
Purinergic Ca2+ Signaling as a Novel Mechanism of Drug Tolerance in BRAF-Mutant Melanoma
 , ,
, ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Drug Treatment
2.4. Dye Loading for the Imaging of Cytoplasmic Calcium
2.5. Live-Cell Imaging of Spontaneous Cytoplasmic Calcium Spikes
2.6. Live-Cell Imaging of ATP-Stimulated Cytoplasmic Calcium Spikes
2.7. Live-Cell Imaging of Store-Operated Calcium Entry (SOCE)
2.8. ppERK Staining
2.9. P2Xi Effects on ppERK
2.10. Ca2+ Mobilization Effects on ppERK
2.11. ATP Solution
ATP Stimulation of ppERK
2.12. Bulk RNAseq
2.12.1. Drug Treatment
2.12.2. RNA Isolation
2.12.3. Transcriptomic Sequencing
2.12.4. Sequence Processing
2.13. Image Processing
2.13.1. ppERK Images
2.13.2. Fura-2 Images
2.13.3. Calbryte Images
2.14. Data Analysis
2.14.1. Differential Expression of Transcript Abundance
2.14.2. ppERK
2.14.3. Fura-2 Spike Detection:
2.14.4. Calbryte-520 Spike Detection
2.14.5. Bootstrapping to Estimate Mean Proportions of Spiking Cells for Each Condition
3. Results
3.1. BRAFi Induces Cell-Intrinsic Ca2+ Signaling in Drug-Tolerant Cells
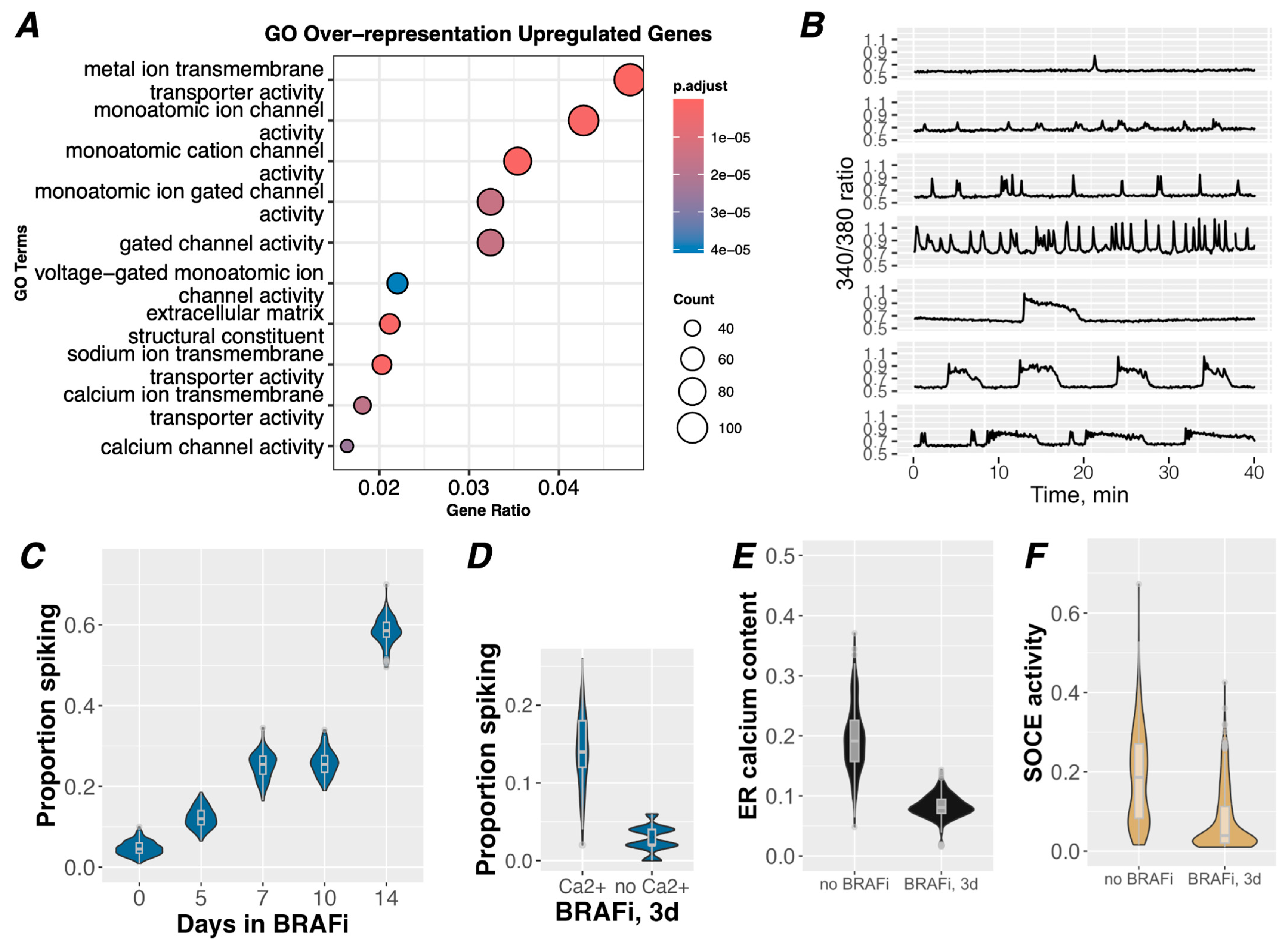
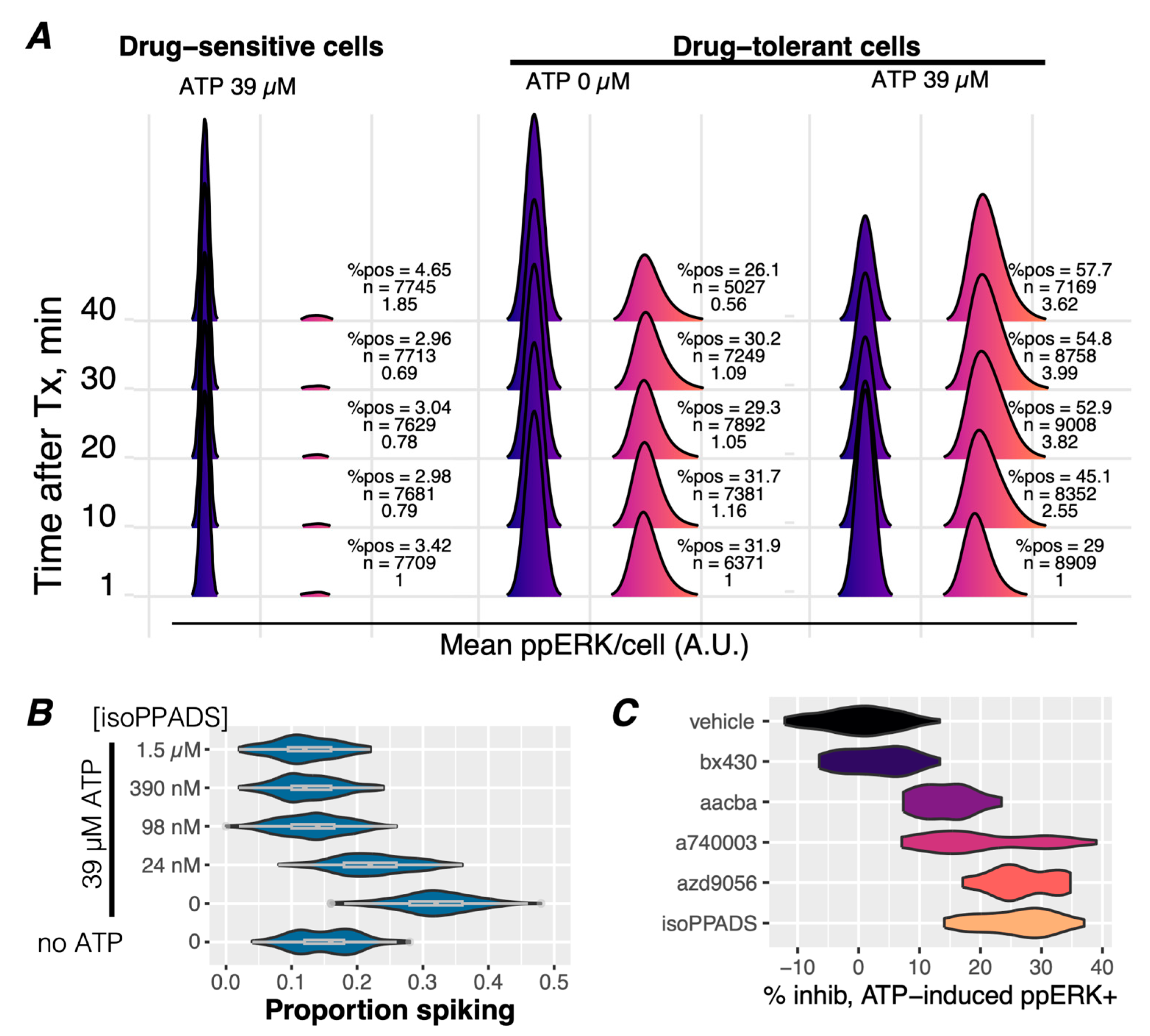
3.2. Purinergic Ca2+ Signaling Is A Mechanism of ERK Reactivation in Drug-Tolerant Idling Cells
3.3. ATP Stimulates Ca2+ Signaling and ERK Reactivation via P2X7 Receptors in Drug-Tolerant Idling Cells
3.4. Ca2+-Mediated Activation of ERK Is Potentiated in Drug-Tolerant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Handwerger, S.; Tomasz, A. Antibiotic Tolerance among Clinical Isolates of Bacteria. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 349–380. [Google Scholar] [CrossRef]
- Meredith, H.R.; Srimani, J.K.; Lee, A.J.; Lopatkin, A.J.; You, L. Collective Antibiotic Tolerance: Mechanisms, Dynamics and Intervention. Nat. Chem. Biol. 2015, 11, 182–188. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype Plasticity as Enabler of Melanoma Progression and Therapy Resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef]
- Marin-Bejar, O.; Rogiers, A.; Dewaele, M.; Femel, J.; Karras, P.; Pozniak, J.; Bervoets, G.; Van Raemdonck, N.; Pedri, D.; Swings, T.; et al. Evolutionary Predictability of Genetic versus Nongenetic Resistance to Anticancer Drugs in Melanoma. Cancer Cell 2021, 39, 1135–1149.e8. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling Cancer Persister Cells Arise from Lineages with Distinct Programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Yang, C.; Tian, C.; Hoffman, T.E.; Jacobsen, N.K.; Spencer, S.L. Melanoma Subpopulations That Rapidly Escape MAPK Pathway Inhibition Incur DNA Damage and Rely on Stress Signalling. Nat. Commun. 2021, 12, 1747. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma Plasticity and Phenotypic Diversity: Therapeutic Barriers and Opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef]
- Huang, S. Genetic and Non-Genetic Instability in Tumor Progression: Link between the Fitness Landscape and the Epigenetic Landscape of Cancer Cells. Cancer Metastasis Rev. 2013, 32, 423–448. [Google Scholar] [CrossRef]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly Targeted Cancer Therapy: Some Lessons from the Past Decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.R.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive Resistance of Melanoma Cells to RAF Inhibition via Reversible Induction of a Slowly Dividing De-Differentiated State. Mol. Syst. Biol. 2017, 13, 905. [Google Scholar] [CrossRef]
- Yan, H.; Chen, X.; Zhang, Q.; Qin, J.; Li, H.; Liu, C.; Calhoun-Davis, T.; Coletta, L.D.; Klostergaard, J.; Fokt, I.; et al. Drug-Tolerant Cancer Cells Show Reduced Tumor-Initiating Capacity: Depletion of CD44 Cells and Evidence for Epigenetic Mechanisms. PLoS ONE 2011, 6, e24397. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Shen, S.; Faouzi, S.; Souquere, S.; Roy, S.; Routier, E.; Libenciuc, C.; André, F.; Pierron, G.; Scoazec, J.Y.; Robert, C. Melanoma Persister Cells Are Tolerant to BRAF/MEK Inhibitors via ACOX1-Mediated Fatty Acid Oxidation. Cell Rep. 2020, 33, 108421. [Google Scholar] [CrossRef]
- Su, Y.; Ko, M.E.; Cheng, H.; Zhu, R.; Xue, M.; Wang, J.; Lee, J.W.; Frankiw, L.; Xu, A.; Wong, S.; et al. Multi-Omic Single-Cell Snapshots Reveal Multiple Independent Trajectories to Drug Tolerance in a Melanoma Cell Line. Nat. Commun. 2020, 11, 2345. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of Development and the Inheritance of Acquired Characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An Inhibitor of KDM5 Demethylases Reduces Survival of Drug-Tolerant Cancer Cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Goldman, A.; Khiste, S.; Freinkman, E.; Dhawan, A.; Majumder, B.; Mondal, J.; Pinkerton, A.B.; Eton, E.; Medhi, R.; Chandrasekar, V.; et al. Targeting Tumor Phenotypic Plasticity and Metabolic Remodeling in Adaptive Cross-Drug Tolerance. Sci. Signal. 2019, 12, eaas8779. [Google Scholar] [CrossRef]
- Liguoro, D.; Fattore, L.; Mancini, R.; Ciliberto, G. Drug Tolerance to Target Therapy in Melanoma Revealed at Single Cell Level: What Next. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188440. [Google Scholar] [CrossRef]
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A Stress-Induced Early Innate Response Causes Multidrug Tolerance in Melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-Mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Emert, B.L.; Reyes Hueros, R.A.; Cote, C.; Harmange, G.; Schaff, D.L.; Sizemore, A.E.; Gupte, R.; Torre, E.; Singh, A.; et al. Memory Sequencing Reveals Heritable Single-Cell Gene Expression Programs Associated with Distinct Cellular Behaviors. Cell 2020, 182, 947–959.e17. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Altschuler, S.J.; Wu, L.F. Cellular Heterogeneity: Do Differences Make a Difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef]
- Dexter, D.L.; Leith, J.T. Tumor Heterogeneity and Drug Resistance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1986, 4, 244–257. [Google Scholar] [CrossRef]
- Lee, M.-C.W.; Lopez-Diaz, F.J.; Khan, S.Y.; Tariq, M.A.; Dayn, Y.; Vaske, C.J.; Radenbaugh, A.J.; Kim, H.J.; Emerson, B.M.; Pourmand, N. Single-Cell Analyses of Transcriptional Heterogeneity during Drug Tolerance Transition in Cancer Cells by RNA Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E4726–E4735. [Google Scholar] [CrossRef]
- Paudel, B.B.; Harris, L.A.; Hardeman, K.N.; Abugable, A.A.; Hayford, C.E.; Tyson, D.R.; Quaranta, V. A Nonquiescent “Idling” Population State in Drug-Treated, BRAF-Mutated Melanoma. Biophys. J. 2018, 114, 1499–1511. [Google Scholar] [CrossRef]
- Jia, D.; Paudel, B.B.; Hayford, C.E.; Hardeman, K.N.; Levine, H.; Onuchic, J.N.; Quaranta, V. Drug-Tolerant Idling Melanoma Cells Exhibit Theory-Predicted Metabolic Low-Low Phenotype. Front. Oncol. 2020, 10, 1426. [Google Scholar] [CrossRef]
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting Minimal Residual Disease: A Path to Cure. Nat. Rev. Cancer 2018, 18, 255–263. [Google Scholar] [CrossRef]
- Pérez-González, A.; Bévant, K.; Blanpain, C. Cancer Cell Plasticity during Tumor Progression, Metastasis and Response to Therapy. Nat. Cancer 2023, 4, 1063–1082. [Google Scholar] [CrossRef]
- Hardeman, K.N.; Peng, C.; Paudel, B.B.; Meyer, C.T.; Luong, T.; Tyson, D.R.; Young, J.D.; Quaranta, V.; Fessel, J.P. Dependence On Glycolysis Sensitizes BRAF-Mutated Melanomas For Increased Response To Targeted BRAF Inhibition. Sci. Rep. 2017, 7, 42604. [Google Scholar] [CrossRef]
- Meyer, C.T.; Wooten, D.J.; Paudel, B.B.; Bauer, J.; Hardeman, K.N.; Westover, D.; Lovly, C.M.; Harris, L.A.; Tyson, D.R.; Quaranta, V. Quantifying Drug Combination Synergy along Potency and Efficacy Axes. Cell Syst. 2019, 8, 97–108.e16. [Google Scholar] [CrossRef]
- Vierra, N.C.; Dadi, P.K.; Milian, S.C.; Dickerson, M.T.; Jordan, K.L.; Gilon, P.; Jacobson, D.A. TALK-1 Channels Control β Cell Endoplasmic Reticulum Ca(2+) Homeostasis. Sci. Signal. 2017, 10, eaan2883. [Google Scholar] [CrossRef] [PubMed]
- Bannon, D.; Moen, E.; Schwartz, M.; Borba, E.; Kudo, T.; Greenwald, N.; Vijayakumar, V.; Chang, B.; Pao, E.; Osterman, E.; et al. DeepCell Kiosk: Scaling Deep Learning–Enabled Cellular Image Analysis with Kubernetes. Nat. Methods 2021, 18, 43–45. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Lubba, C.H.; Sethi, S.S.; Knaute, P.; Schultz, S.R.; Fulcher, B.D.; Jones, N.S. Catch22: CAnonical Time-Series CHaracteristics. Data Min. Knowl. Discov. 2019, 33, 1821–1852. [Google Scholar] [CrossRef]
- Hayford, C.E.; Stauffer, P.E.; Baleami, B.; Paudel, B.B.; Tyson, D.R.; Al’Khafaji, A.; Johnson, K.E.; Harris, L.A.; Brock, A.; Quaranta, V. A Heterogeneous Drug Tolerant Persister State in BRAF-Mutant Melanoma Is Characterized by Ion Channel Dysregulation and Susceptibility to Ferroptosis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Zhang, L.; Barritt, G. TRP Channels in Cancer. Biochim. Biophys. Acta 2007, 1772, 937–946. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The Calcium-Cancer Signalling Nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef]
- Böhme, I.; Schönherr, R.; Eberle, J.; Bosserhoff, A.K. Membrane Transporters and Channels in Melanoma. Rev. Physiol. Biochem. Pharmacol. 2021, 181, 269–374. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 Purinergic Signalling in the Tumour Microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Gross, S.; Mallu, P.; Joshi, H.; Schultz, B.; Go, C.; Soboloff, J. Ca2+ as a Therapeutic Target in Cancer. Adv. Cancer Res. 2020, 148, 233–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Luo, Z.; Hao, Y.; Ba, W.; Wang, R.; Wang, W.; Ding, X.; Li, C. mTOR-Mediated Na+/Ca2+ Exchange Affects Cell Proliferation and Metastasis of Melanoma Cells. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 92, 744–749. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef]
- Parekh, A.B. Store-Operated CRAC Channels: Function in Health and Disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. (V600E)BRAF Is Associated with Disabled Feedback Inhibition of RAF-MEK Signaling and Elevated Transcriptional Output of the Pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-Stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [PubMed]
- North, R.A. Molecular Physiology of P2X Receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef]
- Gachet, C. Regulation of Platelet Functions by P2 Receptors. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.G.; Huang, C.; et al. From Purines to Purinergic Signalling: Molecular Functions and Human Diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single-Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Rosen, N. Mutant BRAF Melanomas--Dependence and Resistance. Cancer Cell 2011, 19, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Allosteric MEK1/2 Inhibitors Including Cobimetanib and Trametinib in the Treatment of Cutaneous Melanomas. Pharmacol. Res. 2017, 117, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J. Decoding Complex Ca2+ Signals through the Modulation of Ras Signaling. Curr. Opin. Cell Biol. 2006, 18, 157–161. [Google Scholar] [CrossRef]
- Cullen, P.J.; Lockyer, P.J. Integration of Calcium and Ras Signalling. Nat. Rev. Mol. Cell Biol 2002, 3, 339–348. [Google Scholar] [CrossRef]
- Kupzig, S.; Walker, S.A.; Cullen, P.J. The Frequencies of Calcium Oscillations Are Optimized for Efficient Calcium-Mediated Activation of Ras and the ERK/MAPK Cascade. Proc. Natl. Acad. Sci. USA 2005, 102, 7577–7582. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Busse, R. Calcium Signaling in Endothelial Cells Involves Activation of Tyrosine Kinases and Leads to Activation of Mitogen-Activated Protein Kinases. Circ. Res. 1995, 76, 522–529. [Google Scholar] [CrossRef]
- Apáti, A.; Jánossy, J.; Brózik, A.; Bauer, P.I.; Magócsi, M. Calcium Induces Cell Survival and Proliferation through the Activation of the MAPK Pathway in a Human Hormone-Dependent Leukemia Cell Line, TF-1. J. Biol. Chem. 2003, 278, 9235–9243. [Google Scholar] [CrossRef]
- Agell, N.; Bachs, O.; Rocamora, N.; Villalonga, P. Modulation of the Ras/Raf/MEK/ERK Pathway by Ca2+, and Calmodulin. Cell Signal. 2002, 14, 649–654. [Google Scholar] [CrossRef]
- Rusanescu, G.; Qi, H.; Thomas, S.M.; Brugge, J.S.; Halegoua, S. Calcium Influx Induces Neurite Growth through a Src-Ras Signaling Cassette. Neuron 1995, 15, 1415–1425. [Google Scholar] [CrossRef]
- Schmidt, M.; Goebeler, M.; Posern, G.; Feller, S.M.; Seitz, C.S.; Brocker, E.B.; Rapp, U.R.; Ludwig, S. Ras-Independent Activation of the Raf/MEK/ERK Pathway upon Calcium-Induced Differentiation of Keratinocytes. J. Biol. Chem. 2000, 275, 41011–41017. [Google Scholar] [CrossRef]
- Tebar, F.; Lladó, A.; Enrich, C. Role of Calmodulin in the Modulation of the MAPK Signalling Pathway and the Transactivation of Epidermal Growth Factor Receptor Mediated by PKC. FEBS Lett. 2002, 517, 206–210. [Google Scholar] [CrossRef]
- Anguita, E.; Villalobo, A. Src-Family Tyrosine Kinases and the Ca2+ Signal. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 915–932. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca(2+)/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef]
- Illes, P.; Müller, C.E.; Jacobson, K.A.; Grutter, T.; Nicke, A.; Fountain, S.J.; Kennedy, C.; Schmalzing, G.; Jarvis, M.F.; Stojilkovic, S.S.; et al. Update of P2X Receptor Properties and Their Pharmacology: IUPHAR Review 30. Br. J. Pharmacol. 2021, 178, 489–514. [Google Scholar] [CrossRef]
- Broom, D.C.; Matson, D.J.; Bradshaw, E.; Buck, M.E.; Meade, R.; Coombs, S.; Matchett, M.; Ford, K.K.; Yu, W.; Yuan, J.; et al. Characterization of N-(Adamantan-1-Ylmethyl)-5-[(3R-Aminopyrrolidin-1-Yl)Methyl]-2-Chloro-Benzamide, a P2X7 Antagonist in Animal Models of Pain and Inflammation. J. Pharmacol. Exp. Ther. 2008, 327, 620–633. [Google Scholar] [CrossRef]
- Gerosa, L.; Chidley, C.; Fröhlich, F.; Sanchez, G.; Lim, S.K.; Muhlich, J.; Chen, J.Y.; Vallabhaneni, S.; Baker, G.J.; Schapiro, D.; et al. Receptor-Driven ERK Pulses Reconfigure MAPK Signaling and Enable Persistence of Drug-Adapted BRAF-Mutant Melanoma Cells. Cell Syst. 2020, 11, 478–494.e9. [Google Scholar] [CrossRef]
- Schenk, U.; Westendorf, A.M.; Radaelli, E.; Casati, A.; Ferro, M.; Fumagalli, M.; Verderio, C.; Buer, J.; Scanziani, E.; Grassi, F. Purinergic Control of T Cell Activation by ATP Released through Pannexin-1 Hemichannels. Sci. Signal. 2008, 1, ra6. [Google Scholar] [CrossRef]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An Autophagy-Driven Pathway of ATP Secretion Supports the Aggressive Phenotype of BRAFV600E Inhibitor-Resistant Metastatic Melanoma Cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Emert, B.L.; Cote, C.J.; Torre, E.A.; Dardani, I.P.; Jiang, C.L.; Jain, N.; Shaffer, S.M.; Raj, A. Variability within Rare Cell States Enables Multiple Paths toward Drug Resistance. Nat. Biotechnol. 2021, 39, 865–876. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of Colon Cancer to BRAF(V600E) Inhibition through Feedback Activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Cutsem, E.V.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Saini, N.; Lakshminarayanan, S.; Kundu, P.; Sarin, A. Notch1 Modulation of Cellular Calcium Regulates Mitochondrial Metabolism and Anti-Apoptotic Activity in T-Regulatory Cells. Front. Immunol. 2022, 13, 832159. [Google Scholar] [CrossRef]
- Mulder, C.; Prust, N.; van Doorn, S.; Reinecke, M.; Kuster, B.; van Bergen En Henegouwen, P.; Lemeer, S. Adaptive Resistance to EGFR-Targeted Therapy by Calcium Signaling in NSCLC Cells. Mol. Cancer Res. MCR 2018, 16, 1773–1784. [Google Scholar] [CrossRef]
- Egea, J.; Espinet, C.; Soler, R.M.; Peiró, S.; Rocamora, N.; Comella, J.X. Nerve Growth Factor Activation of the Extracellular Signal-Regulated Kinase Pathway Is Modulated by Ca(2+) and Calmodulin. Mol. Cell. Biol. 2000, 20, 1931–1946. [Google Scholar] [CrossRef]
- Villalobo, A. Regulation of ErbB Receptors by the Ca2+ Sensor Protein Calmodulin in Cancer. Biomedicines 2023, 11, 661. [Google Scholar] [CrossRef]
- Martín-Nieto, J.; Villalobo, A. The Human Epidermal Growth Factor Receptor Contains a Juxtamembrane Calmodulin-Binding Site. Biochemistry 1998, 37, 227–236. [Google Scholar] [CrossRef]
- Bilbao, P.S.; Katz, S.; Boland, R. Interaction of Purinergic Receptors with GPCRs, Ion Channels, Tyrosine Kinase and Steroid Hormone Receptors Orchestrates Cell Function. Purinergic Signal. 2012, 8, 91–103. [Google Scholar] [CrossRef]
- Amoroso, F.; Capece, M.; Rotondo, A.; Cangelosi, D.; Ferracin, M.; Franceschini, A.; Raffaghello, L.; Pistoia, V.; Varesio, L.; Adinolfi, E. The P2X7 Receptor Is a Key Modulator of the PI3K/GSK3β/VEGF Signaling Network: Evidence in Experimental Neuroblastoma. Oncogene 2015, 34, 5240–5251. [Google Scholar] [CrossRef]
- Hill, L.M.; Gavala, M.L.; Lenertz, L.Y.; Bertics, P.J. Extracellular ATP May Contribute to Tissue Repair by Rapidly Stimulating Purinergic Receptor X7-Dependent Vascular Endothelial Growth Factor Release from Primary Human Monocytes. J. Immunol. 2010, 185, 3028–3034. [Google Scholar] [CrossRef]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 Receptor Increases in Vivo Tumor Growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef]
- Goyal, Y.; Busch, G.T.; Pillai, M.; Li, J.; Boe, R.H.; Grody, E.I.; Chelvanambi, M.; Dardani, I.P.; Emert, B.; Bodkin, N.; et al. Diverse Clonal Fates Emerge upon Drug Treatment of Homogeneous Cancer Cells. Nature 2023, 620, 651–659. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER Stress-Induced Autophagy Overcomes BRAF Inhibitor Resistance in Melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kim, J.Y.; Ahn, Y.; Lee, B.M.; Heo, Y.; Hwang, S.; Lee, S.H.; Lee, J.; Chung, G.; Oh, S.H. Critical Role of ATP-P2X7 Axis in UV-Induced Melanogenesis. J. Investig. Dermatol. 2019, 139, 1554–1563.e6. [Google Scholar] [CrossRef]
- Lara, R.; Adinolfi, E.; Harwood, C.A.; Philpott, M.; Barden, J.A.; Di Virgilio, F.; McNulty, S. P2X7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharmacol. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Randic, T.; Magni, S.; Philippidou, D.; Margue, C.; Grzyb, K.; Preis, J.R.; Wroblewska, J.P.; Nazarov, P.V.; Mittelbronn, M.; Frauenknecht, K.B.M.; et al. Single-Cell Transcriptomics of NRAS-Mutated Melanoma Transitioning to Drug Resistance Reveals P2RX7 as an Indicator of Early Drug Response. Cell. Rep. 2023, 42, 112696. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 2018, 28, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Leli, N.M.; Onorati, A.; Piao, S.; Verginadis, I.I.; Tameire, F.; Rebecca, V.W.; Chude, C.I.; Murugan, S.; Fennelly, C.; et al. ER Translocation of the MAPK Pathway Drives Therapy Resistance in BRAF-Mutant Melanoma. Cancer Discov. 2019, 9, 396–415. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stauffer, P.E.; Brinkley, J.; Jacobson, D.A.; Quaranta, V.; Tyson, D.R. Purinergic Ca2+ Signaling as a Novel Mechanism of Drug Tolerance in BRAF-Mutant Melanoma. Cancers 2024, 16, 2426. https://doi.org/10.3390/cancers16132426
Stauffer PE, Brinkley J, Jacobson DA, Quaranta V, Tyson DR. Purinergic Ca2+ Signaling as a Novel Mechanism of Drug Tolerance in BRAF-Mutant Melanoma. Cancers. 2024; 16(13):2426. https://doi.org/10.3390/cancers16132426
Chicago/Turabian StyleStauffer, Philip E., Jordon Brinkley, David A. Jacobson, Vito Quaranta, and Darren R. Tyson. 2024. "Purinergic Ca2+ Signaling as a Novel Mechanism of Drug Tolerance in BRAF-Mutant Melanoma" Cancers 16, no. 13: 2426. https://doi.org/10.3390/cancers16132426