.; Cheng, X.; Xu, Z.; Chen, C.; Shen, X.; Zhao, J.; Lu, A. A positive feedback loop of β-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis. 2019, 10, 643. [Google Scholar] [CrossRef]
- Dong, N.; Shi, X.; Wang, S.; Gao, Y.; Kuang, Z.; Xie, Q.; Li, Y.; Deng, H.; Wu, Y.; Li, M. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer 2019, 121, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Noel, B.M.; Ouellette, S.B.; Marholz, L.; Dickey, D.; Navis, C.; Yang, T.-Y.; Nguyen, V.; Parker, S.J.; Bernlohr, D.; Sachs, Z. Multiomic profiling of tyrosine kinase inhibitor-resistant K562 cells suggests metabolic reprogramming to promote cell survival. J. Proteome Res. 2019, 18, 1842–1856. [Google Scholar] [CrossRef]
- Niu, X.; Liu, F.; Zhou, Y.; Zhou, Z.; Zhou, D.; Wang, T.; Li, Z.; Ye, X.; Yu, Y.; Weng, X. Genome-wide DNA methylation analysis reveals GABBR2 as a novel epigenetic target for EGFR 19 deletion lung adenocarcinoma with induction erlotinib treatment. Clin. Cancer Res. 2017, 23, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.F.; Chen, L.T.; Wang, H.D.; Liu, Y.H.; Shiah, S.G. Transcriptional suppression of Dicer by HOXB-AS3/EZH2 complex dictates sorafenib resistance and cancer stemness. Cancer Sci. 2022, 113, 1601–1612. [Google Scholar] [CrossRef]
- Liu, S.; Li, Q.; Li, G.; Zhang, Q.; Zhuo, L.; Han, X.; Zhang, M.; Chen, X.; Pan, T.; Yan, L. The mechanism of m6A methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 2020, 11, 969. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papachristos, A.J.; Serrao-Brown, H.; Gill, A.J.; Clifton-Bligh, R.; Sidhu, S.B. Medullary Thyroid Cancer: Molecular Drivers and Immune Cellular Milieu of the Tumour Microenvironment—Implications for Systemic Treatment. Cancers 2024, 16, 2296. https://doi.org/10.3390/cancers16132296
Papachristos AJ, Serrao-Brown H, Gill AJ, Clifton-Bligh R, Sidhu SB. Medullary Thyroid Cancer: Molecular Drivers and Immune Cellular Milieu of the Tumour Microenvironment—Implications for Systemic Treatment. Cancers. 2024; 16(13):2296. https://doi.org/10.3390/cancers16132296
Chicago/Turabian StylePapachristos, Alexander J., Hazel Serrao-Brown, Anthony J. Gill, Roderick Clifton-Bligh, and Stanley B. Sidhu. 2024. "Medullary Thyroid Cancer: Molecular Drivers and Immune Cellular Milieu of the Tumour Microenvironment—Implications for Systemic Treatment" Cancers 16, no. 13: 2296. https://doi.org/10.3390/cancers16132296
APA StylePapachristos, A. J., Serrao-Brown, H., Gill, A. J., Clifton-Bligh, R., & Sidhu, S. B. (2024). Medullary Thyroid Cancer: Molecular Drivers and Immune Cellular Milieu of the Tumour Microenvironment—Implications for Systemic Treatment. Cancers, 16(13), 2296. https://doi.org/10.3390/cancers16132296