Utility of Targeted Sequencing Compared to FISH for Detection of Chronic Lymphocytic Leukemia Copy Number Alterations

 , , , , , ,
, , , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Patients
2.2. DNA Sequencing
2.3. Copy Number Alterations
2.4. Statistical Methods
3. Results
3.1. Patient Characteristics
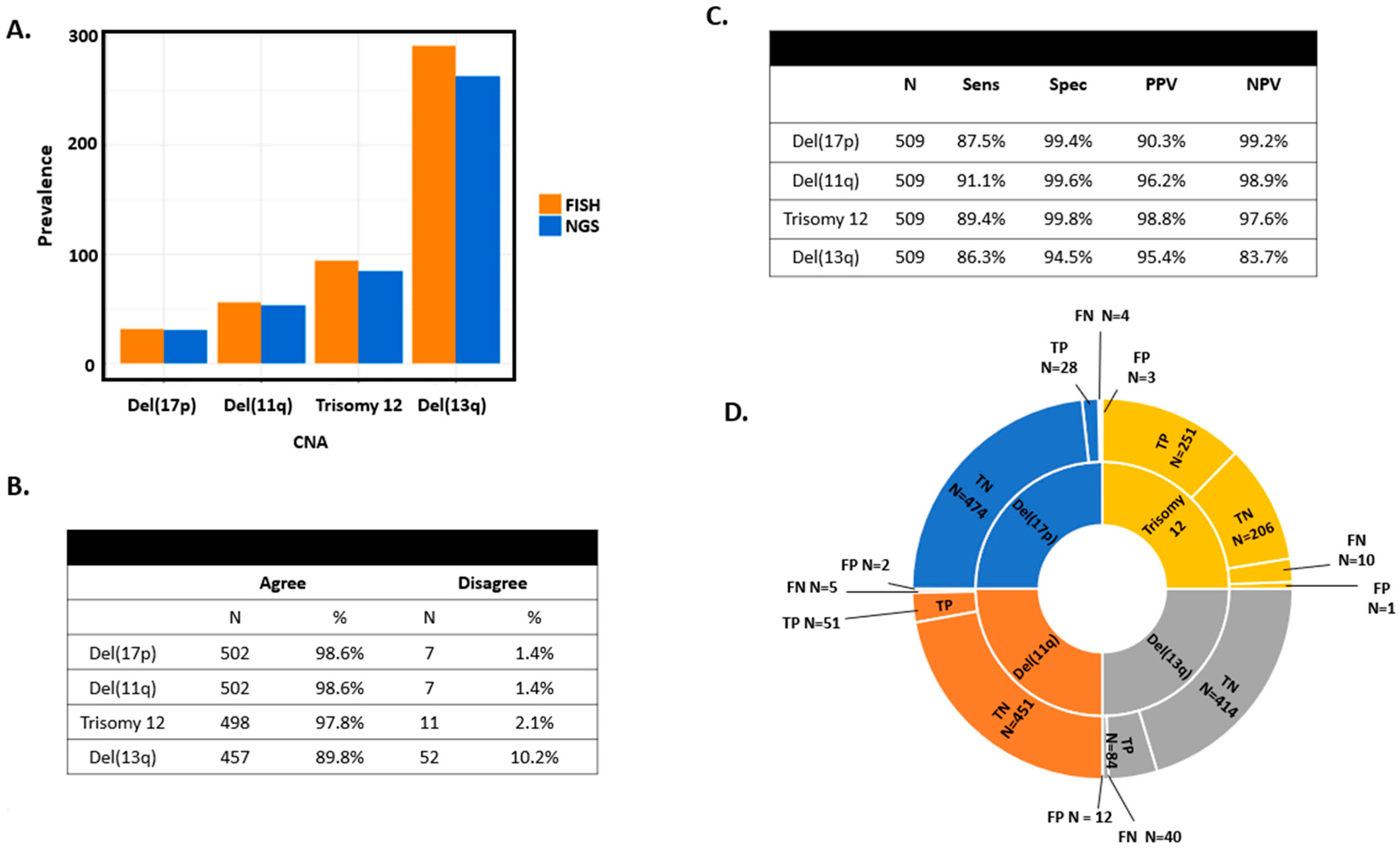
3.2. Sensitivity, Specificity, Positive Predictive Value, and Negative Predictive Value Comparing FISH and Targeted Sequencing
3.3. Discordance between FISH and Targeted Sequencing
3.4. Other CNAs and Genetic Aberrations as Detected by Targeted Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Dyke, D.L.; Werner, L.; Rassenti, L.Z.; Neuberg, D.; Ghia, E.; Heerema, N.A.; Cin, P.D.; Aquila, M.D.; Sreekantaiah, C.; Greaves, A.W.; et al. The Dohner fluorescence in situ hybridization prognostic classification of chronic lymphocytic leukaemia (CLL): The CLL Research Consortium experience. Br. J. Haematol. 2016, 173, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Krober, A.; Bullinger, L.; Dohner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Smoley, S.A.; Van Dyke, D.L.; Kay, N.E.; Heerema, N.A.; Dell’Aquila, M.L.; Cin, P.D.; Koduru, P.; Aviram, A.; Rassenti, L.; Byrd, J.C.; et al. Standardization of fluorescence in situ hybridization studies on chronic lymphocytic leukemia (CLL) blood and marrow cells by the CLL Research Consortium. Cancer Genet. Cytogenet. 2010, 203, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Dewald, G.W.; Brockman, S.R.; Paternoster, S.F.; Bone, N.D.; O’Fallon, J.R.; Allmer, C.; James, C.D.; Jelinek, D.F.; Tschumper, R.C.; Hanson, C.A.; et al. Chromosome anomalies detected by interphase fluorescence in situ hybridization: Correlation with significant biological features of B-cell chronic lymphocytic leukaemia. Br. J. Haematol. 2003, 121, 287–295. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (Version 1.2024). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cll.pdf (accessed on 1 January 2020).
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M.; Cin, P.D.; Campelo, M.D.; Gallego, M.S.; Font, I.G.; Haase, D.T.; et al. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2022, 139, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Acute Myeloma Leukemia (Verson 6.2023). Available online: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf (accessed on 1 November 2023).
- National Comprehensive Cancer Network. Acute Lymphoblastic Leukemia (Version 3.2023). Available online: https://www.nccn.org/professionals/physician_gls/pdf/all.pdf (accessed on 1 November 2023).
- National Comprehensive Cancer Network. Myelodysplastic Syndromes (Version 3.2023). Available online: https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf (accessed on 1 November 2023).
- Landgren, O.; Albitar, M.; Ma, W.; Abbasi, F.; Hayes, R.B.; Ghia, P.; Marti, G.E.; Caporaso, N.E. B-cell clones as early markers for chronic lymphocytic leukemia. N. Engl. J. Med. 2009, 360, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Slager, S.L.; Lanasa, M.C.; Marti, G.E.; Achenbach, S.J.; Camp, N.J.; Abbasi, F.; Kay, N.E.; Vachon, C.M.; Cerhan, J.R.; Johnston, J.B.; et al. Natural history of monoclonal B-cell lymphocytosis among relatives in CLL families. Blood 2021, 137, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, P.M.; Hosnijeh, F.S.; Spath, F.; Hengeveld, P.J.; Agathangelidis, A.; Saleh, M.; Casabonne, D.; Benavente, Y.; Jerkeman, M.; Agudo, A.; et al. High-risk subtypes of chronic lymphocytic leukemia are detectable as early as 16 years prior to diagnosis. Blood 2022, 139, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Kleinstern, G.; O’Brien, D.R.; Li, X.; Tian, S.; Kabat, B.F.; Rabe, K.G.; Norman, A.D.; Yan, H.; Vachon, C.M.; Boddicker, N.J.; et al. Tumor mutational load predicts time to first treatment in chronic lymphocytic leukemia (CLL) and monoclonal B-cell lymphocytosis beyond the CLL international prognostic index. Am. J. Hematol. 2020, 95, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, D.L.; Shanafelt, T.D.; Call, T.G.; Zent, C.S.; Smoley, S.A.; Rabe, K.G.; Schwager, S.M.; Sonbert, J.C.; Slager, S.L.; Kay, N.E. A comprehensive evaluation of the prognostic significance of 13q deletions in patients with B-chronic lymphocytic leukaemia. Br. J. Haematol. 2010, 148, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Pflug, N.; Bahlo, J.; Shanafelt, T.D.; Eichhorst, B.F.; Bergmann, M.A.; Elter, T.; Bauer, K.; Malchau, G.; Rabe, K.G.; Stilgenbauer, S.; et al. Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood 2014, 124, 49–62. [Google Scholar] [CrossRef]
- Parikh, S.A.; Rabe, K.G.; Kay, N.E.; Call, T.G.; Ding, W.; Leis, J.F.; Kenderian, S.S.; Muchtar, E.; Wang, Y.; Koehler, A.B.; et al. The CLL International Prognostic Index predicts outcomes in monoclonal B-cell lymphocytosis and Rai 0 CLL. Blood 2021, 138, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Evans, J.M.; Bhagwate, A.V.; Prodduturi, N.; Sarangi, V.; Middha, M.; Sicotte, H.; Vedell, P.T.; Hart, S.N.; Oliver, G.R.; et al. PatternCNV: A versatile tool for detecting copy number changes from exome sequencing data. Bioinformatics 2014, 30, 2678–2680. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Dicker, F.; Schnittger, S.; Kern, W.; Haferlach, T. Comprehensive genetic characterization of CLL: A study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 2007, 21, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Herling, C.D.; Klaumunzer, M.; Rocha, C.K.; Altmuller, J.; Thiele, H.; Bahlo, J.; Kluth, S.; Crispatzu, G.; Herling, M.; Schiller, J.; et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood 2016, 128, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Rigolin, G.M.; Cavallari, M.; Quaglia, F.M.; Formigaro, L.; Lista, E.; Urso, A.; Guardalben, E.; Liberatore, C.; Faraci, D.; Saccenti, E.; et al. In CLL, comorbidities and the complex karyotype are associated with an inferior outcome independently of CLL-IPI. Blood 2017, 129, 3495–3498. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Espinet, B.; Mellinek, C.; Jarosova, M.; Athanasiadou, A.; Ghia, P.; Kater, A.P.; Oscier, D.; Haferlach, C.; Stamatopoulos, K. Cytogenetics in Chronic Lymphocytic Leukemia: ERIC Perspectives and Recommendations. Hemasphere 2022, 6, e707. [Google Scholar] [CrossRef] [PubMed]
- Leeksma, A.C.; Baliakas, P.; Moysiadis, T.; Puiggros, A.; Plevova, K.; Van der Kevie-Kersemaekers, A.M.; Posthuma, H.; Rodriguez-Vicente, A.E.; Tran, A.N.; Barbany, G.; et al. Genomic arrays identify high-risk chronic lymphocytic leukemia with genomic complexity: A multi-center study. Haematologica 2021, 106, 87–97. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stilgenbauer, S.; Bullinger, L.; Lichter, P.; Döhner, H. Genetics of chronic lymphocytic leukemia: Genomic aberrations and V(H) gene mutation status in pathogenesis and clinical course. Leukemia 2002, 16, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Montserrat, E. New prognostic markers in CLL. Hematol. Am. Soc. Hematol. Educ. Program 2006, 2006, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.; Wiedmeier-Nutor, J.E.; Parikh, S.A.; McCabe, C.E.; O’Brien, D.R.; Boddicker, N.J.; Kleinstern, G.; Rabe, K.G.; Bruins, L.; Brown, S.; et al. Differential prognosis of single and multiple TP53 abnormalities in high-count MBL and untreated CLL. Blood Adv. 2023, 7, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wu, C.J. SF3B1 mutations in chronic lymphocytic leukemia. Blood 2013, 121, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laruenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Tausch, E.; Stilgenbauer, S. BIRC3 mutations in chronic lymphocytic leukemia—Uncommon and unfavorable. Haematologica 2020, 105, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, S.J.; Clifford, R.; Parker, H.; Antoniou, P.; Stec-Dziedzic, E.; Larrayoz, M.; Davis, Z.; Kadalyayil, L.; Colin, A.; Robbe, P.; et al. Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: Data from the randomised UK LRF CLL4 trial. Leukemia 2020, 34, 1760–1774. [Google Scholar] [CrossRef]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, J.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Smardova, J.; Rocnova, L.; Tichy, B.; Kuglik, P.; Vranova, V.; Cejkova, S.; Svitakova, M.; Skuhrova Francova, H.; Brychtova, Y.; et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: Selection, impact on survival, and response to DNA damage. Blood 2009, 114, 5307–5314. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Cerri, M.; Deambrogi, C.; Sozzi, E.; Cresta, S.; Rasi, S.; De Paoli, L.; Spina, V.; Gattei, V.; Capello, D.; et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: Implications for overall survival and chemorefractoriness. Clin. Cancer Res. 2009, 15, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Rigolin, G.M.; Del Giudice, I.; Bardi, A.; Melandri, A.; García-Jacobo, R.E.; Cura, F.; Raponi, S.; Ilari, C.; Cafforio, L.; Piciocchi, A.; et al. Complex karyotype in unfit patients with CLL treated with ibrutinib and rituximab: The GIMEMA LLC1114 phase 2 study. Blood 2021, 138, 2727–2730. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLL/SLL (N = 379) | MBL (N = 130) | Total (N = 509) | |
|---|---|---|---|
| Age at Diagnosis | |||
| Missing | 0 | 0 | 0 |
| Mean (SD) | 61.8 (10.4) | 64.2 (9.9) | 62.4 (10.3) |
| Median | 61.4 | 64.0 | 62.0 |
| Q1, Q3 | 55.0, 69.0 | 58.0, 72.0 | 55.0, 70.0 |
| Range | (31.0–89.0) | (42.0–86.0) | (31.0–89.0) |
| Gender | |||
| F | 105 (27.7%) | 46 (35.4%) | 151 (29.7%) |
| M | 274 (72.3%) | 84 (64.6%) | 358 (70.3%) |
| CLL-IPI | |||
| Missing | 27 | 17 | 44 |
| Low risk (0–1) | 109 (31.0%) | 65 (57.5%) | 174 (37.4%) |
| Intermediate (2–3) | 127 (36.1%) | 31 (27.4%) | 158 (34.0%) |
| High (4–6) | 81 (23.0%) | 15 (13.3%) | 96 (20.6%) |
| Very high (7–10) | 35 (9.9%) | 2 (1.8%) | 37 (8.0%) |
| Rai Category | |||
| Missing | 1 | 0 | 1 |
| Rai 0: Low risk | 287 (75.9%) | 130 (100.0%) | 417 (82.1%) |
| Rai 1 or 2: Intermediate risk | 59 (15.6%) | 0 (0.0%) | 59 (11.6%) |
| Rai 3 or 4: High risk | 32 (8.5%) | 0 (0.0%) | 32 (6.3%) |
| B2M | |||
| Missing | 12 | 3 | 15 |
| Mean (SD) | 3.2 (1.9) | 2.4 (1.2) | 3.0 (1.8) |
| Median | 2.6 | 2.0 | 2.4 |
| Q1, Q3 | 2.0, 3.5 | 1.7, 2.6 | 1.9, 3.3 |
| Range | (1.1–16.2) | (1.2–9.9) | (1.1–16.2) |
| IGHV | |||
| Missing | 11 | 12 | 23 |
| Mutated | 158 (42.9%) | 84 (71.2%) | 242 (49.8%) |
| Unmutated | 210 (57.1%) | 34 (28.8%) | 244 (50.2%) |
| FISH del(17p) | |||
| Normal | 350 (92.3%) | 127 (97.7%) | 477 (93.7%) |
| Abnormal | 29 (7.7%) | 3 (2.3%) | 32 (6.3%) |
| FISH del(11q) | |||
| Normal | 328 (86.5%) | 125 (96.2%) | 453 (89.0%) |
| Abnormal | 51 (13.5%) | 5 (3.8%) | 56 (11.0%) |
| FISH del(13q) | |||
| Normal | 151 (39.8%) | 67 (51.5%) | 218 (42.8%) |
| Abnormal | 228 (60.2%) | 63 (48.5%) | 291 (57.2%) |
| FISH Trisomy 12 | |||
| Normal | 312 (82.3%) | 103 (79.2%) | 415 (81.5%) |
| Abnormal | 67 (17.7%) | 27 (20.8%) | 94 (18.5%) |
| Del(17p) | Sample # | Microarray Result | Notes |
|---|---|---|---|
| NGS+/FISH− | 2238 | absent | Tetraploid |
| 3046 | absent | ||
| 3283 | present | subclonaldel(17p) | |
| Del(11q) | |||
| NGS−/FISH+ | 1856 | absent | |
| Trisomy 12 | |||
| NGS+/FISH− | WC2760 | present | |
| Del(13q) | |||
| NGS−/FISH+ | 1370 | absent | |
| 1446 | present | Del(13q) is centromeric of miRNAs | |
| 3091 | absent | ||
| 3670 | present | small del(13q)—865 kb | |
| 3968 | present | small del(13q)—718 kb | |
| WC2760 | absent | ||
| NGS+/FISH− | 2433 | present | |
| 2513 | present | ||
| 3046 | absent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiedmeier-Nutor, J.E.; McCabe, C.E.; O’Brien, D.R.; Jessen, E.; Bonolo de Campos, C.; Boddicker, N.J.; Griffin, R.; Allmer, C.; Rabe, K.G.; Cerhan, J.R.; et al. Utility of Targeted Sequencing Compared to FISH for Detection of Chronic Lymphocytic Leukemia Copy Number Alterations. Cancers 2024, 16, 2450. https://doi.org/10.3390/cancers16132450
Wiedmeier-Nutor JE, McCabe CE, O’Brien DR, Jessen E, Bonolo de Campos C, Boddicker NJ, Griffin R, Allmer C, Rabe KG, Cerhan JR, et al. Utility of Targeted Sequencing Compared to FISH for Detection of Chronic Lymphocytic Leukemia Copy Number Alterations. Cancers. 2024; 16(13):2450. https://doi.org/10.3390/cancers16132450
Chicago/Turabian StyleWiedmeier-Nutor, J. Erin, Chantal E. McCabe, Daniel R. O’Brien, Erik Jessen, Cecilia Bonolo de Campos, Nicholas J. Boddicker, Rosalie Griffin, Cristine Allmer, Kari G. Rabe, James R. Cerhan, and et al. 2024. "Utility of Targeted Sequencing Compared to FISH for Detection of Chronic Lymphocytic Leukemia Copy Number Alterations" Cancers 16, no. 13: 2450. https://doi.org/10.3390/cancers16132450
APA StyleWiedmeier-Nutor, J. E., McCabe, C. E., O’Brien, D. R., Jessen, E., Bonolo de Campos, C., Boddicker, N. J., Griffin, R., Allmer, C., Rabe, K. G., Cerhan, J. R., Parikh, S. A., Kay, N. E., Yan, H., Van Dyke, D. L., Slager, S. L., & Braggio, E. (2024). Utility of Targeted Sequencing Compared to FISH for Detection of Chronic Lymphocytic Leukemia Copy Number Alterations. Cancers, 16(13), 2450. https://doi.org/10.3390/cancers16132450