
Preclinical Therapeutic Efficacy of RAF/MEK/ERK and IGF1R/AKT/mTOR Inhibition in Neuroblastoma
 , , , , , , , , ,
, , , , , , , , ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
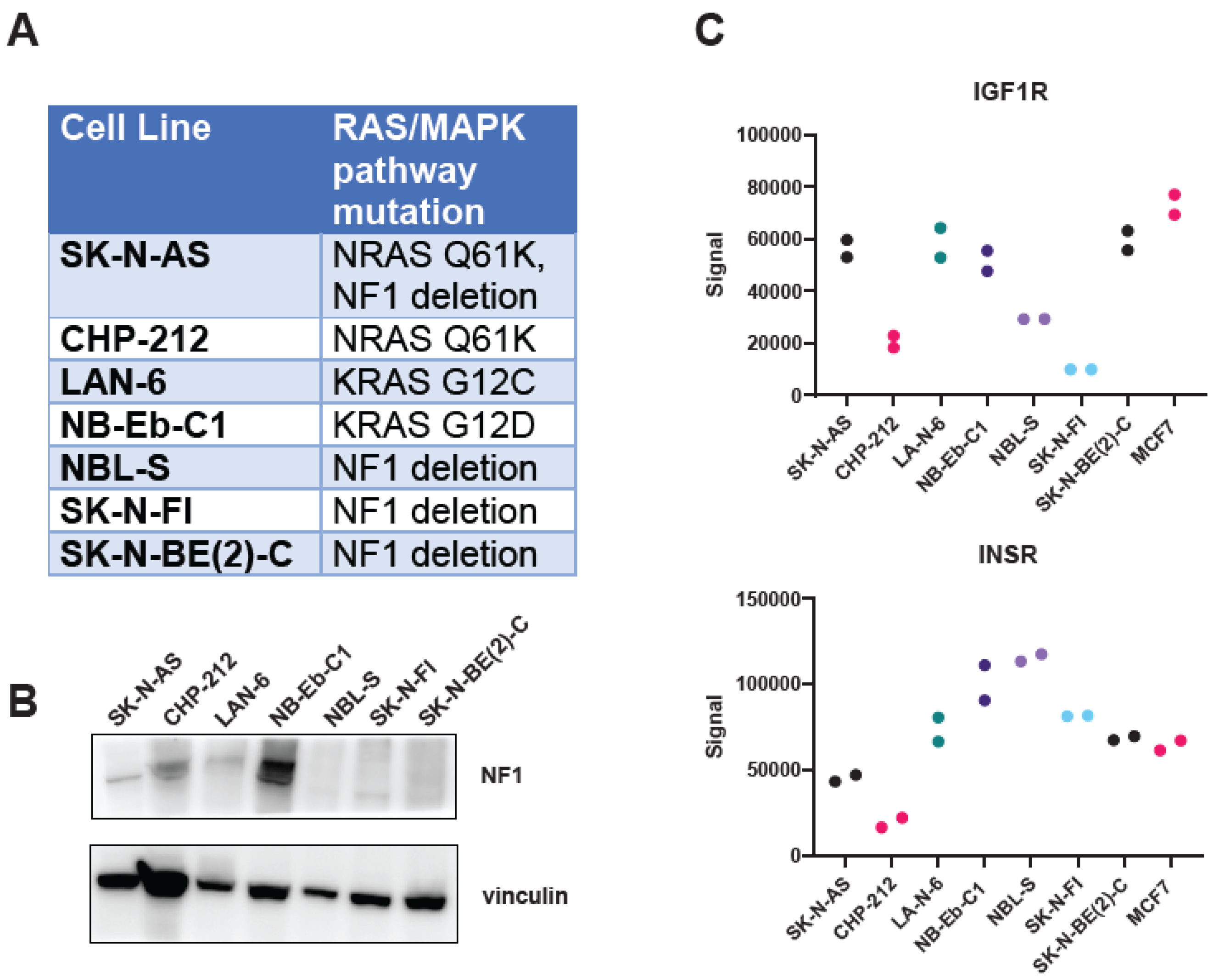
2.1. Cell Lines and Reagents
2.2. Whole-Exome Sequencing
2.3. Cell Viability Assay
2.4. Matrix Combination Assay
2.5. Apoptosis Assays
2.6. Capillary Immunoassays
2.7. Immunoblot Assays
2.8. Subcutaneous Xenograft Experiments
2.9. Orthotopic Xenograft Experiments
2.10. Mouse Imaging Preparation
2.11. Ultrasound
2.12. Magnetic Resonance Imaging
2.13. Electrochemiluminescence Assays
2.14. Histological Analysis
3. Results
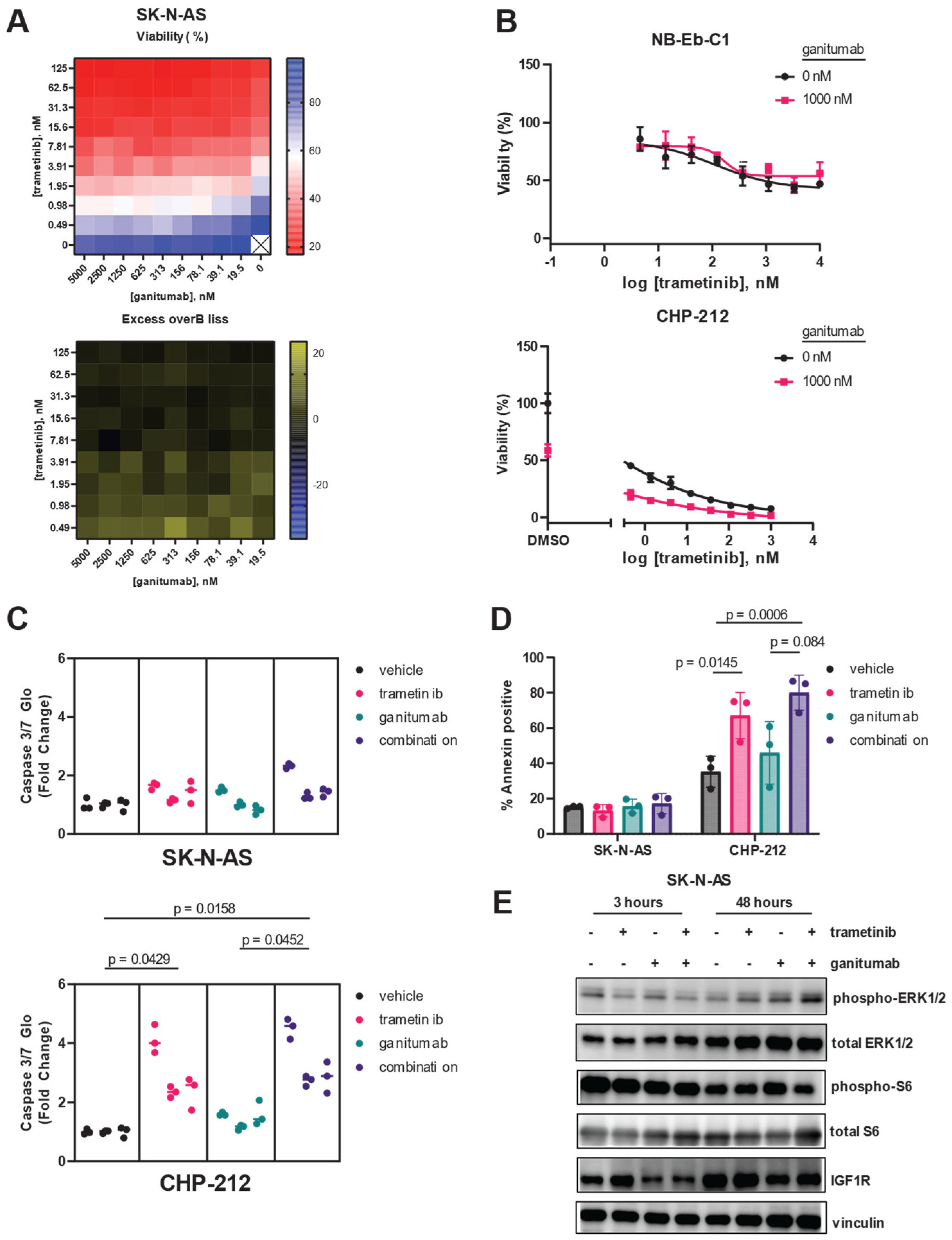
3.1. Drugs That Specifically Inhibit MEK1/2 and IGF1R Synergistically Inhibit Proliferation of RAS-Mutated Neuroblastoma Cells
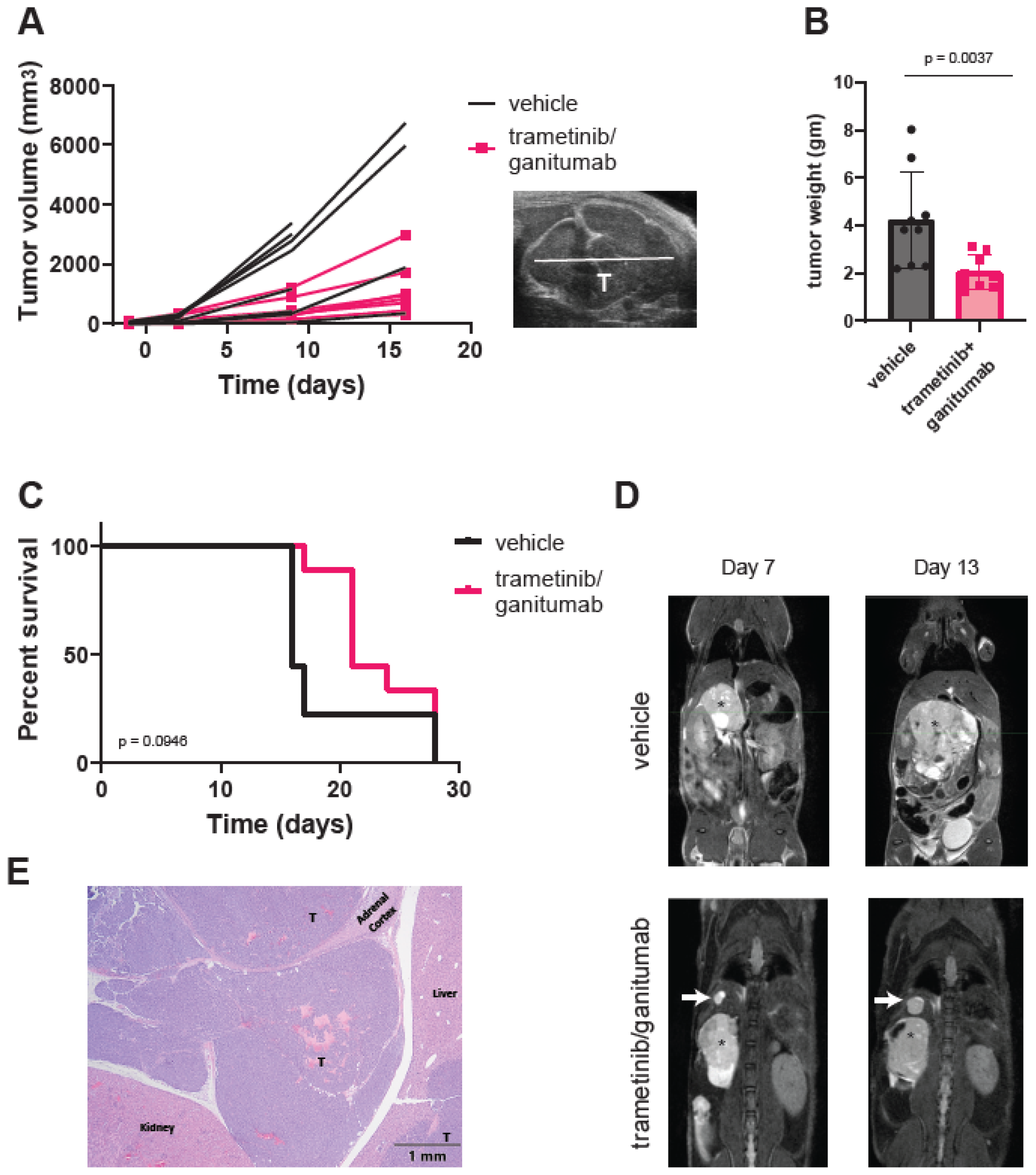
3.2. Combined Trametinib and Ganitumab Treatment Is Efficacious in Murine Xenograft Models of RAS-Mutated Neuroblastoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Yohe, M.E. Targeting RAS in pediatric cancer: Is it becoming a reality? Curr. Opin. Pediatr. 2020, 32, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ruan, Y.; Tippett, T.; Narendran, A. Targeted inhibition of MEK1 by cobimetinib leads to differentiation and apoptosis in neuroblastoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 104. [Google Scholar] [CrossRef]
- Tanaka, T.; Higashi, M.; Kimura, K.; Wakao, J.; Fumino, S.; Iehara, T.; Hosoi, H.; Sakai, T.; Tajiri, T. MEK inhibitors as a novel therapy for neuroblastoma: Their in vitro effects and predicting their efficacy. J. Pediatr. Surg. 2016, 51, 2074–2079. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci. Signal 2017, 10, eaam7550. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Kim, J.Y.; Qiao, J.; Clark, R.A.; Powers, C.M.; Correa, H.; Chung, D.H. Dual-Targeting AKT2 and ERK in cancer stem-like cells in neuroblastoma. Oncotarget 2019, 10, 5645–5659. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.Q.; Toyoda, H.; Qi, L.; Morimoto, M.; Hanaki, R.; Iwamoto, S.; Komada, Y.; Hirayama, M. Induction of MEK/ERK activity by AZD8055 confers acquired resistance in neuroblastoma. Biochem. Biophys. Res. Commun. 2018, 499, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef]
- Duffy, D.J.; Krstic, A.; Halasz, M.; Schwarzl, T.; Fey, D.; Iljin, K.; Mehta, J.P.; Killick, K.; Whilde, J.; Turriziani, B.; et al. Integrative omics reveals MYCN as a global suppressor of cellular signalling and enables network-based therapeutic target discovery in neuroblastoma. Oncotarget 2015, 6, 43182–43201. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Huang, S.; Koster, J.; Ora, I.; Lakeman, A.; Caron, H.; Nijkamp, W.; Xie, J.; Callens, T.; Asgharzadeh, S.; et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 2010, 142, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Mayes, P.A.; Degenhardt, Y.Y.; Wood, A.; Toporovskya, Y.; Diskin, S.J.; Haglund, E.; Moy, C.; Wooster, R.; Maris, J.M. Mitogen-activated protein kinase (MEK/ERK) inhibition sensitizes cancer cells to centromere-associated protein E inhibition. Int. J. Cancer 2013, 132, E149–E157. [Google Scholar] [CrossRef] [PubMed]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. NRAS Status Determines Sensitivity to SHP2 Inhibitor Combination Therapies Targeting the RAS-MAPK Pathway in Neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef]
- Malone, C.F.; Kim, M.; Alexe, G.; Engel, K.; Forman, A.B.; Robichaud, A.; Conway, A.S.; Goodale, A.; Meyer, A.; Khalid, D.; et al. Transcriptional Antagonism by CDK8 Inhibition Improves Therapeutic Efficacy of MEK Inhibitors. Cancer Res. 2023, 83, 285–300. [Google Scholar] [CrossRef]
- Healy, J.R.; Hart, L.S.; Shazad, A.L.; Gagliardi, M.E.; Tsang, M.; Elias, J.; Ruden, J.; Farrel, A.; Rokita, J.L.; Li, Y.; et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28267. [Google Scholar] [CrossRef]
- El-Badry, O.M.; Helman, L.J.; Chatten, J.; Steinberg, S.M.; Evans, A.E.; Israel, M.A. Insulin-like growth factor II-mediated proliferation of human neuroblastoma. J. Clin. Investig. 1991, 87, 648–657. [Google Scholar] [CrossRef]
- El-Badry, O.M.; Romanus, J.A.; Helman, L.J.; Cooper, M.J.; Rechler, M.M.; Israel, M.A. Autonomous growth of a human neuroblastoma cell line is mediated by insulin-like growth factor II. J. Clin. Investig. 1989, 84, 829–839. [Google Scholar] [CrossRef]
- Leventhal, P.S.; Randolph, A.E.; Vesbit, T.E.; Schenone, A.; Windebank, A.; Feldman, E.L. Insulin-like growth factor-II as a paracrine growth factor in human neuroblastoma cells. Exp. Cell Res. 1995, 221, 179–186. [Google Scholar] [CrossRef]
- Singleton, J.R.; Randolph, A.E.; Feldman, E.L. Insulin-like growth factor I receptor prevents apoptosis and enhances neuroblastoma tumorigenesis. Cancer Res. 1996, 56, 4522–4529. [Google Scholar]
- Kim, B.; van Golen, C.M.; Feldman, E.L. Insulin-like growth factor-I signaling in human neuroblastoma cells. Oncogene 2004, 23, 130–141. [Google Scholar] [CrossRef]
- Meyer, G.E.; Shelden, E.; Kim, B.; Feldman, E.L. Insulin-like growth factor I stimulates motility in human neuroblastoma cells. Oncogene 2001, 20, 7542–7550. [Google Scholar] [CrossRef]
- Misawa, A.; Hosoi, H.; Arimoto, A.; Shikata, T.; Akioka, S.; Matsumura, T.; Houghton, P.J.; Sawada, T. N-Myc induction stimulated by insulin-like growth factor I through mitogen-activated protein kinase signaling pathway in human neuroblastoma cells. Cancer Res. 2000, 60, 64–69. [Google Scholar]
- van Golen, C.M.; Castle, V.P.; Feldman, E.L. IGF-I receptor activation and BCL-2 overexpression prevent early apoptotic events in human neuroblastoma. Cell Death Differ. 2000, 7, 654–665. [Google Scholar] [CrossRef]
- van Golen, C.M.; Schwab, T.S.; Kim, B.; Soules, M.E.; Su Oh, S.; Fung, K.; van Golen, K.L.; Feldman, E.L. Insulin-like growth factor-I receptor expression regulates neuroblastoma metastasis to bone. Cancer Res. 2006, 66, 6570–6578. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Naraparaju, K.; Iyer, R.; Guan, P.; Kolla, V.; Hu, Y.; Tan, K.; Brodeur, G.M. Mechanisms of Entrectinib Resistance in a Neuroblastoma Xenograft Model. Mol. Cancer Ther. 2020, 19, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Gaetano, C.; Daughaday, W.H.; Thiele, C.J. Retinoic acid regulates insulin-like growth factor II expression in a neuroblastoma cell line. Endocrinology 1992, 130, 3669–3676. [Google Scholar] [CrossRef]
- Matsumoto, K.; Lucarelli, E.; Minniti, C.; Gaetano, C.; Thiele, C.J. Signals transduced via insulin-like growth factor I receptor (IGF(R)) mediate resistance to retinoic acid-induced cell growth arrest in a human neuroblastoma cell line. Cell Death Differ. 1994, 1, 49–58. [Google Scholar] [PubMed]
- Wang, X.H.; Wu, H.Y.; Gao, J.; Wang, X.H.; Gao, T.H.; Zhang, S.F. IGF1R facilitates epithelial-mesenchymal transition and cancer stem cell properties in neuroblastoma via the STAT3/AKT axis. Cancer Manag. Res. 2019, 11, 5459–5472. [Google Scholar] [CrossRef]
- Coulter, D.W.; Wilkie, M.B.; Moats-Staats, B.M. Inhibition of IGF-I receptor signaling in combination with rapamycin or temsirolimus increases MYC-N phosphorylation. Anticancer. Res. 2009, 29, 1943–1949. [Google Scholar]
- DeNardo, B.D.; Holloway, M.P.; Ji, Q.; Nguyen, K.T.; Cheng, Y.; Valentine, M.B.; Salomon, A.; Altura, R.A. Quantitative phosphoproteomic analysis identifies activation of the RET and IGF-1R/IR signaling pathways in neuroblastoma. PLoS ONE 2013, 8, e82513. [Google Scholar] [CrossRef]
- Geoerger, B.; Brasme, J.F.; Daudigeos-Dubus, E.; Opolon, P.; Venot, C.; Debussche, L.; Vrignaud, P.; Vassal, G. Anti-insulin-like growth factor 1 receptor antibody EM164 (murine AVE1642) exhibits anti-tumour activity alone and in combination with temozolomide against neuroblastoma. Eur. J. Cancer 2010, 46, 3251–3262. [Google Scholar] [CrossRef]
- Guerreiro, A.S.; Boller, D.; Shalaby, T.; Grotzer, M.A.; Arcaro, A. Protein kinase B modulates the sensitivity of human neuroblastoma cells to insulin-like growth factor receptor inhibition. Int. J. Cancer 2006, 119, 2527–2538. [Google Scholar] [CrossRef]
- Houghton, P.J.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Wu, J.; Smith, M.A. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2010, 54, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Kolb, E.A.; Gorlick, R.; Lock, R.; Carol, H.; Morton, C.L.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Billups, C.; et al. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2011, 56, 595–603. [Google Scholar] [CrossRef]
- Pappano, W.N.; Jung, P.M.; Meulbroek, J.A.; Wang, Y.C.; Hubbard, R.D.; Zhang, Q.; Grudzien, M.M.; Soni, N.B.; Johnson, E.F.; Sheppard, G.S.; et al. Reversal of oncogene transformation and suppression of tumor growth by the novel IGF1R kinase inhibitor A-928605. BMC Cancer 2009, 9, 314. [Google Scholar] [CrossRef]
- Singh, A.; Meier-Stephenson, V.; Jayanthan, A.; Narendran, A. In Vitro Sensitivity Profiling of Neuroblastoma Cells Against A Comprehensive Small Molecule Kinase Inhibitor Library to Identify Agents for Future Therapeutic Studies. Curr. Cancer Drug Targets 2017, 17, 569–584. [Google Scholar] [CrossRef]
- Tanno, B.; Mancini, C.; Vitali, R.; Mancuso, M.; McDowell, H.P.; Dominici, C.; Raschella, G. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin. Cancer Res. 2006, 12, 6772–6780. [Google Scholar] [CrossRef]
- Wojtalla, A.; Salm, F.; Christiansen, D.G.; Cremona, T.; Cwiek, P.; Shalaby, T.; Gross, N.; Grotzer, M.A.; Arcaro, A. Novel agents targeting the IGF-1R/PI3K pathway impair cell proliferation and survival in subsets of medulloblastoma and neuroblastoma. PLoS ONE 2012, 7, e47109. [Google Scholar] [CrossRef]
- Zhao, Q.; Tran, H.; Dimitrov, D.S.; Cheung, N.K. A dual-specific anti-IGF-1/IGF-2 human monoclonal antibody alone and in combination with temsirolimus for therapy of neuroblastoma. Int. J. Cancer 2015, 137, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Boller, D.; Schramm, A.; Doepfner, K.T.; Shalaby, T.; von Bueren, A.O.; Eggert, A.; Grotzer, M.A.; Arcaro, A. Targeting the phosphoinositide 3-kinase isoform p110delta impairs growth and survival in neuroblastoma cells. Clin. Cancer Res. 2008, 14, 1172–1181. [Google Scholar] [CrossRef]
- Chanthery, Y.H.; Gustafson, W.C.; Itsara, M.; Persson, A.; Hackett, C.S.; Grimmer, M.; Charron, E.; Yakovenko, S.; Kim, G.; Matthay, K.K.; et al. Paracrine signaling through MYCN enhances tumor-vascular interactions in neuroblastoma. Sci. Transl. Med. 2012, 4, 115ra113. [Google Scholar] [CrossRef]
- Chesler, L.; Schlieve, C.; Goldenberg, D.D.; Kenney, A.; Kim, G.; McMillan, A.; Matthay, K.K.; Rowitch, D.; Weiss, W.A. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006, 66, 8139–8146. [Google Scholar] [CrossRef]
- Johnsen, J.I.; Segerstrom, L.; Orrego, A.; Elfman, L.; Henriksson, M.; Kagedal, B.; Eksborg, S.; Sveinbjornsson, B.; Kogner, P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 2008, 27, 2910–2922. [Google Scholar] [CrossRef] [PubMed]
- Mohlin, S.; Hamidian, A.; von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Pahlman, S. PI3K-mTORC2 but not PI3K-mTORC1 regulates transcription of HIF2A/EPAS1 and vascularization in neuroblastoma. Cancer Res. 2015, 75, 4617–4628. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, L.; Clarke, P.A.; Barker, K.; Chanthery, Y.; Gustafson, C.W.; Tucker, E.; Renshaw, J.; Raynaud, F.; Li, X.; Burke, R.; et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget 2016, 7, 57525–57544. [Google Scholar] [CrossRef] [PubMed]
- Hebron, K.E.; Wan, X.; Roth, J.S.; Liewehr, D.J.; Sealover, N.E.; Frye, W.J.E.; Kim, A.; Stauffer, S.; Perkins, O.L.; Sun, W.; et al. The Combination of Trametinib and Ganitumab is Effective in RAS-Mutated PAX-Fusion Negative Rhabdomyosarcoma Models. Clin. Cancer Res. 2023, 29, 472–487. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Hancock, D.C.; Sheridan, C.; Kumar, M.S.; Downward, J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013, 3, 548–563. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Nonami, A.; Chen, Z.; Nelson, E.; Chen, Y.; Liu, F.; Cho, H.; Zhang, J.; Sattler, M.; Mitsiades, C.; et al. Upregulation of IGF1R by mutant RAS in leukemia and potentiation of RAS signaling inhibitors by small-molecule inhibition of IGF1R. Clin. Cancer Res. 2014, 20, 5483–5495. [Google Scholar] [CrossRef]
- Wilky, B.A.; Rudek, M.A.; Ahmed, S.; Laheru, D.A.; Cosgrove, D.; Donehower, R.C.; Nelkin, B.; Ball, D.; Doyle, L.A.; Chen, H.; et al. A phase I trial of vertical inhibition of IGF signalling using cixutumumab, an anti-IGF-1R antibody, and selumetinib, an MEK 1/2 inhibitor, in advanced solid tumours. Br. J. Cancer 2015, 112, 24–31. [Google Scholar] [CrossRef]
- Yohe, M.E.; Gryder, B.E.; Shern, J.F.; Song, Y.K.; Chou, H.C.; Sindiri, S.; Mendoza, A.; Patidar, R.; Zhang, X.; Guha, R.; et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018, 10, eaan4470. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef]
- Beltran, P.J.; Calzone, F.J.; Mitchell, P.; Chung, Y.A.; Cajulis, E.; Moody, G.; Belmontes, B.; Li, C.M.; Vonderfecht, S.; Velculescu, V.E.; et al. Ganitumab (AMG 479) inhibits IGF-II-dependent ovarian cancer growth and potentiates platinum-based chemotherapy. Clin. Cancer Res. 2014, 20, 2947–2958. [Google Scholar] [CrossRef]
- Tatum, J.L.; Kalen, J.D.; Jacobs, P.M.; Ileva, L.V.; Riffle, L.A.; Hollingshead, M.G.; Doroshow, J.H. A spontaneously metastatic model of bladder cancer: Imaging characterization. J. Transl. Med. 2019, 17, 425. [Google Scholar] [CrossRef] [PubMed]
- Kalen, J.D.; Ileva, L.V.; Riffle, L.A.; Keita, S.; Tatum, J.L.; Jacobs, P.M.; Sanders, C.; James, A.; Difilippantonio, S.; Thang, L.; et al. Serial Non-Contrast Non-Gated T2w MRI Datasets of Patient Derived Xenograft Cancer Models for Development of Tissue Characterization Algorithms (PDMR-Texture Analysis) (Version 1) [Data Set]. The Cancer Imaging Archive. 2023. Available online: https://www.cancerimagingarchive.net/collection/pdmr-texture-analysis/ (accessed on 25 April 2024).
- Thiele, C.J. Neuroblastoma. In Human Cell Culture: Cancer Cell Lines Part 1; Masters, J.R.W., Palsson, B., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 21–53. [Google Scholar]
- Gao, M.-Z.; Wang, H.-B.; Chen, X.-L.; Cao, W.-T.; Fu, L.; Li, Y.; Quan, H.-T.; Xie, C.-Y.; Lou, L.-G. Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma. Acta Pharmacol. Sin. 2019, 40, 268–278. [Google Scholar] [CrossRef]
- Khanna, C.; Jaboin, J.J.; Drakos, E.; Tsokos, M.; Thiele, C.J. Biologically relevant orthotopic neuroblastoma xenograft models: Primary adrenal tumor growth and spontaneous distant metastasis. In Vivo 2002, 16, 77–85. [Google Scholar] [PubMed]
- Kugel III, C.H.; Aplin, A.E. Adaptive resistance to RAF inhibitors in melanoma. Pigment Cell Melanoma Res. 2014, 27, 1032–1038. [Google Scholar] [CrossRef]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Theurillat, J.P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef]
- See, W.L.; Tan, I.L.; Mukherjee, J.; Nicolaides, T.; Pieper, R.O. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012, 72, 3350–3359. [Google Scholar] [CrossRef]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A first-generation pediatric cancer dependency map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef]
- Hua, Z.; Gu, X.; Dong, Y.; Tan, F.; Liu, Z.; Thiele, C.J.; Li, Z. PI3K and MAPK pathways mediate the BDNF/TrkB-increased metastasis in neuroblastoma. Tumour Biol. 2016, 37, 16227–16236. [Google Scholar] [CrossRef]
- Li, Z.; Oh, D.Y.; Nakamura, K.; Thiele, C.J. Perifosine-induced inhibition of Akt attenuates brain-derived neurotrophic factor/TrkB-induced chemoresistance in neuroblastoma in vivo. Cancer 2011, 117, 5412–5422. [Google Scholar] [CrossRef]
- Rozen, E.J.; Shohet, J.M. Systematic review of the receptor tyrosine kinase superfamily in neuroblastoma pathophysiology. Cancer Metastasis Rev. 2022, 41, 33–52. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes. Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef]
- Ferguson, J.; Arozarena, I.; Ehrhardt, M.; Wellbrock, C. Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene 2013, 32, 86–96. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stauffer, S.; Roth, J.S.; Hernandez, E.R.; Kowalczyk, J.T.; Sealover, N.E.; Hebron, K.E.; James, A.; Isanogle, K.A.; Riffle, L.A.; Ileva, L.; et al. Preclinical Therapeutic Efficacy of RAF/MEK/ERK and IGF1R/AKT/mTOR Inhibition in Neuroblastoma. Cancers 2024, 16, 2320. https://doi.org/10.3390/cancers16132320
Stauffer S, Roth JS, Hernandez ER, Kowalczyk JT, Sealover NE, Hebron KE, James A, Isanogle KA, Riffle LA, Ileva L, et al. Preclinical Therapeutic Efficacy of RAF/MEK/ERK and IGF1R/AKT/mTOR Inhibition in Neuroblastoma. Cancers. 2024; 16(13):2320. https://doi.org/10.3390/cancers16132320
Chicago/Turabian StyleStauffer, Stacey, Jacob S. Roth, Edjay R. Hernandez, Joshua T. Kowalczyk, Nancy E. Sealover, Katie E. Hebron, Amy James, Kristine A. Isanogle, Lisa A. Riffle, Lilia Ileva, and et al. 2024. "Preclinical Therapeutic Efficacy of RAF/MEK/ERK and IGF1R/AKT/mTOR Inhibition in Neuroblastoma" Cancers 16, no. 13: 2320. https://doi.org/10.3390/cancers16132320