
Efficacy of Cisplatin–CXCR4 Antagonist Combination Therapy in Oral Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Reagent
2.3. Cell Proliferation Assay (MTS Assay)
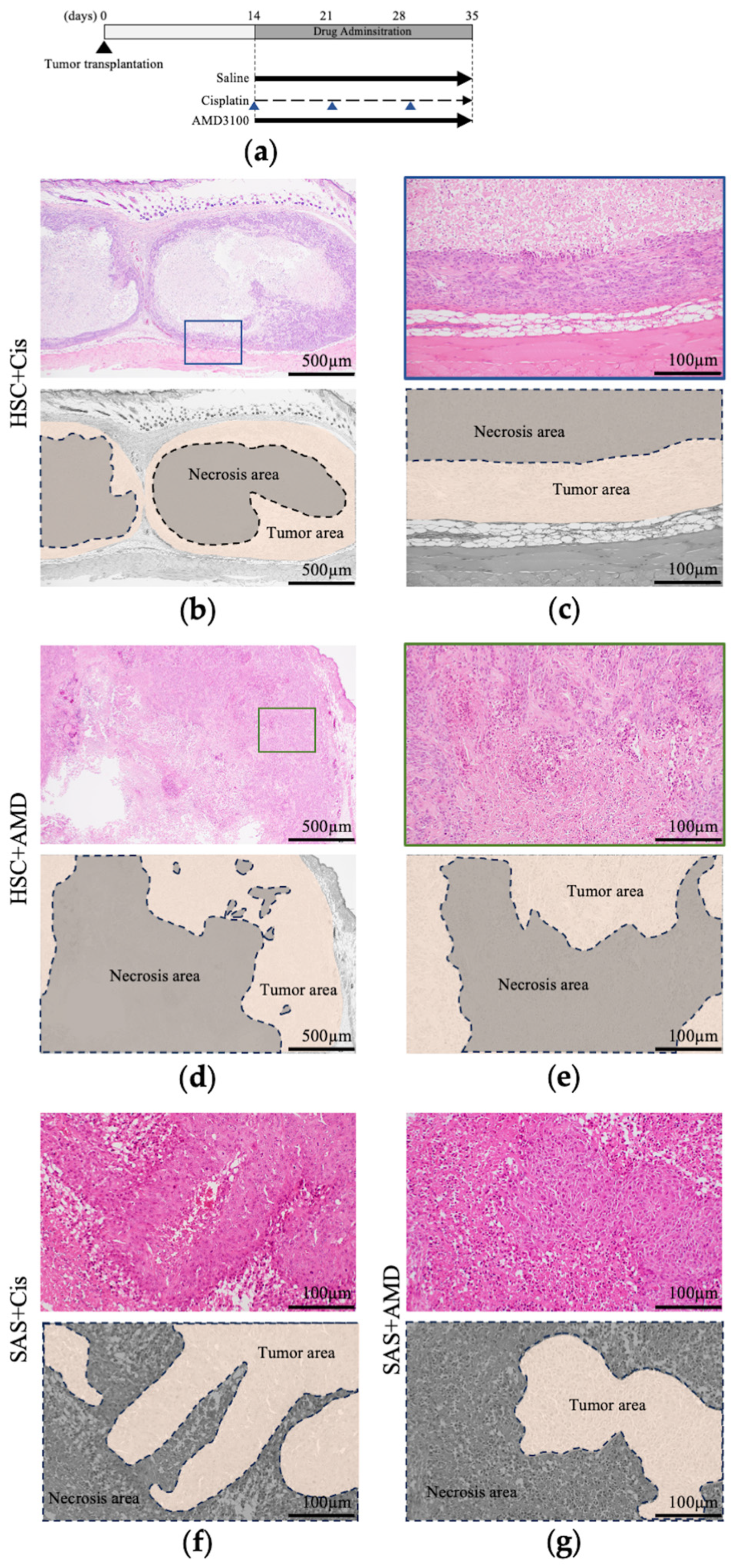
2.4. Xenografts and Administration of AMD3100 and Cisplatin to Mice
2.5. Immunohistochemistry (IHC)
2.6. Statistical Analysis
3. Results
3.1. MTS Assay
3.2. Administration of AMD3100 and Cisplatin to Tumor Cell-Implanted Mice
3.2.1. Histological Features of HSC-2 and SAS Cells
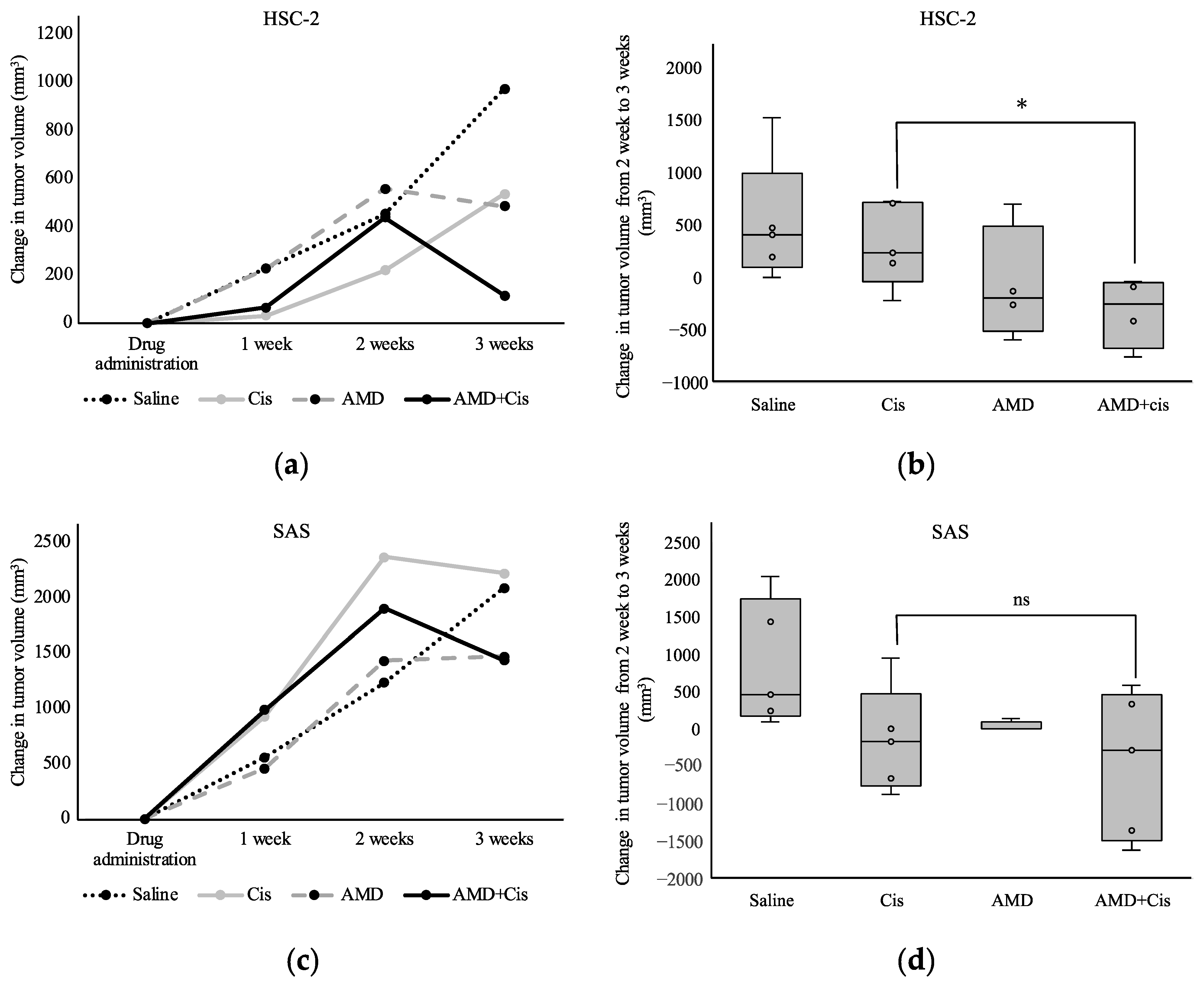
3.2.2. Tumor Volume
3.2.3. Histological Findings of Resected Tumors
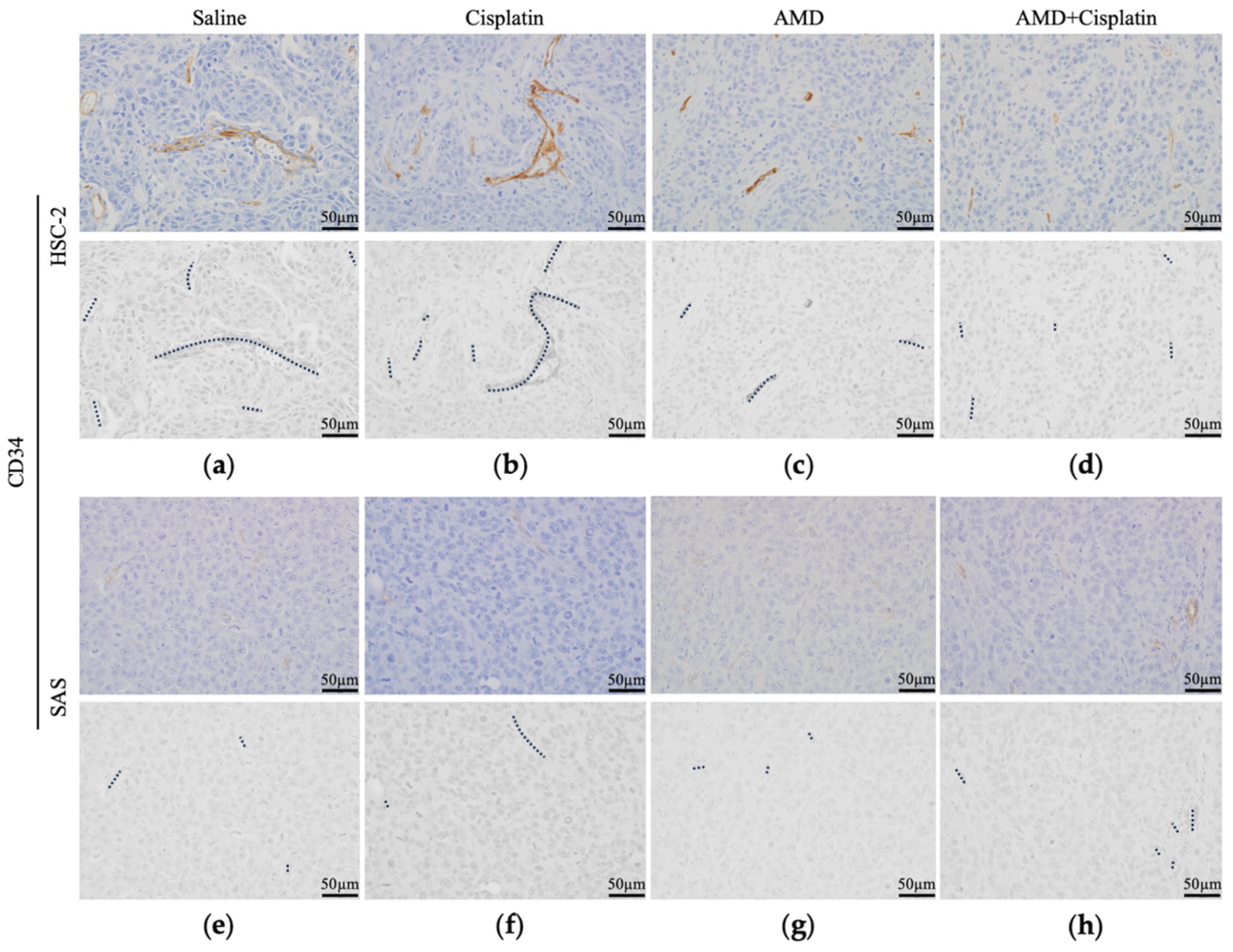
3.2.4. Comparison of Intratumoral Blood Vessels
3.2.5. Comparison of Intratumoral Blood Vessel Length
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonig, H.; Papayannopoulou, T. Hematopoietic stem cell mobilization: Updated conceptual renditions. Leukemia 2013, 27, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.J.; Clay, D.; Dupuy, C.; Rigal, S.; Jasmin, C.; Bourin, P.; Le Bousse-Kerdilès, M.C. Chemokine SDF-1 enhances circulating CD34+ cell proliferation in synergy with cytokines: Possible role in progenitor survival. Blood 2000, 95, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Tavian, M.; Cipponi, A.; Ficara, F.; Zappone, E.; Hoxie, J.; Peault, B.; Bordignon, C. Expression of CXCR4, the receptor for stromal cell-derived factor-1 on fetal and adult human lymphohematopoietic progenitors. Eur. J. Immunol. 1999, 29, 1823–1831. [Google Scholar] [CrossRef]
- Tachibana, K.; Hirota, S.; Iizasa, H.; Yoshida, H.; Kawabata, K.; Kataoka, Y.; Kitamura, Y.; Matsushima, K.; Yoshida, N.; Nishikawa, S.; et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 1998, 393, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Kruizinga, R.C.; Bestebroer, J.; Berghuis, P.; de Haas, C.J.C.; Links, T.P.; de Vries, E.G.E.; Walenkamp, A.M.E. Role of chemokines and their receptors in cancer. Curr. Pharm. Des. 2009, 15, 3396–3416. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, P.; Moser, B.; Baggiolini, M. Chemokines and their receptors in lymphocyte traffic and HIV infection. Adv. Immunol. 1999, 74, 127–180. [Google Scholar] [CrossRef]
- Sun, Y.; Mao, X.; Fan, C.; Liu, C.; Guo, A.; Guan, S.; Jin, Q.; Li, B.; Yao, F.; Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumour Biol. 2014, 35, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Schimanski, C.C.; Bahre, R.; Gockel, I.; Müller, A.; Frerichs, K.; Hörner, V.; Teufel, A.; Simiantonaki, N.; Biesterfeld, S.; Wehler, T.; et al. Dissemination of hepatocellular carcinoma is mediated via chemokine receptor CXCR4. Br. J. Cancer 2006, 95, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Yoshida, T.; Yamamoto, Y.; Furukita, Y.; Inoue, S.; Fujiwara, S.; Kawakita, N.; Nishino, T.; Minato, T.; Yuasa, Y.; et al. CXCR4 expression is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2017, 24, 832–840. [Google Scholar] [CrossRef]
- Rios, A.; Hsu, S.H.; Blanco, A.; Buryanek, J.; Day, A.L.; McGuire, M.F.; Brown, R.E. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience 2016, 3, 156–163. [Google Scholar] [CrossRef]
- Cui, K.; Zhao, W.; Wang, C.; Wang, A.; Zhang, B.; Zhou, W.; Yu, J.; Sun, Z.; Li, S. The CXCR4-CXCL12 pathway facilitates the progression of pancreatic cancer via induction of angiogenesis and lymphangiogenesis. J. Surg. Res. 2011, 171, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Righetti, A.; Giulietti, M.; Šabanović, B.; Occhipinti, G.; Principato, G.; Piva, F. CXCL12 and its isoforms: Different roles in pancreatic cancer? J. Oncol. 2019, 2019, 9681698. [Google Scholar] [CrossRef] [PubMed]
- Billadeau, D.D.; Chatterjee, S.; Bramati, P.; Sreekumar, R.; Shah, V.; Hedin, K.; Urrutia, R. Characterization of the CXCR4 signaling in pancreatic cancer cells. Int. J. Gastrointest. Cancer 2006, 37, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Almofti, A.; Uchida, D.; Begum, N.M.; Tomizuka, Y.; Iga, H.; Yoshida, H.; Sato, M. The clinicopathological significance of the expression of CXCR4 protein in oral squamous cell carcinoma. Int. J. Oncol. 2004, 25, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Rave-Fränk, M.; Tehrany, N.; Kitz, J.; Leu, M.; Weber, H.E.; Burfeind, P.; Schliephake, H.; Canis, M.; Beissbarth, T.; Reichardt, H.M.; et al. Prognostic value of CXCL12 and CXCR4 in inoperable head and neck squamous cell carcinoma. Strahlenther. Onkol. 2016, 192, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kawai, H.; Eguchi, T.; Sukegawa, S.; Oo, M.W.; Anqi, C.; Takabatake, K.; Nakano, K.; Okamoto, K.; Nagatsuka, H. Tumor angiogenic inhibition triggered necrosis (TAITN) in oral cancer. Cells 2019, 8, 761. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Li, R. Contemporary treatment of locally advanced oral cancer. Curr. Treat. Options Oncol. 2019, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Baru, J.; Huo, D.; Haraf, D.J.; Crowley, M.; Witt, M.E.; Blair, E.A.; Weichselbaum, R.R.; Rosen, F.; Vokes, E.E.; et al. Efficacy and safety of treating T4 oral cavity tumors with primary chemoradiotherapy. Head Neck 2009, 31, 1013–1021. [Google Scholar] [CrossRef]
- Harris, S.L.; Kimple, R.J.; Hayes, D.N.; Couch, M.E.; Rosenman, J.G. Never-smokers, never-drinkers: Unique clinical subgroup of young patients with head and neck squamous cell cancers. Head Neck 2010, 32, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Sakata, S.; Larson, D.W. Targeted therapy for colorectal cancer. Surg. Oncol. Clin. N. Am. 2022, 31, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, S.; Deng, J.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; Li, X.; et al. VEGF/VEGFR-targeted therapy and immunotherapy in non-small cell lung cancer: Targeting the tumor microenvironment. Int. J. Biol. Sci. 2022, 18, 3845–3858. [Google Scholar] [CrossRef] [PubMed]
- Al Kawas, H.; Saaid, I.; Jank, P.; Westhoff, C.C.; Denkert, C.; Pross, T.; Weiler, K.B.S.; Karsten, M.M. How VEGF-A and its splice variants affect breast cancer development—Clinical implications. Cell. Oncol. 2022, 45, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Passardi, A.; Scarpi, E.; Cavanna, L.; Dall’Agata, M.; Tassinari, D.; Leo, S.; Bernardini, I.; Gelsomino, F.; Tamberi, S.; Brandes, A.A.; et al. Inflammatory indexes as predictors of prognosis and bevacizumab efficacy in patients with metastatic colorectal cancer. Oncotarget 2016, 7, 33210–33219. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro. Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Dineen, S.P.; Lynn, K.D.; Sullivan, L.A.; Dellinger, M.T.; Sadegh, L.; Sullivan, J.P.; Shames, D.S.; Brekken, R.A. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol. Cancer Ther. 2009, 8, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Jan, K.; Klaudyna, K.; Patrycja, K.; Patrycja, K.; Barbara, G.S.; Izabela, G.; Dariusz, C.; Irena, B.B. The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors-A Review of Literature. Int. J. Mol. Sci. 2021, 22, 843. [Google Scholar] [CrossRef]
- Shannon, O.; Gwyn, B. The CXCR4/SDF-1 Chemokine Receptor Axis: A New Target Therapeutic for Non-small Cell Lung Cancer. J. Thorac. Oncol. 2008, 3, 1379–1383. [Google Scholar] [CrossRef]
- DiPersio, J.F.; Micallef, I.N.; Stiff, P.J.; Bolwell, B.J.; Maziarz, R.T.; Jacobsen, E.; Nademanee, A.; McCarty, J.; Bridger, G.; Calandra, G.; et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J. Clin. Oncol. 2009, 27, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Devine, S.M.; Vij, R.; Rettig, M.; Todt, L.; McGlauchlen, K.; Fisher, N.; Devine, H.; Link, D.C.; Calandra, G.; Bridger, G.; et al. Rapid mobilization of functional donor hematopoietic cells without G-CSF using AMD3100, an antagonist of the CXCR4/SDF-1 interaction. Blood 2008, 112, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Itashiki, Y.; Harada, K.; Takenawa, T.; Ferdous, T.; Ueyama, Y.; Mishima, K. Antitumor effects of bevacizumab in combination with fluoropyrimidine drugs on human oral squamous cell carcinoma. Oncol. Lett. 2021, 22, 730. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, S.; Kawai, H.; Soe, Y.; Eain, H.S.; Sanou, S.; Takabatake, K.; Takeshita, Y.; Hisatomi, M.; Nagatsuka, H.; Asaumi, J.; et al. Efficacy of Cisplatin–CXCR4 Antagonist Combination Therapy in Oral Cancer. Cancers 2024, 16, 2326. https://doi.org/10.3390/cancers16132326
Yoshida S, Kawai H, Soe Y, Eain HS, Sanou S, Takabatake K, Takeshita Y, Hisatomi M, Nagatsuka H, Asaumi J, et al. Efficacy of Cisplatin–CXCR4 Antagonist Combination Therapy in Oral Cancer. Cancers. 2024; 16(13):2326. https://doi.org/10.3390/cancers16132326
Chicago/Turabian StyleYoshida, Saori, Hotaka Kawai, Yamin Soe, Htoo Shwe Eain, Sho Sanou, Kiyofumi Takabatake, Yohei Takeshita, Miki Hisatomi, Hitoshi Nagatsuka, Junichi Asaumi, and et al. 2024. "Efficacy of Cisplatin–CXCR4 Antagonist Combination Therapy in Oral Cancer" Cancers 16, no. 13: 2326. https://doi.org/10.3390/cancers16132326
APA StyleYoshida, S., Kawai, H., Soe, Y., Eain, H. S., Sanou, S., Takabatake, K., Takeshita, Y., Hisatomi, M., Nagatsuka, H., Asaumi, J., & Yanagi, Y. (2024). Efficacy of Cisplatin–CXCR4 Antagonist Combination Therapy in Oral Cancer. Cancers, 16(13), 2326. https://doi.org/10.3390/cancers16132326