
The Paradox of Ribosomal Insufficiency Coupled with Increased Cancer: Shifting the Perspective from the Cancer Cell to the Microenvironment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
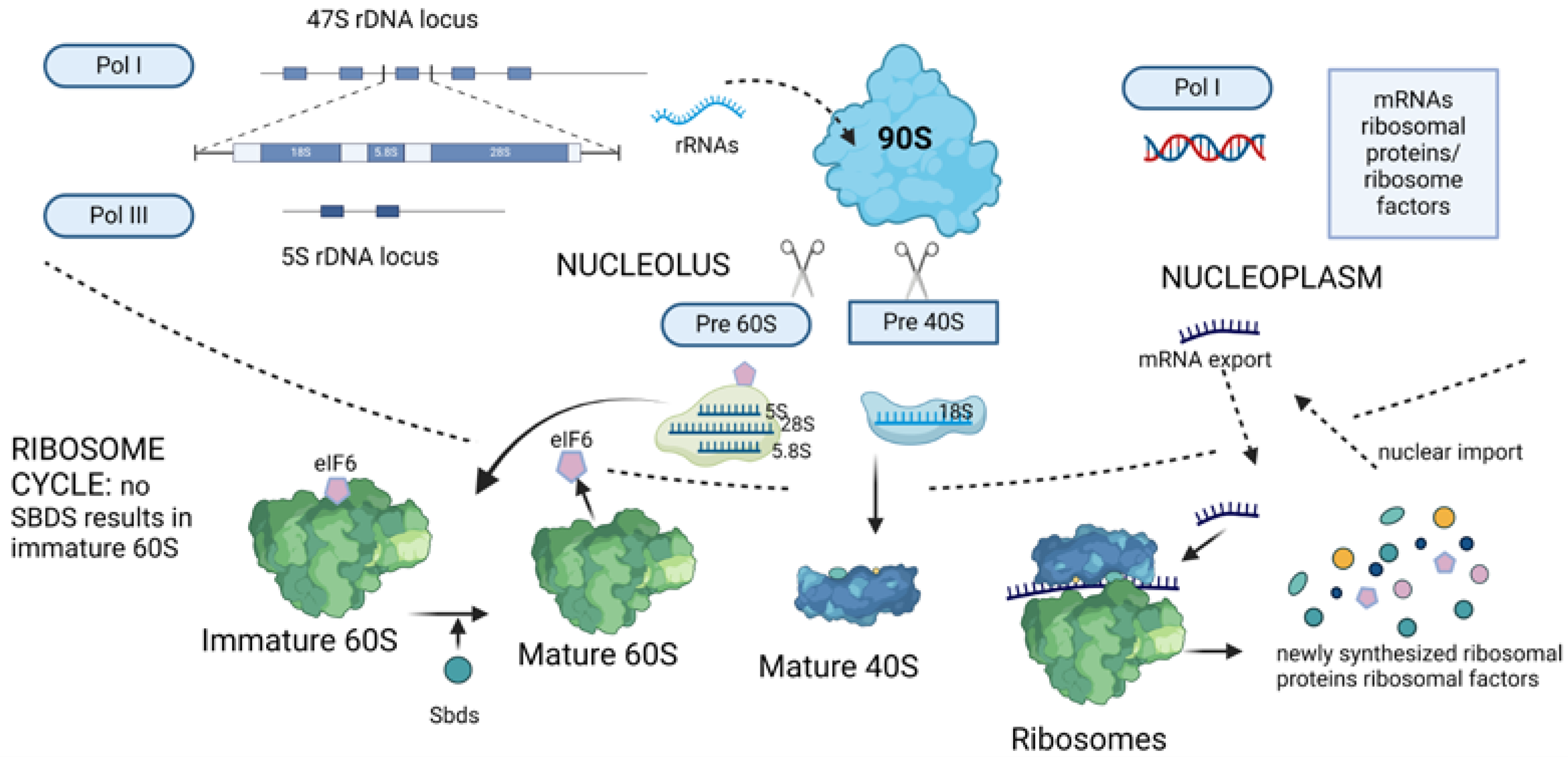
2. Ribosome Biogenesis and Translation
3. Ribosomopathies and Cancer at First Sight: A Simplified View
4. Abundant Ribosomes and Translation Factors Favor Cancer Development in a Cell-Autonomous Fashion
5. Loss of Ribosomes and Cancer: Do Alterations in Ribosomal Stoichiometry Lead to Oncoribosomes?
6. Loss of Ribosomes and Cancer: Are Ribosomal Proteins Tumor Suppressors?
7. Immunological Defects Driven by Translation and Cancer as the Result of a Poor Immunological Response
8. Bone Marrow Deficits and Tumors
9. Concluding Remarks and Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lempiäinen, H.; Shore, D. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Sulima, S.O.; Dinman, J.D. Ribosomopathies and the paradox of cellular hypo-to hyperproliferation. Blood J. Am. Soc. Hematol. 2015, 125, 1377–1382. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Dörner, K.; Ruggeri, C.; Zemp, I.; Kutay, U. Ribosome biogenesis factors—From names to functions. EMBO J. 2023, 42, e112699. [Google Scholar]
- Penzo, M.; Montanaro, L.; Treré, D.; Derenzini, M. The ribosome biogenesis—Cancer connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar]
- Blagden, S.P.; Willis, A.E. The biological and therapeutic relevance of mRNA translation in cancer. Nat. Rev. Clin. Oncol. 2011, 8, 280–291. [Google Scholar] [CrossRef]
- Yang, K.; Yang, J.; Yi, J. Nucleolar Stress: Hallmarks, sensing mechanism and diseases. Cell Stress 2018, 2, 125. [Google Scholar]
- Schimmel, P. The emerging complexity of the tRNA world: Mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 2018, 19, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Iborra, F.J.; Jackson, D.A.; Cook, P.R. The case for nuclear translation. J. Cell Sci. 2004, 117, 5713–5720. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.E.; Loughran, G.; Fedorova, A.D.; Mikhaylova, M.S.; Shatsky, I.N.; Baranov, P.V. Non-AUG translation initiation in mammals. Genome Biol. 2022, 23, 111. [Google Scholar] [CrossRef]
- Fabbri, L.; Chakraborty, A.; Robert, C.; Vagner, S. The plasticity of mRNA translation during cancer progression and therapy resistance. Nat. Rev. Cancer 2021, 21, 558–577. [Google Scholar] [CrossRef]
- Roux, P.P.; Topisirovic, I. Signaling pathways involved in the regulation of mRNA translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Farley-Barnes, K.I.; Ogawa, L.M.; Baserga, S.J. Ribosomopathies: Old concepts, new controversies. Trends Genet. 2019, 35, 754–767. [Google Scholar] [CrossRef]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef]
- Ban, N.; Beckmann, R.; Cate, J.H.; Dinman, J.D.; Dragon, F.; Ellis, S.R.; Lafontaine, D.L.; Lindahl, L.; Liljas, A.; Lipton, J.M.; et al. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014, 24, 165–169. [Google Scholar] [CrossRef]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.-N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef]
- Danilova, N.; Gazda, H.T. Ribosomopathies: How a common root can cause a tree of pathologies. Dis. Models Mech. 2015, 8, 1013–1026. [Google Scholar] [CrossRef]
- Marszałek-Kruk, B.A.; Wójcicki, P.; Dowgierd, K.; Śmigiel, R. Treacher Collins syndrome: Genetics, clinical features and management. Genes 2021, 12, 1392. [Google Scholar] [CrossRef]
- Wise, C.A.; Chiang, L.C.; Paznekas, W.A.; Sharma, M.; Musy, M.M.; Ashley, J.A.; Lovett, M.; Jabs, E.W. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl. Acad. Sci. USA 1997, 94, 3110–3115. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.S.; Giri, N.; Gianferante, D.M.; Jones, K.; Savage, S.A.; Alter, B.P.; McReynolds, L.J. Shwachman Diamond syndrome: Narrow genotypic spectrum and variable clinical features. Pediatr. Res. 2022, 92, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Venturi, G.; Montanaro, L. How altered ribosome production can cause or contribute to human disease: The spectrum of ribosomopathies. Cells 2020, 9, 2300. [Google Scholar] [CrossRef]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q-syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef]
- Valli, R.; Penzo, M. Inherited and acquired ribosomopathies: Missing puzzle pieces. Front. Genet. 2023, 14, 1194788. [Google Scholar] [CrossRef]
- Savage, S.A. Dyskeratosis congenita and telomere biology disorders. Hematology 2022, 2022, 637–648. [Google Scholar] [CrossRef]
- Sulima, S.O.; Kampen, K.R.; De Keersmaecker, K. Cancer biogenesis in ribosomopathies. Cells 2019, 8, 229. [Google Scholar] [CrossRef]
- Loreni, F.; Mancino, M.; Biffo, S. Translation factors and ribosomal proteins control tumor onset and progression: How? Oncogene 2014, 33, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.; Ferreira-Cerca, S.; Hurt, E. Eukaryotic ribosome biogenesis at a glance. J. Cell Sci. 2013, 126, 4815–4821. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.; Riback, J.A.; Bascetin, R.; Brangwynne, C.P. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182. [Google Scholar] [CrossRef]
- Derenzini, M.; Montanaro, L.; Treré, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, H.; Hsu, T. Identification of nucleolus organizer regions (NORs) in normal and neoplastic human cells by the silver-staining technique. Cytogenet. Genome Res. 1977, 19, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R. In the absence of ribosomal RNA synthesis, the ribosomal proteins of HeLa cells are synthesized normally and degraded rapidly. J. Mol. Biol. 1977, 115, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Pagano, M.; Russo, G.; Russo, A. Ribosome biogenesis and cancer: Overview on ribosomal proteins. Int. J. Mol. Sci. 2021, 22, 5496. [Google Scholar] [CrossRef] [PubMed]
- Ebright, R.Y.; Lee, S.; Wittner, B.S.; Niederhoffer, K.L.; Nicholson, B.T.; Bardia, A.; Truesdell, S.; Wiley, D.F.; Wesley, B.; Li, S.; et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 2020, 367, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, M.H.; Porvari, K.S.; Kyllönen, A.P.; Mustonen, M.V.; Lukkarinen, O.; Vihko, P.T. Several genes encoding ribosomal proteins are over-expressed in prostate-cancer cell lines: Confirmation of L7a and L37 over-expression in prostate-cancer tissue samples. Int. J. Cancer 1998, 78, 27–32. [Google Scholar] [CrossRef]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.N.; Lafita-Navarro, M.C.; Conacci-Sorrell, M. Regulation of nucleolar activity by MYC. Cells 2022, 11, 574. [Google Scholar] [CrossRef] [PubMed]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef]
- Ceci, M.; Gaviraghi, C.; Gorrini, C.; Sala, L.A.; Offenhäuser, N.; Carlo Marchisio, P.; Biffo, S. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 2003, 426, 579–584. [Google Scholar] [CrossRef]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef]
- Volta, V.; Beugnet, A.; Gallo, S.; Magri, L.; Brina, D.; Pesce, E.; Calamita, P.; Sanvito, F.; Biffo, S. RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiency. Cell. Mol. Life Sci. 2013, 70, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Xu, J.D.; Liu, X.L.; Xu, J.W.; Wang, W.J.; Li, Q.Q.; Chen, Q.; Xu, Z.D.; Liu, X.P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int. J. Cancer 2010, 127, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Deng, Y.-Z.; Zhao, J.-S.; Ji, X.-D.; Shi, J.; Feng, Y.-X.; Li, G.; Li, J.-J.; Zhu, D.; Koeffler, H.P.; et al. RACK1 promotes non-small-cell lung cancer tumorigenicity through activating sonic hedgehog signaling pathway. J. Biol. Chem. 2012, 287, 7845–7858. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, D. RACK1, a versatile hub in cancer. Oncogene 2015, 34, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; You, K.R.; Kim, I.H.; Cho, B.H.; Kim, C.Y.; Kim, D.G. Over-expression of the ribosomal protein L36a gene is associated with cellular proliferation in hepatocellular carcinoma. Hepatology 2004, 39, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.L.; Coggin, D.L.; King, C.R. High-Level Expression of the Ribosomal Protein L19 in Human Breast Tumors That Overexpress erb B-2. Cancer Res. 1993, 53, 1403–1408. [Google Scholar]
- Duff, D.; Long, A. Roles for RACK1 in cancer cell migration and invasion. Cell. Signal. 2017, 35, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; You, K.S.; Park, J.-S.; Lee, S.-G.; Seong, Y.-S. Ribosomal protein S6: A potential therapeutic target against cancer? Int. J. Mol. Sci. 2021, 23, 48. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef]
- Scagliola, A.; Miluzio, A.; Biffo, S. Translational control of metabolism and cell cycle progression in hepatocellular carcinoma. Int. J. Mol. Sci. 2023, 24, 4885. [Google Scholar] [CrossRef] [PubMed]
- Pradet-Balade, B.; Boulmé, F.; Beug, H.; Müllner, E.W.; Garcia-Sanz, J.A. Translation control: Bridging the gap between genomics and proteomics? Trends Biochem. Sci. 2001, 26, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Loreni, F.; Gandin, V.; Sala, L.A.; Sonenberg, N.; Marchisio, P.C.; Biffo, S. Fibronectin controls cap-dependent translation through β1 integrin and eukaryotic initiation factors 4 and 2 coordinated pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 9200–9205. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Miluzio, A.; Barbieri, A.M.; Beugnet, A.; Kiyokawa, H.; Marchisio, P.C.; Biffo, S. Eukaryotic initiation factor 6 is rate-limiting in translation, growth and transformation. Nature 2008, 455, 684–688. [Google Scholar] [CrossRef]
- Miluzio, A.; Beugnet, A.; Grosso, S.; Brina, D.; Mancino, M.; Campaner, S.; Amati, B.; de Marco, A.; Biffo, S. Impairment of cytoplasmic eIF6 activity restricts lymphomagenesis and tumor progression without affecting normal growth. Cancer Cell 2011, 19, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Scagliola, A.; Miluzio, A.; Ventura, G.; Oliveto, S.; Cordiglieri, C.; Manfrini, N.; Cirino, D.; Ricciardi, S.; Valenti, L.; Baselli, G.; et al. Targeting of eIF6-driven translation induces a metabolic rewiring that reduces NAFLD and the consequent evolution to hepatocellular carcinoma. Nat. Commun. 2021, 12, 4878. [Google Scholar] [CrossRef]
- Jones, R.M.; Branda, J.; Johnston, K.A.; Polymenis, M.; Gadd, M.; Rustgi, A.; Callanan, L.; Schmidt, E.V. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol. Cell. Biol. 1996, 16, 4754–4764. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational control in cancer. Cold Spring Harb. Perspect. Biol. 2019, 11, a032896. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Deo, R.; Hough, R.P.; Liu, Y.; Horn, J.L.; Wright, J.L.; Lam, H.M.; Webster, K.R.; Chiang, G.G.; Sonenberg, N.; et al. mRNA translation is a therapeutic vulnerability necessary for bladder epithelial transformation. JCI Insight 2021, 6, e144920. [Google Scholar] [CrossRef]
- Karampelias, C.; Watt, K.; Mattsson, C.L.; Ruiz, A.F.; Rezanejad, H.; Mi, J.; Liu, X.; Chu, L.; Locasale, J.W.; Korbutt, G.S.; et al. MNK2 deficiency potentiates beta-cell regeneration via translational regulation. Nat. Chem. Biol. 2022, 18, 942–953. [Google Scholar] [CrossRef]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell. Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Anand, N.; Murthy, S.; Amann, G.; Wernick, M.; Porter, L.A.; Cukier, I.H.; Collins, C.; Gray, J.W.; Diebold, J.; Demetrick, D.J.; et al. Protein elongation factor EEF1A2 is a putative oncogene in ovarian cancer. Nat. Genet. 2002, 31, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Sadler, K.C.; Lai, K.; Farrington, S.; Bronson, R.T.; Lees, J.A.; Hopkins, N. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004, 2, e139. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; De Keersmaecker, K. Rise of the specialized onco-ribosomes. Oncotarget 2018, 9, 35205. [Google Scholar] [CrossRef] [PubMed]
- Gelfo, V.; Venturi, G.; Zacchini, F.; Montanaro, L. Decoding Ribosome Heterogeneity: A New Horizon in Cancer Therapy. Biomedicines 2024, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 2017, 67, 71–83.e7. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Xue, S.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef]
- Li, H.; Huo, Y.; He, X.; Yao, L.; Zhang, H.; Cui, Y.; Xiao, H.; Xie, W.; Zhang, D.; Wang, Y.; et al. A male germ-cell-specific ribosome controls male fertility. Nature 2022, 612, 725–731. [Google Scholar] [CrossRef]
- Geiger, T.; Wehner, A.; Schaab, C.; Cox, J.; Mann, M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteom. 2012, 11, M111.014050. [Google Scholar] [CrossRef]
- Reschke, M.; Clohessy, J.G.; Seitzer, N.; Goldstein, D.P.; Breitkopf, S.B.; Schmolze, D.B.; Ala, U.; Asara, J.M.; Beck, A.H.; Pandolfi, P.P. Characterization and analysis of the composition and dynamics of the mammalian riboproteome. Cell Rep. 2013, 4, 1276–1287. [Google Scholar] [CrossRef]
- Sulima, S.O.; Patchett, S.; Advani, V.M.; De Keersmaecker, K.; Johnson, A.W.; Dinman, J.D. Bypass of the pre-60S ribosomal quality control as a pathway to oncogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5640–5645. [Google Scholar] [CrossRef]
- Molavi, G.; Samadi, N.; Hosseingholi, E.Z. The roles of moonlight ribosomal proteins in the development of human cancers. J. Cell. Physiol. 2019, 234, 8327–8341. [Google Scholar] [CrossRef]
- Gallo, S.; Ricciardi, S.; Manfrini, N.; Pesce, E.; Oliveto, S.; Calamita, P.; Mancino, M.; Maffioli, E.; Moro, M.; Crosti, M.; et al. RACK1 specifically regulates translation through its binding to ribosomes. Mol. Cell. Biol. 2018, 38, e00230-18. [Google Scholar] [CrossRef]
- Calamita, P.; Gatti, G.; Miluzio, A.; Scagliola, A.; Biffo, S. Translating the game: Ribosomes as active players. Front. Genet. 2018, 9, 418681. [Google Scholar] [CrossRef]
- Weinberg, R.A. Tumor suppressor genes. Science 1991, 254, 1138–1146. [Google Scholar] [CrossRef]
- Boutelle, A.M.; Attardi, L.D. p53 and tumor suppression: It takes a network. Trends Cell Biol. 2021, 31, 298–310. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
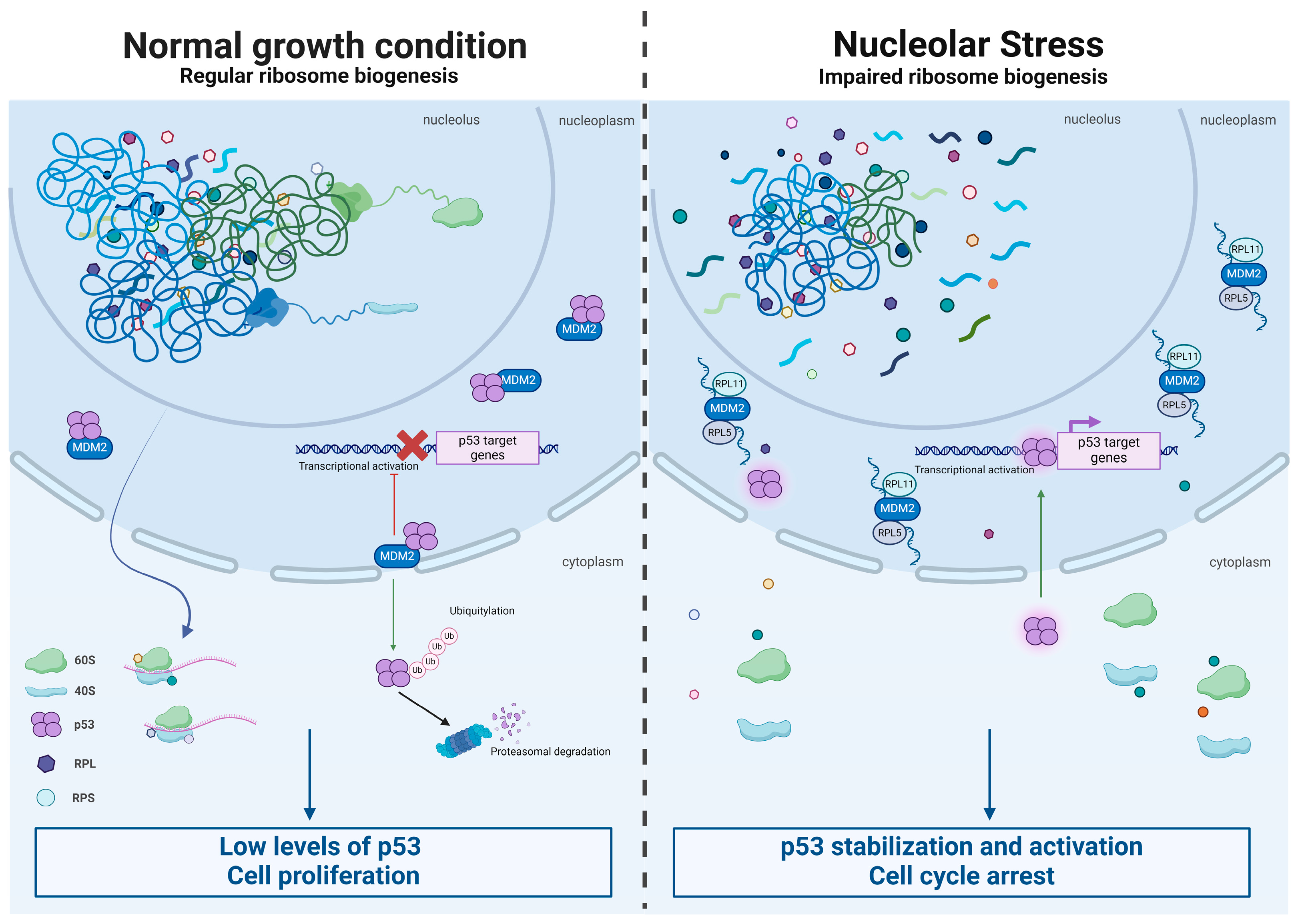
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef]
- Freiman, S.; Chen, J.; Flygare, J.; Kampman, C.; Pike-Overzet, K.; Mohseny, A.; Ha, T.-C.; Lidgren, M.; Soneji, S.; Karlsson, S.; et al. Single-Cell-Multiomics Demonstrates Molecular Efficacy of a Clinical Lentiviral Vector for Gene Therapy of RPS19-Deficient Diamond-Blackfan Anemia. Blood 2023, 142, 943. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, J.; Chen, Y.; Wang, S.; Cao, M.; Lu, H.; Zhou, X. Dual regulation of p53 by the ribosome maturation factor SBDS. Cell Death Dis. 2020, 11, 197. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, M.; Ma, L.; Jariyasakulroj, S.; Chang, Q.; Lin, Z.; Lu, Z.; Chen, J.-F. Recapitulating and reversing human brain ribosomopathy defects via the maladaptive integrated stress response. Sci. Adv. 2024, 10, eadk1034. [Google Scholar] [CrossRef]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; De Walque, R.; De Vleeschouwer, C.; Lafontaine, D.L. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef]
- Donati, G.; Peddigari, S.; Mercer, C.A.; Thomas, G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013, 4, 87–98. [Google Scholar] [CrossRef]
- Teng, T.; Mercer, C.A.; Hexley, P.; Thomas, G.; Fumagalli, S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol. Cell. Biol. 2013, 33, 4660–4671. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Atak, Z.K.; Li, N.; Vicente, C.; Patchett, S.; Girardi, T.; Gianfelici, V.; Geerdens, E.; Clappier, E.; Porcu, M.; et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 186–190. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Biggar, R.J.; Chaturvedi, A.K.; Goedert, J.J.; Engels, E.A. AIDS-related cancer and severity of immunosuppression in persons with AIDS. J. Natl. Cancer Inst. 2007, 99, 962–972. [Google Scholar] [CrossRef]
- Al-Herz, W.; Bousfiha, A.; Casanova, J.-L.; Chatila, T.; Conley, M.E.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Gaspar, H.B.; Holland, S.M. Primary immunodeficiency diseases: An update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front. Immunol. 2014, 5, 162. [Google Scholar]
- Mayor, P.C.; Eng, K.H.; Singel, K.L.; Abrams, S.I.; Odunsi, K.; Moysich, K.B.; Fuleihan, R.; Garabedian, E.; Lugar, P.; Ochs, H.D.; et al. Cancer in primary immunodeficiency diseases: Cancer incidence in the United States Immune Deficiency Network Registry. J. Allergy Clin. Immunol. 2018, 141, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Tsilifis, C.; Slatter, M.A.; Gennery, A.R. Too much of a good thing: A review of primary immune regulatory disorders. Front. Immunol. 2023, 14, 1279201. [Google Scholar] [CrossRef]
- Oliveira, G.; Wu, C.J. Dynamics and specificities of T cells in cancer immunotherapy. Nat. Rev. Cancer 2023, 23, 295–316. [Google Scholar] [CrossRef]
- Sigel, K.; Makinson, A.; Thaler, J. Lung cancer in persons with HIV. Curr. Opin. HIV AIDS 2017, 12, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 challenges in cancer immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Storb, U.; Martin, T.E. Number and activity of free and membrane-bound spleen ribosomes during the course of the immune response. Biochim. Biophys. Acta (BBA)—Nucleic Acids Protein Synth. 1972, 281, 406–415. [Google Scholar] [CrossRef]
- Piccirillo, C.A.; Bjur, E.; Topisirovic, I.; Sonenberg, N.; Larsson, O. Translational control of immune responses: From transcripts to translatomes. Nat. Immunol. 2014, 15, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, M.; Clavarino, G.; Gatti, E.; Schmidt, E.K.; de Gassart, A.; Blankenship, D.; Ogola, G.; Banchereau, J.; Chaussabel, D.; Pierre, P. Ribosomal protein mRNAs are translationally-regulated during human dendritic cells activation by LPS. Immunome Res. 2009, 5, 5. [Google Scholar] [CrossRef]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Morita, M.; Bederman, A.G.; Konieczny, B.T.; Kissick, H.T.; Sonenberg, N.; Ahmed, R. Translation is actively regulated during the differentiation of CD8+ effector T cells. Nat. Immunol. 2017, 18, 1046–1057. [Google Scholar] [CrossRef]
- Istomine, R.; Al-Aubodah, T.-A.; Alvarez, F.; Smith, J.A.; Wagner, C.; Piccirillo, C.A. The eIF4EBP-eIF4E axis regulates CD4+ T cell differentiation through modulation of T cell activation and metabolism. iScience 2023, 26, 106683. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Manfrini, N.; Alfieri, R.; Calamita, P.; Crosti, M.C.; Gallo, S.; Müller, R.; Pagani, M.; Abrignani, S.; Biffo, S. The translational machinery of human CD4+ T cells is poised for activation and controls the switch from quiescence to metabolic remodeling. Cell Metab. 2018, 28, 895–906.e5. [Google Scholar] [CrossRef] [PubMed]
- Herdy, B.; Jaramillo, M.; Svitkin, Y.V.; Rosenfeld, A.B.; Kobayashi, M.; Walsh, D.; Alain, T.; Sean, P.; Robichaud, N.; Topisirovic, I. Translational control of the activation of transcription factor NF-κB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat. Immunol. 2012, 13, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.; Jackson, L.V.; Oh, K.I.; Mai, A.; Ronai, Z.e.A.; Ruggero, D.; Fruman, D.A. The mTORC1/4E-BP/eIF4E axis promotes antibody class switching in B lymphocytes. J. Immunol. 2019, 202, 579–590. [Google Scholar] [CrossRef]
- Manfrini, N.; Ricciardi, S.; Miluzio, A.; Fedeli, M.; Scagliola, A.; Gallo, S.; Brina, D.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; et al. High levels of eukaryotic Initiation Factor 6 (eIF6) are required for immune system homeostasis and for steering the glycolytic flux of TCR-stimulated CD4+ T cells in both mice and humans. Dev. Comp. Immunol. 2017, 77, 69–76. [Google Scholar] [CrossRef]
- De Ponte Conti, B.; Miluzio, A.; Grassi, F.; Abrignani, S.; Biffo, S.; Ricciardi, S. mTOR-dependent translation drives tumor infiltrating CD8+ effector and CD4+ Treg cells expansion. eLife 2021, 10, e69015. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, J.; Zhou, Q.; Philippot, Q.; Ogishi, M.; Rinchai, D.; Nieminen, T.; Seyedpour, S.; Parvaneh, N.; Rezaei, N.; Yazdanpanah, N.; et al. Human MCTS1-dependent translation of JAK2 is essential for IFN-γ immunity to mycobacteria. Cell 2023, 186, 5114–5134.e5127. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Jin, W.; Zoppi, G.; Vogel, I.A.; Akhmedov, M.; Bleck, C.K.; Beltraminelli, T.; Rieckmann, J.C.; Ramirez, N.J.; Benevento, M.; et al. Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 2020, 21, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J. Neuronal cell lineages in the nematode Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol. 1983, 48, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Woloszynek, J.R.; Rothbaum, R.J.; Rawls, A.S.; Minx, P.J.; Wilson, R.K.; Mason, P.J.; Bessler, M.; Link, D.C. Mutations of the SBDS gene are present in most patients with Shwachman-Diamond syndrome. Blood 2004, 104, 3588–3590. [Google Scholar] [CrossRef]
- Calamita, P.; Miluzio, A.; Russo, A.; Pesce, E.; Ricciardi, S.; Khanim, F.; Cheroni, C.; Alfieri, R.; Mancino, M.; Gorrini, C.; et al. SBDS-deficient cells have an altered homeostatic equilibrium due to translational inefficiency which explains their reduced fitness and provides a logical framework for intervention. PLoS Genet. 2017, 13, e1006552. [Google Scholar] [CrossRef]
- Myers, K.C.; Bolyard, A.A.; Otto, B.; Wong, T.E.; Jones, A.T.; Harris, R.E.; Davies, S.M.; Dale, D.C.; Shimamura, A. Variable clinical presentation of Shwachman–Diamond syndrome: Update from the North American Shwachman–Diamond syndrome registry. J. Pediatr. 2014, 164, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Link, D.C. Mechanisms of leukemic transformation in congenital neutropenia. Curr. Opin. Hematol. 2019, 26, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Rupec, R.A.; Jundt, F.; Rebholz, B.; Eckelt, B.; Herzinger, T.; Flaig, M.J.; Moosmann, S.; Plewig, G.; Dörken, B.; Förster, I. Stroma-mediated dysregulation of myelopoiesis in mice lacking IκBα. Immunity 2005, 22, 479–491. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Myers, K.C.; Bowman, J.; Gibson, C.J.; Camarda, N.D.; Furutani, E.; Muscato, G.M.; Klein, R.H.; Ballotti, K.; Liu, S. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat. Commun. 2021, 12, 1334. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Kermasson, L.; Hilcenko, C.; Kargas, V.; Traynor, D.; Boukerrou, A.Z.; Escudero-Urquijo, N.; Faille, A.; Bertrand, A.; Rossmann, M. Somatic genetic rescue of a germline ribosome assembly defect. Nat. Commun. 2021, 12, 5044. [Google Scholar] [CrossRef]
- Menne, T.F.; Goyenechea, B.; Sánchez-Puig, N.; Wong, C.C.; Tonkin, L.M.; Ancliff, P.J.; Brost, R.L.; Costanzo, M.; Boone, C.; Warren, A.J. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 2007, 39, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.R.; Shimamura, A. Predisposition to myeloid malignancies in Shwachman-Diamond syndrome: Biological insights and clinical advances. Blood 2023, 141, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers 2017, 3, 17032. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Bezzerri, V.; Vella, A.; Gennaro, G.D.; Ortolani, R.; Nicolis, E.; Cesaro, S.; Fabrizzi, B.; Bronte, V.; Corey, S.J.; Cipolli, M. Peripheral blood immunophenotyping in a large cohort of patients with Shwachman–Diamond syndrome. Pediatr. Blood Cancer 2019, 66, e27597. [Google Scholar] [CrossRef]
- Rawls, A.S.; Gregory, A.D.; Woloszynek, J.R.; Liu, F.; Link, D.C. Lentiviral-mediated RNAi inhibition of Sbds in murine hematopoietic progenitors impairs their hematopoietic potential. Blood J. Am. Soc. Hematol. 2007, 110, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.S.; Pérez-Baos, S.; Walters, B.; Orlen, M.; Volkova, A.; Ruggles, K.; Park, C.Y.; Schneider, R.J. Translational regulation of TFH cell differentiation and autoimmune pathogenesis. Sci. Adv. 2022, 8, eabo1782. [Google Scholar] [CrossRef]
- Lipton, J.M.; Molmenti, C.L.; Desai, P.; Lipton, A.; Ellis, S.R.; Vlachos, A. Early onset colorectal cancer: An emerging cancer risk in patients with diamond blackfan anemia. Genes 2021, 13, 56. [Google Scholar] [CrossRef]
- Vlachos, A.; Rosenberg, P.S.; Atsidaftos, E.; Kang, J.; Onel, K.; Sharaf, R.N.; Alter, B.P.; Lipton, J.M. Increased risk of colon cancer and osteogenic sarcoma in Diamond-Blackfan anemia. Blood J. Am. Soc. Hematol. 2018, 132, 2205–2208. [Google Scholar] [CrossRef] [PubMed]
- Iskander, D.; Roberts, I.; Rees, C.; Szydlo, R.; Alikian, M.; Neale, M.; Harrington, Y.; Kelleher, P.; Karadimitris, A.; de la Fuente, J. Impaired cellular and humoral immunity is a feature of Diamond-Blackfan anaemia; experience of 107 unselected cases in the United Kingdom. Br. J. Haematol. 2019, 186, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.A.; Bagnara, G.P.; Strippoli, P.; Bonsi, L.; Vitale, L.; Tonelli, R.; Locatelli, F.; Gabutti, V.; Ramenghi, U.; D’Avanzo, M.; et al. Long-term bone marrow cultures in Diamond-Blackfan anemia reveal a defect of both granulomacrophage and erythroid progenitors. Exp. Hematol. 1999, 27, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Giri, N.; Alter, B.P.; Penrose, K.; Falk, R.T.; Pan, Y.; Savage, S.A.; Williams, M.; Kemp, T.J.; Pinto, L.A. Immune status of patients with inherited bone marrow failure syndromes. Am. J. Hematol. 2015, 90, 702–708. [Google Scholar] [CrossRef]
- Myers, K.C.; Furutani, E.; Weller, E.; Siegele, B.; Galvin, A.; Arsenault, V.; Alter, B.P.; Boulad, F.; Bueso-Ramos, C.; Burroughs, L. Myelodysplastic syndrome and acute myeloid leukemia in patients with Shwachman Diamond syndrome: A multicentre, retrospective, cohort study. Lancet Haematol. 2020, 7, e238. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Andrea, G.; Deroma, G.; Miluzio, A.; Biffo, S. The Paradox of Ribosomal Insufficiency Coupled with Increased Cancer: Shifting the Perspective from the Cancer Cell to the Microenvironment. Cancers 2024, 16, 2392. https://doi.org/10.3390/cancers16132392
D’Andrea G, Deroma G, Miluzio A, Biffo S. The Paradox of Ribosomal Insufficiency Coupled with Increased Cancer: Shifting the Perspective from the Cancer Cell to the Microenvironment. Cancers. 2024; 16(13):2392. https://doi.org/10.3390/cancers16132392
Chicago/Turabian StyleD’Andrea, Giacomo, Giorgia Deroma, Annarita Miluzio, and Stefano Biffo. 2024. "The Paradox of Ribosomal Insufficiency Coupled with Increased Cancer: Shifting the Perspective from the Cancer Cell to the Microenvironment" Cancers 16, no. 13: 2392. https://doi.org/10.3390/cancers16132392