
Fibroblasts Promote Resistance to KRAS Silencing in Colorectal Cancer Cells
 , , , , and
, , , , and
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Production of Conditioned Media for Fibroblasts
2.3. Cell Culture with Conditioned Media
2.4. siRNA Transfection
2.5. Protein Extraction and Western Blotting
2.6. Flow Cytometry
2.7. Sphere-Forming Assay
2.8. RNA Extraction
2.9. Library Preparation and Transcriptome Sequencing
2.10. Database for Annotation, Visualization, and Integrated Discovery (DAVID)
2.11. Gene Set Enrichment Analysis (GSEA)
2.12. Quantification of G0 Arrest
2.13. Statistical Analysis
3. Results
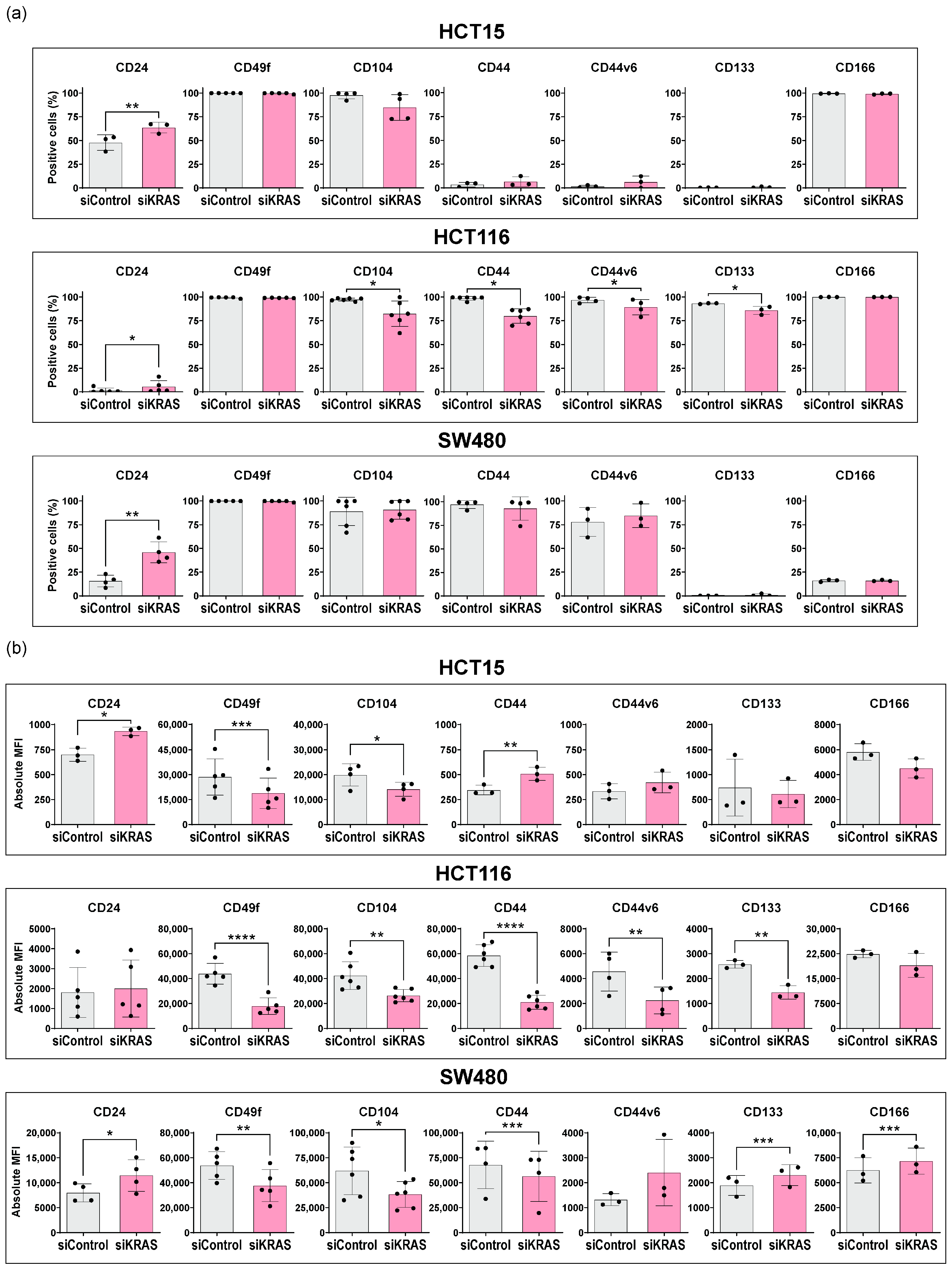
3.1. Colorectal Cancer Cells Express Variable Basal Levels of Membrane Cancer Stem Cell Markers
3.2. KRAS Silencing Up-Regulates CD24 and Down-Regulates CD49f and CD104 Stemness Markers across Cell Lines
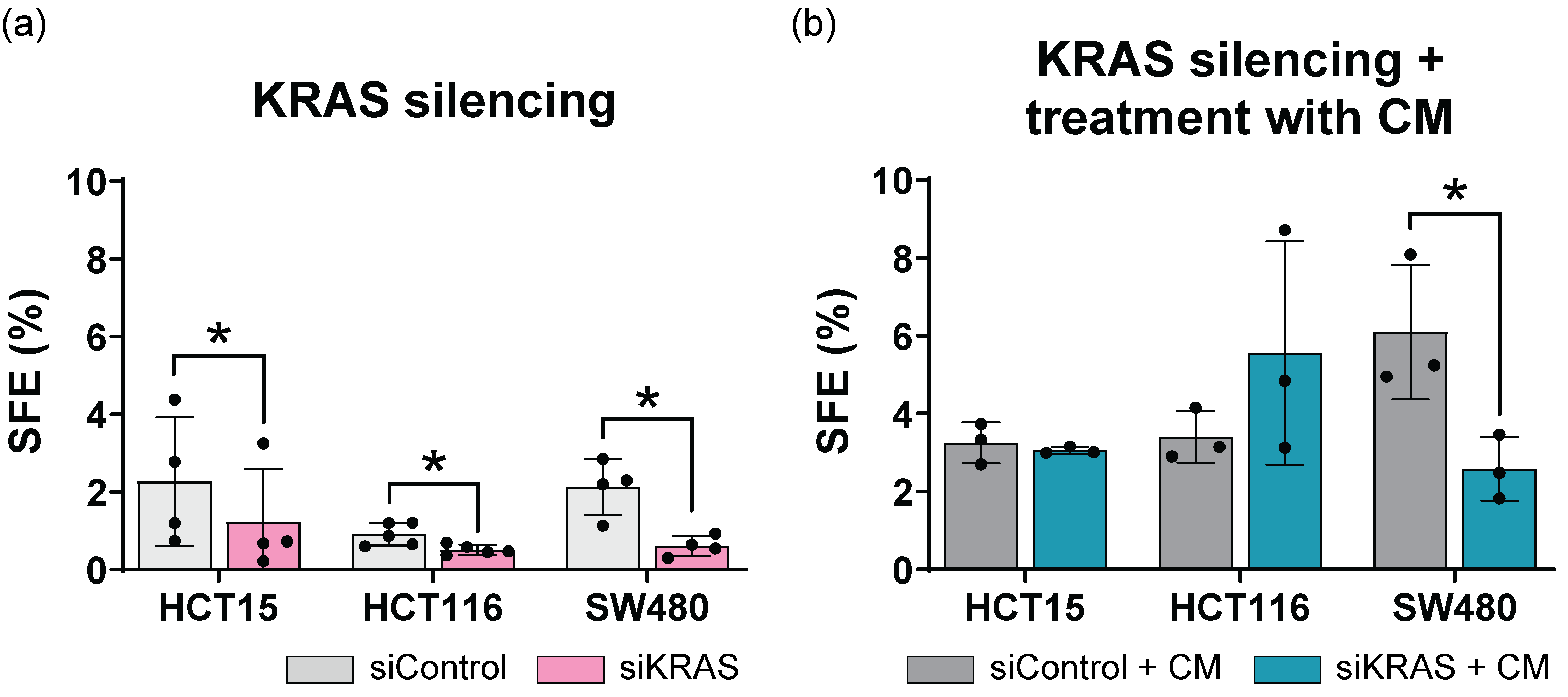
3.3. Fibroblast-Secreted Factors Attenuate the Capacity of KRAS Silencing to Regulate the Expression of Cancer Stem Cell Markers
3.4. Fibroblast-Secreted Factors Attenuate the Inhibitory Effect Promoted by KRAS Silencing in the Sphere Formation Assay
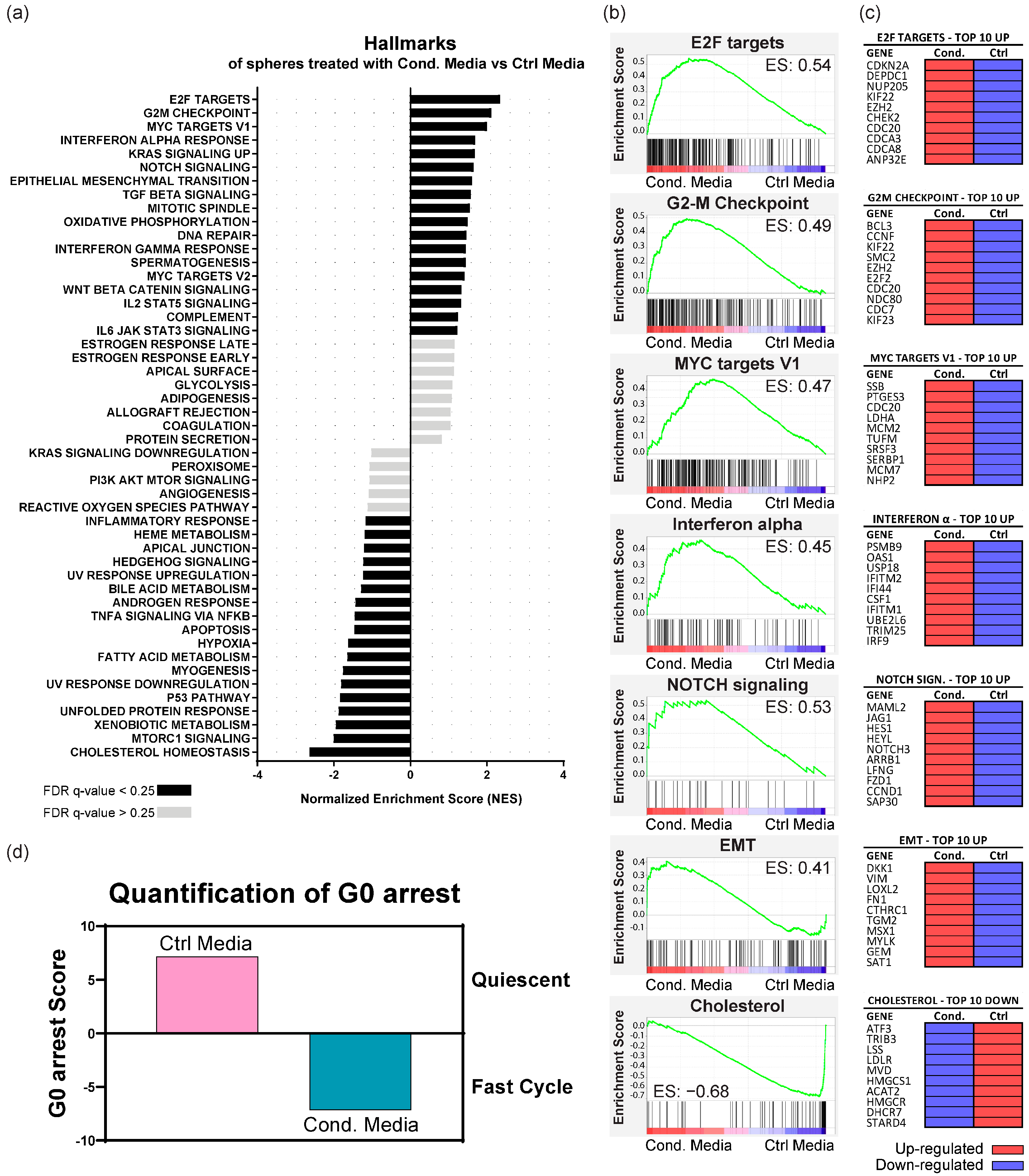
3.5. Fibroblast-Secreted Factors Up-Regulate Proliferation, Pro-Tumorigenic, EMT, and Immune System Regulation Pathways in KRAS-Silenced Cells
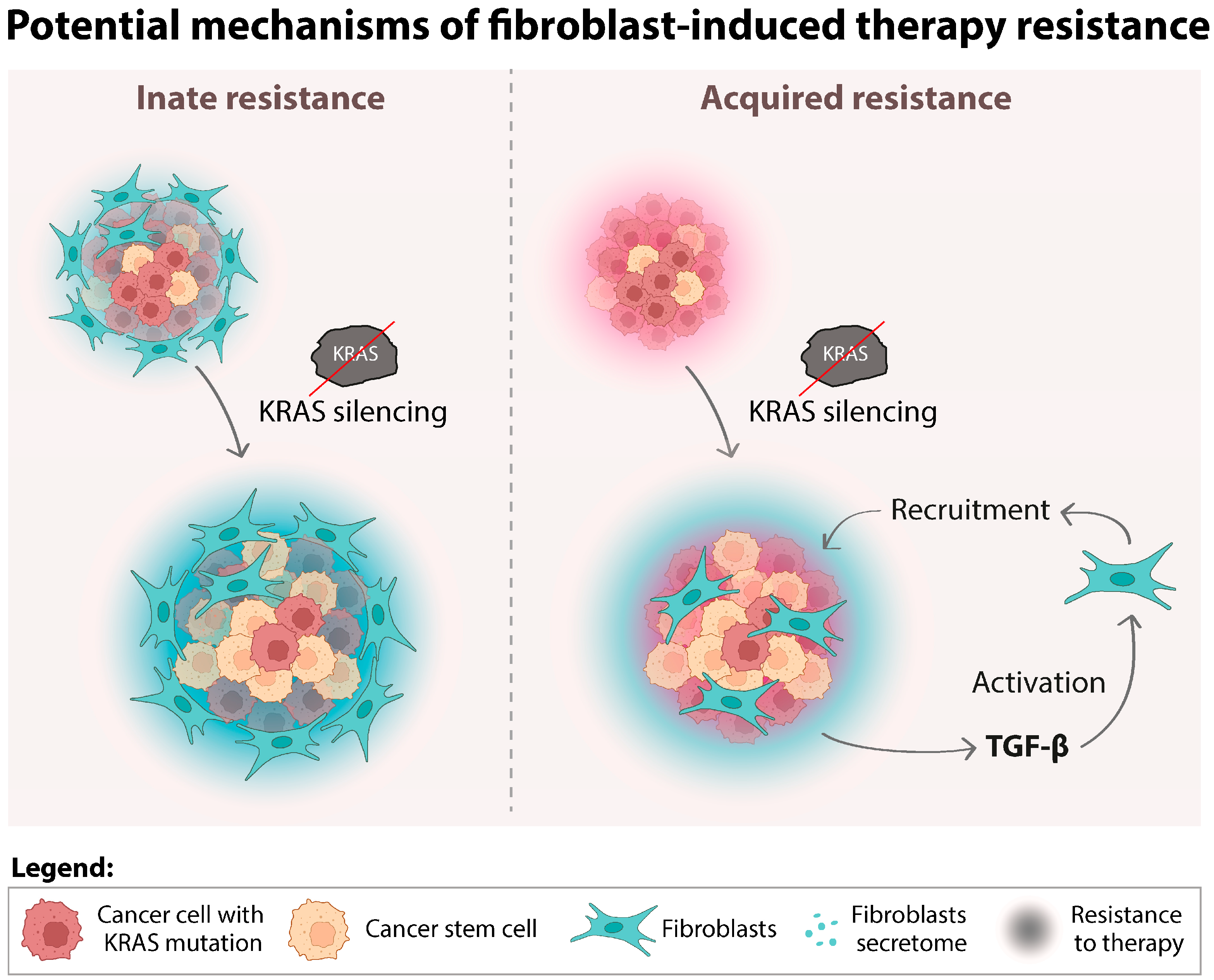
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The Improbable Targeted Therapy: KRAS as an Emerging Target in Non-Small Cell Lung Cancer (NSCLC). Cell Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted Therapies: Is the Undruggable Drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. Mechanisms of Resistance to Krasg12c-Targeted Therapy. Cancer Discov. 2021, 11, 1345–1352. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C Inhibitor: Activity and Resistance. Cancer Gene Ther. 2022, 29, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal Transition Is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to Krasg12c Inhibition through a Novel Kras Switch-Ii Pocket Mutation and Polyclonal Alterations Converging on Ras–Mapk Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of Pancreatic Cancer Cells Lacking KRAS Function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef]
- Scholl, C.; Fröhling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic Lethal Interaction between Oncogenic KRAS Dependency and STK33 Suppression in Human Cancer Cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. TAK1 Inhibition Promotes Apoptosis in KRAS-Dependent Colon Cancers. Cell 2012, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.-H.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor Microenvironment and Therapeutic Response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting Signalling Pathways and the Immune Microenvironment of Cancer Stem Cells—A Clinical Update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Chippalkatti, R.; Abankwa, D. Promotion of Cancer Cell Stemness by Ras. Biochem. Soc. Trans. 2021, 49, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The Cancer Stem Cell: Premises, Promises and Challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Ohta, K.; Uemura, M.; Nishimura, J.; Hata, T.; Takemasa, I.; Mizushima, T.; Yamamoto, H.; et al. CD49f-Positive Cell Population Efficiently Enriches Colon Cancer-Initiating Cells. Int. J. Oncol. 2013, 43, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic Characterization of Human Colorectal Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenço, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem Cell Functionality Is Microenvironmentally Defined during Tumour Expansion and Therapy Response in Colon Cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling Pathways in Cancer-Associated Fibroblasts and Targeted Therapy for Cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-Associated Fibroblasts Induce Epithelial-Mesenchymal Transition of Breast Cancer Cells through Paracrine TGF-β Signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef]
- Valcz, G.; Buzás, E.I.; Szállási, Z.; Kalmár, A.; Krenács, T.; Tulassay, Z.; Igaz, P.; Molnár, B. Perspective: Bidirectional Exosomal Transport between Cancer Stem Cells and Their Fibroblast-Rich Microenvironment during Metastasis Formation. NPJ Breast Cancer 2018, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.; Marín-Jiménez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and Functions of CAFs Secretome During Cancer Progression and Therapy. Front. Cell Dev. Biol. 2021, 8, 621070. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Martins, F.; Carvalho, J.; Oliveira, M.J.; Velho, S. Mutant KRAS-Associated Proteome Is Mainly Controlled by Exogenous Factors. Cells 2022, 11, 1988. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and Genetic Features of 24 Colon Cancer Cell Lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Kurobe, M.; Furukawi, S.; Hayashi, K. Synthesis and Secretion of an Epidermal Growth Factor (EGF) by Human Fibroblast Cells in Culture. Biochem. Biophys. Res. Commun. 1985, 131, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.J.; Mcleskey, S.W.; Wellstein, A. Fibroblast Growth Factors, Their Receptors and Signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef]
- Pratsinis, H.; Kletsas, D. Growth Factors in Fetal and Adult Wound Healing. In Wound Healing Biomaterials; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 1, pp. 41–68. ISBN 9780081006054. [Google Scholar]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Wiecek, A.J.; Cutty, S.J.; Kornai, D.; Parreno-Centeno, M.; Gourmet, L.E.; Tagliazucchi, G.M.; Jacobson, D.H.; Zhang, P.; Xiong, L.; Bond, G.L.; et al. Genomic Hallmarks and Therapeutic Implications of G0 Cell Cycle Arrest in Cancer. Genome Biol. 2023, 24, 128. [Google Scholar] [CrossRef]
- Krebsbach, P.H.; Villa-Diaz, L.G. The Role of Integrin A6 (CD49f) in Stem Cells: More than a Conserved Biomarker. Stem Cells Dev. 2017, 26, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms That Link the Oncogenic Epithelial- Mesenchymal Transition to Suppression of Anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Mendonça, S.; Martins, F.; Oliveira, M.J.; Velho, S. Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS. Cells 2022, 11, 2490. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.K.; Lee, Y.M.; Seong, I.O.; Choi, S.Y.; Suh, K.S.; Song, K.S.; Lee, C.S.; Kim, J.M.; Kim, K.H. Up-Regulation of Cytoplasmic CD24 Expression Is Associated with Malignant Transformation but Favorable Prognosis of Colorectal Adenocarcinoma. Anticancer. Res. 2016, 36, 6593–6598. [Google Scholar] [CrossRef] [PubMed]
- Paschall, A.V.; Yang, D.; Lu, C.; Redd, P.S.; Choi, J.-H.; Heaton, C.M.; Lee, J.R.; Nayak-Kapoor, A.; Liu, K. CD133 + CD24 Lo Defines a 5-Fluorouracil-Resistant Colon Cancer Stem Cell-like Phenotype. Oncotarget 2016, 7, 78698–78712. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Lee, H.W.; Hur, K.Y.; Kim, J.J.; Park, G.S.; Jang, S.H.; Song, Y.S.; Jang, K.S.; Paik, S.S. Cancer Stem Cell Markers CD133 and CD24 Correlate with Invasiveness and Differentiation in Colorectal Adenocarinoma. World J. Gastroenterol. 2009, 15, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.H.; Al-Attar, A.; Kim, J.; Watson, N.F.S.; Scholefield, J.H.; Durrant, L.G.; Ilyas, M. CD24 Shows Early Upregulation and Nuclear Expression but Is Not a Prognostic Marker in Colorectal Cancer. J. Clin. Pathol. 2009, 62, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, J.K.; Chen, C.T.; Wu, C.; Marco, M.R.; Barriga, F.M.; O’Rourke, K.; Pelossof, R.; Qu, X.; Chang, Q.; et al. KRAS Mutants Upregulate Integrin Β4 to Promote Invasion and Metastasis in Colorectal Cancer. Mol. Cancer Res. 2022, 20, 1305–1319. [Google Scholar] [CrossRef]
- Beaulieu, J.F. Integrin A6β4 in Colorectal Cancer: Expression, Regulation, Functional Alterations and Use as a Biomarker. Cancers 2020, 12, 41. [Google Scholar] [CrossRef]
- Tammaccaro, S.L.; Prigent, P.; Le Bail, J.C.; Dos-Santos, O.; Dassencourt, L.; Eskandar, M.; Buzy, A.; Venier, O.; Guillemot, J.C.; Veeranagouda, Y.; et al. TEAD Inhibitors Sensitize KRASG12C Inhibitors via Dual Cell Cycle Arrest in KRASG12C-Mutant NSCLC. Pharmaceuticals 2023, 16, 553. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargi, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-Associated Fibroblasts as Abettors of Tumor Progression at the Crossroads of EMT and Therapy Resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 Secreted by Cancer-Associated Fibroblasts Induces Epithelial-Mesenchymal Transition of Bladder Cancer Cells through LncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Kumar Jolly, M.; Rai, K.; et al. Identification of EMT Signaling Cross-Talk and Gene Regulatory Networks by Single-Cell RNA Sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, N.; Afshinpour, M.; Fakhr, S.S.; Kalkhoran, P.G.; Shadman-Manesh, V.; Adelian, S.; Beiranvand, S.; Rezaei-Tazangi, F.; Khorram, R.; Hamblin, M.R.; et al. Cancer Stem Cells in Colorectal Cancer: Signaling Pathways Involved in Stemness and Therapy Resistance. Crit. Rev. Oncol. Hematol. 2023, 182, 103920. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Nicolas, A.; Petrocelli, V.; Pesic, M.; Mahmoud, A.; Michels, B.E.; Etlioglu, E.; Yepes, D.; Haüpl, B.; Ziegler, P.K.; et al. AKT-Dependent NOTCH3 Activation Drives Tumor Progression in a Model of Mesenchymal Colorectal Cancer. J. Exp. Med. 2020, 217, e20191515. [Google Scholar] [CrossRef]
- Xiu, M.; Wang, Y.; Li, B.; Wang, X.; Xiao, F.; Chen, S.; Zhang, L.; Zhou, B.; Hua, F. The Role of Notch3 Signaling in Cancer Stemness and Chemoresistance: Molecular Mechanisms and Targeting Strategies. Front. Mol. Biosci. 2021, 8, 694141. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ Controls Smad Nucleocytoplasmic Shuttling and Regulates Human Embryonic Stem-Cell Self-Renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Huang, H.Y.; Lin, Z.; Ranieri, M.; Li, S.; Sahu, S.; Liu, Y.; Ban, Y.; Guidry, K.; Hu, H.; et al. Genome-Wide CRISPR Screens Identify Multiple Synthetic Lethal Targets That Enhance KRASG12C Inhibitor Efficacy. Cancer Res. 2023, 83, 4095–4111. [Google Scholar] [CrossRef] [PubMed]
- Karta, J.; Bossicard, Y.; Kotzamanis, K.; Dolznig, H.; Letellier, E. Mapping the Metabolic Networks of Tumor Cells and Cancer-associated Fibroblasts. Cells 2021, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras Decouples Glucose and Glutamine Metabolism to Support Cancer Cell Growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | KRAS Mutation | Disease | Derived from | Reference |
|---|---|---|---|---|
| HCT116 | G13D | Colorectal Carcinoma | Primary tumor | [34] |
| HCT15 | G13D | Colorectal Adenocarcinoma | DLD-1 misclassified | |
| SW480 | G12V | Colorectal Adenocarcinoma | Primary tumor |
| Ab 1 | MW 2 (kDA) 3 | Blocking | Dilution | Species | Brand | Catalog no. | Storage (°C) | Secondary Dilution |
|---|---|---|---|---|---|---|---|---|
| α-SMA | 42 | 5% milk in 0.5% PBS-T | 1:250 | Mouse | Abcam | ab7817 | −20 | 1:3000 |
| GAPDH | 37 | 5% milk in 0.5% PBS-T | 1:10,000 | Mouse | SantaCruz | sc-47724 | 4 | 1:20,000 |
| KRAS | 21 | 5% milk in 0.5% PBS-T | 1:4000 | Mouse | LSBio | C175665 | −20 | 1:8000 |
| Antibody | Conjugate | Clone | Catalog no. | Brand |
|---|---|---|---|---|
| CD24 | PE | 32D12 | 130-098-861 | Miltenyi Biotec |
| CD49f | APC | GoH3 | 130-100-147 | Miltenyi Biotec |
| CD104 | FITC | REA236 | 130-124-266 | Miltenyi Biotec |
| CD44 | FITC | DB105 | 130-113-896 | Miltenyi Biotec |
| CD44.V6 | APC | REA706 | 130-111-425 | Miltenyi Biotec |
| CD133 | PE | AC133 | 130-098-826 | Miltenyi Biotec |
| CD166 | APC | REA442 | 130-106-619 | Miltenyi Biotec |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, S.M.; Carvalho, P.D.; Serra-Roma, A.; Oliveira, P.; Ribeiro, A.; Carvalho, J.; Martins, F.; Machado, A.L.; Oliveira, M.J.; Velho, S. Fibroblasts Promote Resistance to KRAS Silencing in Colorectal Cancer Cells. Cancers 2024, 16, 2595. https://doi.org/10.3390/cancers16142595
Oliveira SM, Carvalho PD, Serra-Roma A, Oliveira P, Ribeiro A, Carvalho J, Martins F, Machado AL, Oliveira MJ, Velho S. Fibroblasts Promote Resistance to KRAS Silencing in Colorectal Cancer Cells. Cancers. 2024; 16(14):2595. https://doi.org/10.3390/cancers16142595
Chicago/Turabian StyleOliveira, Susana Mendonça, Patrícia Dias Carvalho, André Serra-Roma, Patrícia Oliveira, Andreia Ribeiro, Joana Carvalho, Flávia Martins, Ana Luísa Machado, Maria José Oliveira, and Sérgia Velho. 2024. "Fibroblasts Promote Resistance to KRAS Silencing in Colorectal Cancer Cells" Cancers 16, no. 14: 2595. https://doi.org/10.3390/cancers16142595
APA StyleOliveira, S. M., Carvalho, P. D., Serra-Roma, A., Oliveira, P., Ribeiro, A., Carvalho, J., Martins, F., Machado, A. L., Oliveira, M. J., & Velho, S. (2024). Fibroblasts Promote Resistance to KRAS Silencing in Colorectal Cancer Cells. Cancers, 16(14), 2595. https://doi.org/10.3390/cancers16142595