

The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies

Abstract
Simple Summary
Abstract
1. Introduction
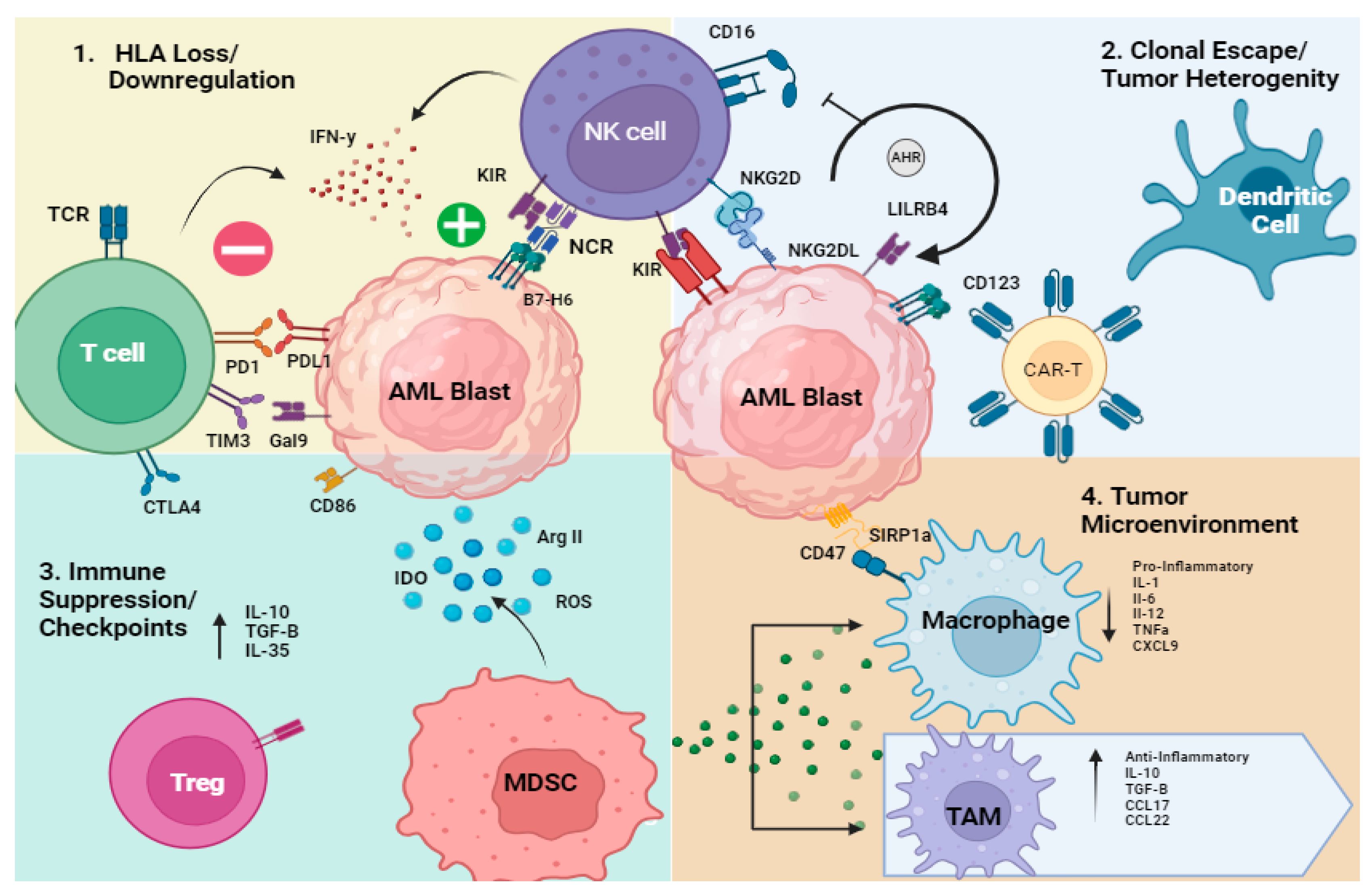
1.1. Overview of Mechanisms of Immune Evasion in AML
- Genetic or transcriptional loss of HLA
- 2.
- Clonal escape/tumor heterogeneity/oncogenesis
- 3.
- Immune Suppression (T and NK cells, MDSCs, dendritic cells)/Checkpoint
1.2. T Cell Dysfunction in AML
1.3. NK Cell Dysfunction in AML
- 4.
- Tumor Microenvironment
2. Current Immunotherapies in AML
2.1. T-Cell-Based Immunotherapy
Chimeric Antigen Receptor T Cells
2.2. Antibody-Based Therapies
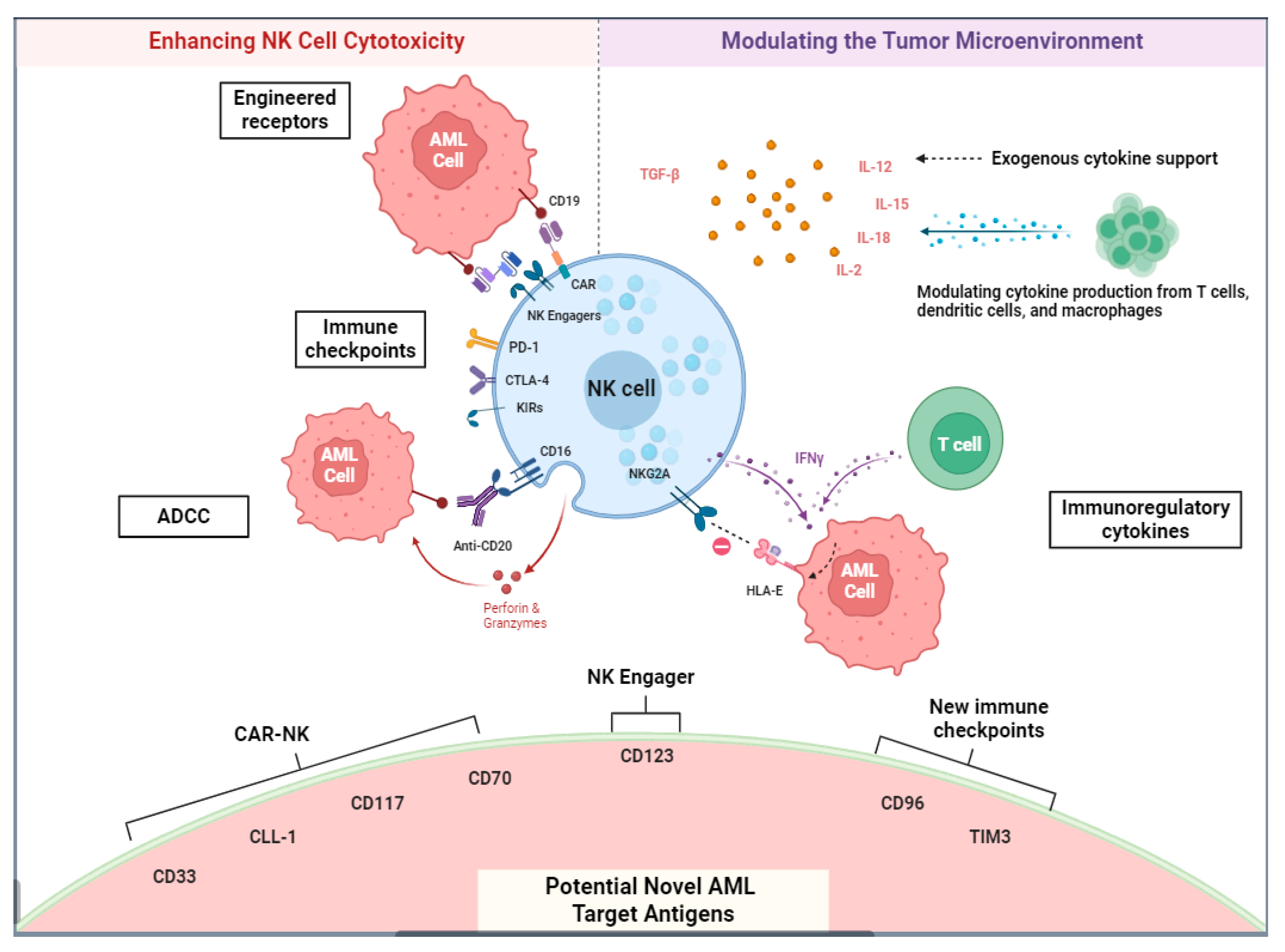
2.3. NK Cell-Based Immunotherapy
NK Chimeric Antigen Receptors and Other Methods of Adoptive NK Cell Transfer
2.4. NK Cell Engagers
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Saultz, J.N.; Tyner, J.W. Chasing leukemia differentiation through induction therapy, relapse and transplantation. Blood Rev. 2023, 57, 101000. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; de Wreede, L.C.; van Biezen, A.; Finke, J.; Ehninger, G.; Ganser, A.; Volin, L.; Niederwieser, D.; Beelen, D.; Alessandrino, P.; et al. Outcome after relapse of myelodysplastic syndrome and secondary acute myeloid leukemia following allogeneic stem cell transplantation: A retrospective registry analysis on 698 patients by the Chronic Malignancies Working Party of the European Society of Blood and Marrow Transplantation. Haematologica 2018, 103, 237–245. [Google Scholar] [PubMed]
- Sharpe, A.H. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol. Rev. 2017, 276, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Longo, D.L.; Wilson, W.H. CAR T-Cell Therapy for Large B-Cell Lymphoma—Who, When, and How? N. Engl. J. Med. 2022, 386, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Acheampong, D.O.; Adokoh, C.K.; Asante, D.B.; Asiamah, E.A.; Barnie, P.A.; Bonsu, D.O.M.; Kyei, F. Immunotherapy for acute myeloid leukemia (AML): A potent alternative therapy. Biomed. Pharmacother. 2018, 97, 225–232. [Google Scholar] [CrossRef]
- Daver, N. The Emerging Profile of Immunotherapy Approaches in the Treatment of AML. Oncology 2019, 33, 28–32. [Google Scholar] [PubMed]
- Haddad, F.; Daver, N. An Update on Immune Based Therapies in Acute Myeloid Leukemia: 2021 and Beyond! Adv. Exp. Med. Biol. 2021, 1342, 273–295. [Google Scholar]
- Killock, D. Immunotherapy: Exploiting natural killers in AML. Nat. Rev. Clin. Oncol. 2016, 13, 654. [Google Scholar] [CrossRef]
- Chen, C.; Liang, C.; Wang, S.; Chio, C.L.; Zhang, Y.; Zeng, C.; Chen, S.; Wang, C.; Li, Y. Expression patterns of immune checkpoints in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Lü, M.; Cao, F.; Wu, G.; Gao, F.; Pang, H.; Li, Y.; Zhang, Y.; Xing, H.; Liang, C.; et al. Single-cell map of diverse immune phenotypes in the acute myeloid leukemia microenvironment. Biomark. Res. 2021, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Brück, O.; Dufva, O.; Hohtari, H.; Blom, S.; Turkki, R.; Ilander, M.; Kovanen, P.; Pallaud, C.; Ramos, P.M.; Lähteenmäki, H.; et al. Immune profiles in acute myeloid leukemia bone marrow associate with patient age, T-cell receptor clonality, and survival. Blood Adv. 2020, 4, 274–286. [Google Scholar] [CrossRef] [PubMed]
- MacNabb, B.W.; Kline, J. MHC cross-dressing in antigen presentation. Adv. Immunol. 2023, 159, 115–147. [Google Scholar] [PubMed]
- Antohe, I.; Tanasa, M.P.; Dascalescu, A.; Danaila, C.; Titieanu, A.; Zlei, M.; Ivanov, I.; Sireteanu, A.; Cianga, P. The MHC-II antigen presentation machinery and B7 checkpoint ligands display distinctive patterns correlated with acute myeloid leukaemias blast cells HLA-DR expression. Immunobiology 2021, 226, 152049. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H–2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Leventhal, M.J.; Morgan, E.A.; Wengrod, J.C.; Nag, A.; Drinan, S.D.; Wollison, B.M.; Ducar, M.D.; Thorner, A.R.; Leppanen, S.; et al. Recurrent genetic HLA loss in AML relapsed after matched unrelated allogeneic hematopoietic cell transplantation. Blood Adv. 2019, 3, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Magenau, J.M.; Peltier, D.; Riwes, M.; Pawarode, A.; Parkin, B.; Braun, T.; Anand, S.; Ghosh, M.; Maciejewski, J.; Yanik, G.; et al. Type 1 interferon to prevent leukemia relapse after allogeneic transplantation. Blood Adv. 2021, 5, 5047–5056. [Google Scholar] [CrossRef]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Austin, R.J.; Straube, J.; Halder, R.; Janardhanan, Y.; Bruedigam, C.; Witkowski, M.; Cooper, L.; Porter, A.; Braun, M.; Souza-Fonseca-Guimaraes, F.; et al. Oncogenic drivers dictate immune control of acute myeloid leukemia. Nat. Commun. 2023, 14, 2155. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef]
- Vago, L.; Perna, S.K.; Zanussi, M.; Mazzi, B.; Barlassina, C.; Stanghellini, M.T.; Perrelli, N.F.; Cosentino, C.; Torri, F.; Angius, A.; et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N. Engl. J. Med. 2009, 361, 478–488. [Google Scholar] [CrossRef]
- Ito, S.; Krakow, E.F.; Ventura, K.; Rodger, A.; Geramita, E.; Moore, E.; Hill, G.R.; Shlomchik, W.D. Pilot Trial of IFN-γ and Donor Lymphocyte Infusion to Treat Relapsed AML and MDS after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2022, 140 (Suppl. S1), 7678–7679. [Google Scholar] [CrossRef]
- Freedman, A.S.; Freeman, G.J.; Rhynhart, K.; Nadler, L.M. Selective induction of B7/BB-1 on interferon-γ stimulated monocytes: A potential mechanism for amplification of T cell activation through the CD28 pathway. Cell. Immunol. 1991, 137, 429–437. [Google Scholar] [CrossRef]
- Wang, B.; Reville, P.K.; Yassouf, M.Y.; Jelloul, F.Z.; Ly, C.; Desai, P.N.; Wang, Z.; Borges, P.; Veletic, I.; Dasdemir, E.; et al. Comprehensive characterization of IFNγ signaling in acute myeloid leukemia reveals prognostic and therapeutic strategies. Nat. Commun. 2024, 15, 1821. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Mooyaart, J.E.; Devillier, R.; Koc, Y.; Vydra, J.; Castagna, L.; Gülbas, Z.; Martin, J.D.; Araujo, M.C.; Kulagin, A.; et al. Donor lymphocyte infusions after haploidentical stem cell transplantation with PTCY: A study on behalf of the EBMT cellular therapy & immunobiology working party. Bone Marrow Transpl. 2023, 58, 54–60. [Google Scholar]
- van Galen, P.; Hovestadt, V.; Wadsworth, M.H., II; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pollyea, D.A.; Konopleva, M.; Hong, K.; Huang, T.; Song, A.; Wieland, E.; Woodard, P.; Liao, C.; Zhang, C.; et al. A First-in-Human (FIH) Phase 1 Study of the Anti-LILRB4 Antibody IO-202 in Relapsed/Refractory (R/R) Myelomonocytic and Monocytic Acute Myeloid Leukemia (AML) and R/R Chronic Myelomonocytic Leukemia (CMML). Blood 2020, 136 (Suppl. S1), 19–20. [Google Scholar] [CrossRef]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Guo, Y.; Taniguchi, S. An emerging role for cellular crosstalk in the cancer stem cell niche. J. Pathol. 2021, 254, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Luciano, M.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. 2018, 43, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Sholy, C.; Wang, C.; Cao, M.; Kang, X. The Role of T Cell Immunotherapy in Acute Myeloid Leukemia. Cells 2021, 10, 3376. [Google Scholar] [CrossRef] [PubMed]
- Sendker, S.; Reinhardt, D.; Niktoreh, N. Redirecting the Immune Microenvironment in Acute Myeloid Leukemia. Cancers 2021, 13, 1423. [Google Scholar] [CrossRef] [PubMed]
- Dama, P.; Tang, M.; Fulton, N.; Kline, J.; Liu, H. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J. Immunother. Cancer 2019, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Qian, Y.; Liang, X.; Wu, J.; Zou, M.; Deng, M. LILRB4, an immune checkpoint on myeloid cells. Blood Sci. 2022, 4, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Radwan, S.; Elleboudy, N.; Nabih, N.; Kamal, A. AML-273: The Immune checkpoints CTLA-4 and LAG-3 expression is up-regulated in acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, S198. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Taghiloo, S.; Asgarian-Omran, H. Immune evasion mechanisms in acute myeloid leukemia: A focus on immune checkpoint pathways. Crit. Rev. Oncol. Hematol. 2021, 157, 103164. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Q.; Shi, J.; Guo, W.; Jiang, X.; Zhou, B.; Ren, C. LILRB4, from the immune system to the disease target. Am. J. Transl. Res. 2020, 12, 3149–3166. [Google Scholar] [PubMed]
- Wang, L.; Jia, B.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncol. Immunol. 2018, 7, e1469594. [Google Scholar] [CrossRef] [PubMed]
- Lamble, A.J.; Kosaka, Y.; Laderas, T.; Maffit, A.; Kaempf, A.; Brady, L.K.; Wang, W.; Long, N.; Saultz, J.N.; Mori, M.; et al. Reversible suppression of T cell function in the bone marrow microenvironment of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2020, 117, 14331–14341. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Workman, C.J.; Vignali, D.A. Targeting regulatory T cells in tumors. FEBS J. 2016, 283, 2731–2748. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Ahmed, R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 2003, 4, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.E.; Crawford, J.C.; Thomas, P.G. Hitting the Target: How T Cells Detect and Eliminate Tumors. J. Immunol. 2018, 200, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhao, Y.; Lai, W.; Tan, J.; Zheng, X.; Zha, X.; Li, Y.; Chen, S. Higher PD-1/Tim-3 expression on IFN-gamma+ T cells is associated with poor prognosis in patients with acute myeloid leukemia. Cancer Biol. Ther. 2023, 24, 2278229. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Asgarian-Omran, H.; Najafi, B.; Najafi, A.; Valadan, R.; Karami, H.; Naderisoraki, M.; Alizadeforoutan, M.; Shekarriz, R.; Tehrani, M. Evaluation of mRNA Expressions of TOX and NR4As in CD8+ T cells in Acute Leukemia. Iran J. Immunol. 2023, 20, 438–445. [Google Scholar]
- Wysota, M.; Konopleva, M.; Mitchell, S. Novel Therapeutic Targets in Acute Myeloid Leukemia (AML). Curr. Oncol. Rep. 2024, 26, 409–420. [Google Scholar] [CrossRef]
- Perzolli, A.; Koedijk, J.B.; Zwaan, C.M.; Heidenreich, O. Targeting the innate immune system in pediatric and adult AML. Leukemia 2024, 38, 1191–1201. [Google Scholar] [CrossRef]
- Xu, Y.; Mou, J.; Wang, Y.; Zhou, W.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Wang, M.; Wang, J. Regulatory T cells promote the stemness of leukemia stem cells through IL10 cytokine-related signaling pathway. Leukemia 2022, 36, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Tang, Z.; Liang, X.; Shi, Z.; Yao, Y.; Huang, X.; Zhu, S.; Wu, M.; Li, J.; Zhao, W.; et al. An immune-related gene prognostic index for acute myeloid leukemia associated with regulatory T cells infiltration. Hematology 2022, 27, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Daver, N. Immune checkpoint inhibitors in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2021, 34, 101247. [Google Scholar] [CrossRef] [PubMed]
- Brauneck, F.; Haag, F.; Woost, R.; Wildner, N.; Tolosa, E.; Rissiek, A.; Vohwinkel, G.; Wellbrock, J.; Bokemeyer, C.; Schulze Zur Wiesch, J.; et al. Increased frequency of TIGIT+CD73−CD8+ T cells with a TOX+ TCF-1low profile in patients with newly diagnosed and relapsed AML. Oncoimmunology 2021, 10, 1930391. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Jimbu, L.; Mesaros, O.; Popescu, C.; Neaga, A.; Berceanu, I.; Dima, D.; Gaman, M.; Zdrenghea, M. Is There a Place for PD-1-PD-L Blockade in Acute Myeloid Leukemia? Pharmaceuticals 2021, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Hendrickson, S.; Pierce, S.; Ning, J.; Konopleva, M.; et al. Phase IB/II Study of Nivolumab in Combination with Azacytidine (AZA) in Patients (pts) with Relapsed Acute Myeloid Leukemia (AML). Blood 2016, 128, 763. [Google Scholar] [CrossRef]
- Héninger, E.; Krueger, T.E.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, K.E.; Thompson, J.; Gui, G.; Valdez, J.; Worthy, T.; Tekleab, H.; Hughes, T.; Goswami, M.; Oetjen, K.; Kim, D.-Y.; et al. Pembrolizumab and Decitabine for Refractory or Relapsed Acute Myeloid Leukemia. Blood 2018, 132 (Suppl. S1), 1437. [Google Scholar] [CrossRef]
- Liu, H.; Sharon, E.; Karrison, T.G.; Zha, Y.; Fulton, N.; Streicher, H.; Sweet, K.; Yaghmour, G.; Liu, J.J.; Jonas, B.A.; et al. Randomized Phase II Study to Assess the Role of Nivolumab As Single Agent to Eliminate Minimal Residual Disease and Maintain Remission in Acute Myelogenous Leukemia (AML) Patients after Chemotherapy (NCI9706 protocol; REMAIN Trial). Blood 2022, 140 (Suppl. S1), 1716–1719. [Google Scholar] [CrossRef]
- Schroeder, T.; Czibere, A.; Platzbecker, U.; Bug, G.; Uharek, L.; Luft, T.; Giagounidis, A.; Zohren, F.; Bruns, I.; Wolschke, C.; et al. Azacitidine and donor lymphocyte infusions as first salvage therapy for relapse of AML or MDS after allogeneic stem cell transplantation. Leukemia 2013, 27, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.-i.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 2015, 17, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Chakraborty, A.K.; Shaw, A.S. Understanding the structure and function of the immunological synapse. Cold Spring Harb. Perspect. Biol. 2010, 2, a002311. [Google Scholar] [CrossRef] [PubMed]
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 2009, 114, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013, 132, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Zhang, S.; Farag, S.S. Natural killer cell activity and killer immunoglobulin-like receptors in hematopoietic stem cell transplantation. Cancer Treat. Res. 2009, 144, 47–69. [Google Scholar] [PubMed]
- Ruggeri, L.; Parisi, S.; Urbani, E.; Curti, A. Alloreactive Natural Killer Cells for the Treatment of Acute Myeloid Leukemia: From Stem Cell Transplantation to Adoptive Immunotherapy. Front. Immunol. 2015, 6, 479. [Google Scholar] [CrossRef]
- Ruggeri, L.; Mancusi, A.; Perruccio, K.; Burchielli, E.; Martelli, M.F.; Velardi, A. Natural killer cell alloreactivity for leukemia therapy. J. Immunother. 2005, 28, 175–182. [Google Scholar] [CrossRef]
- Xu, J.; Niu, T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 167. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef]
- Mehta, R.S.; Randolph, B.; Daher, M.; Rezvani, K. NK cell therapy for hematologic malignancies. Int. J. Hematol. 2018, 107, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar]
- Wu, Z.; Zhang, H.; Wu, M.; Peng, G.; He, Y.; Wan, N.; Zeng, Y. Targeting the NKG2D/NKG2D-L axis in acute myeloid leukemia. Biomed. Pharmacother. 2021, 137, 111299. [Google Scholar] [CrossRef]
- Nguyen, S.; Beziat, V.; Dhedin, N.; Kuentz, M.; Vernant, J.P.; Debre, P.; Vieillard, V. HLA-E upregulation on IFN-γ-activated AML blasts impairs CD94/NKG2A-dependent NK cytolysis after haplo-mismatched hematopoietic SCT. Bone Marrow Transplant. 2008, 43, 693. [Google Scholar] [CrossRef]
- Hecht, M.L.; Rosental, B.; Horlacher, T.; Hershkovitz, O.; De Paz, J.L.; Noti, C.; Schauer, S.; Porgador, A.; Seeberger, P.H. Natural cytotoxicity receptors NKp30, NKp44 and NKp46 bind to different heparan sulfate/heparin sequences. J. Proteome Res. 2009, 8, 712–720. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557. [Google Scholar] [CrossRef]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; et al. CD56(dim)CD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Elliott, J.M.; Keyel, P.A.; Yang, L.; Carrero, J.A.; Yokoyama, W.M. Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 1915–1919. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Parisi, S.; Ruggeri, L.; Dan, E.; Rizzi, S.; Sinigaglia, B.; Ocadlikova, D.; Bontadini, A.; Giudice, V.; Urbani, E.; Ciardelli, S.; et al. Long-Term Outcome After Adoptive Immunotherapy With Natural Killer Cells: Alloreactive NK Cell Dose Still Matters. Front. Immunol. 2021, 12, 804988. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine activation induces human memory-like NK cells. Blood 2012, 120, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Borrego, D.; Moreno-Lafont, M.C.; Vazquez-Sanchez, E.A.; Gutierrez-Hoya, A.; López-Santiago, R.; Montiel-Cervantes, L.A.; Ramírez-Saldaña, M.; Vela-Ojeda, J. Overexpression of CD158 and NKG2A Inhibitory Receptors and Underexpression of NKG2D and NKp46 Activating Receptors on NK Cells in Acute Myeloid Leukemia. Arch. Med. Res. 2016, 47, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Martner, A.; Rydstrom, A.; Riise, R.E.; Aurelius, J.; Brune, M.; Foa, R.; Hellstrand, K.; Thoren, F.B. NK cell expression of natural cytotoxicity receptors may determine relapse risk in older AML patients undergoing immunotherapy for remission maintenance. Oncotarget 2015, 6, 42569–42574. [Google Scholar] [CrossRef]
- Kim, S.; Choi, J. Restoring NK cell functions in AML relapse. Blood 2022, 140, 2765–2766. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.S.; Fauriat, C.; Orlanducci, F.; Galseran, C.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Granjeaud, S.; Hamel-Broza, J.F.; Demerle, C.; et al. Natural Killer Defective Maturation Is Associated with Adverse Clinical Outcome in Patients with Acute Myeloid Leukemia. Front. Immunol. 2017, 8, 573. [Google Scholar] [CrossRef]
- Mundy-Bosse, B.L.; Scoville, S.D.; Chen, L.; McConnell, K.; Mao, H.C.; Ahmed, E.H.; Zorko, N.; Harvey, S.; Cole, J.; Zhang, X.; et al. MicroRNA-29b mediates altered innate immune development in acute leukemia. J. Clin. Investig. 2016, 126, 4404–4416. [Google Scholar] [CrossRef]
- Chretien, A.S.; Devillier, R.; Granjeaud, S.; Cordier, C.; Demerle, C.; Salem, N.; Wlosik, J.; Orlanducci, F.; Gorvel, L.; Fattori, S.; et al. High-dimensional mass cytometry analysis of NK cell alterations in AML identifies a subgroup with adverse clinical outcome. Proc. Natl. Acad. Sci. USA 2021, 118, e2020459118. [Google Scholar] [CrossRef]
- Scoville, S.D.; Nalin, A.P.; Chen, L.; Chen, L.; Zhang, M.; McConnell, K.; Beceiro Casas, S.; Ernst, G.; Traboulsi, A.A.-R.; Hashi, N.; et al. Human AML activates the AHR pathway to impair NK cell development and function. Blood 2018, 132, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Wang, E.C.; Man, S.; Hills, R.K.; Burnett, A.K.; Tonks, A.; Darley, R.L. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia 2011, 25, 792–799. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Q.; Yang, J.; Li, X.; Xian, L.; Li, W.; Lin, T.; Cheng, J.; Lin, Q.; Xu, X.; et al. Increased TIGIT expressing NK cells with dysfunctional phenotype in AML patients correlated with poor prognosis. Cancer Immunol. Immunother. 2022, 71, 277–287. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, A.E.; Warshaw, J.N.; Kyalwazi, B.L.; Matsui, H.; Jepsen, K.; Panopoulos, A.D. An iPSC line derived from a human acute myeloid leukemia cell line (HL-60-iPSC) retains leukemic abnormalities and displays myeloid differentiation defects. Stem Cell Res 2020, 49, 102096. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Yano, H.; Workman, C.J.; Vignali, D.A.A. Interleukin-35: Structure, Function and Its Impact on Immune-Related Diseases. J. Interferon Cytokine Res. 2021, 41, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297.e5. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages in Hematologic Malignancies: New Insights and Targeted Therapies. Cells 2019, 8, 1256. [Google Scholar] [CrossRef]
- Leisch, M.; Greil, R.; Pleyer, L. IDO in MDS/AML disease progression and its role in resistance to azacitidine: A potential new drug target? Br. J. Haematol. 2020, 190, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef]
- Pan, J. Chimeric Antigen Receptor T Cell Therapy for Acute Leukemia. Blood Cell Ther. 2023, 6, 145–150. [Google Scholar] [PubMed]
- Wang, X.; Zhang, Y.; Xue, S. Recent progress in chimeric antigen receptor therapy for acute myeloid leukemia. Ann. Hematol. 2024, 103, 1843–1857. [Google Scholar] [CrossRef] [PubMed]
- Atilla, E.; Benabdellah, K. The Black Hole: CAR T Cell Therapy in AML. Cancers 2023, 15, 2713. [Google Scholar] [CrossRef] [PubMed]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef] [PubMed]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasmfollowing treatmentwith Cd123-specific car T cells: A first-in-Human clinical trial. Blood 2017, 130, 811. [Google Scholar] [CrossRef]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-directed CAR-T cell therapy: A novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, M.; Sun, R.; Lyu, H.; Xiao, X.; Zhang, X.; Li, F.; Xie, D.; Xiong, X.; Wang, J.; et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 88. [Google Scholar] [CrossRef]
- Venditti, A.; Del Poeta, G.; Buccisano, F.; Tamburini, A.; Cox-Froncillo, M.C.; Aronica, G.; Bruno, A.; Del Moro, B.; Epiceno, A.M.; Battaglia, A.; et al. Prognostic relevance of the expression of Tdt and CD7 in 335 cases of acute myeloid leukemia. Leukemia 1998, 12, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.S.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2019, 27, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, B.; Li, S.; Wei, W.; Liu, X.; Cui, X.; Zhang, X.; Liu, N.; Yan, L.; Deng, Y.; et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci. Transl. Med. 2023, 15, eadd1951. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; He, S.; Yu, L. Clinical Benefits and Safety of Gemtuzumab Ozogamicin in Treating Acute Myeloid Leukemia in Various Subgroups: An Updated Systematic Review, Meta-Analysis, and Network Meta-Analysis. Front. Immunol. 2021, 12, 683595. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol 2021, 11, 656218. [Google Scholar] [CrossRef]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Müller-Tidow, C.; et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Pabst, T.; Vey, N.; Adès, L.; Bacher, U.; Bargetzi, M.; Fung, S.; Gaidano, G.; Gandini, D.; Hultberg, A.; Johnson, A.; et al. Results from a phase I/II trial of cusatuzumab combined with azacitidine in patients with newly diagnosed acute myeloid leukemia who are ineligible for intensive chemotherapy. Haematologica 2023, 108, 1793–1802. [Google Scholar] [CrossRef]
- Allen, C.; Zeidan, A.M.; Bewersdorf, J.P. BiTEs, DARTS, BiKEs and TriKEs-Are Antibody Based Therapies Changing the Future Treatment of AML? Life 2021, 11, 465. [Google Scholar] [CrossRef]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Volkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-bispecific T-cell engaging (BiTE(R)) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef]
- Epperly, R.; Gottschalk, S.; Velasquez, M.P. Harnessing T Cells to Target Pediatric Acute Myeloid Leukemia: CARs, BiTEs, and Beyond. Children 2020, 7, 14. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, T.; Ahmadvand, M.; Yaghmaie, M.; Sadeghi, A.; Mousavi, S.A.; Rostami, T.; Ganjalikhani-Hakemi, M. Investigation of KIR/HLA relationship and other clinical variables after T-cell-replete haploidentical bone marrow transplantation in patients with acute myeloid leukemia (AML). BMC Immunol. 2023, 24, 10. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Horwitz, M.E.; Rein, L.A.M. Leveraging Natural Killer Cell Innate Immunity against Hematologic Malignancies: From Stem Cell Transplant to Adoptive Transfer and Beyond. Int. J. Mol. Sci. 2022, 24, 204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, Y.; Yao, H.; Zhan, R.; Chen, S.; Miao, W.; Ma, S.; Xu, X.; Li, Y.; Yu, M.; et al. CAR-NK cells for acute myeloid leukemia immunotherapy: Past, present and future. Am. J. Cancer Res. 2023, 13, 5559–5576. [Google Scholar] [PubMed]
- Garg, S.; Ni, W.; Griffin, J.D.; Sattler, M. Chimeric Antigen Receptor T Cell Therapy in Acute Myeloid Leukemia: Trials and Tribulations. Hematol. Rep. 2023, 15, 608–626. [Google Scholar] [CrossRef]
- Shao, R.; Li, Z.; Xin, H.; Jiang, S.; Zhu, Y.; Liu, J.; Huang, R.; Xu, K.; Shi, X. Biomarkers as targets for CAR-T/NK cell therapy in AML. Biomark. Res. 2023, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Xie, S.; Chen, M.; Li, Y.; Yue, J.; Ma, J.; Shu, X.; He, Y.; Xiao, W.; Tian, Z. Advances in NK cell production. Cell Mol. Immunol. 2022, 19, 460–481. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Santos, S.; Vesga, M.A.; Anguita, J.; Martin-Ruiz, I.; Carrascosa, T.; Juan, M.; Eguizabal, C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci. Rep. 2019, 9, 18729. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.H.; Zhu, H.; Wang, Y.M.; Heragu, N.; Bernareggi, D.; Ruiz-Cisneros, A.; Bahena, A.; Ask, E.H.; Hoel, H.J.; Malmberg, K.J.; et al. Umbilical Cord Blood and iPSC-Derived Natural Killer Cells Demonstrate Key Differences in Cytotoxic Activity and KIR Profiles. Front. Immunol. 2020, 11, 561553. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. High expression of NKG2A/CD94 and low expression of granzyme B are associated with reduced cord blood NK cell activity. Cell Mol. Immunol. 2007, 4, 377–382. [Google Scholar] [PubMed]
- Lin, X.; Sun, Y.; Dong, X.; Liu, Z.; Sugimura, R.; Xie, G. IPSC-derived CAR-NK cells for cancer immunotherapy. Biomed. Pharmacother. 2023, 165, 115123. [Google Scholar] [CrossRef] [PubMed]
- Chiu, E.; Felices, M.; Cichocki, F.; Davis, Z.; Wang, H.; Tuninga, K.; Vallera, D.A.; Lee, T.; Bjordahl, R.; Malmberg, K.J.; et al. Anti-NKG2C/IL-15/anti-CD33 killer engager directs primary and iPSC-derived NKG2C+ NK cells to target myeloid leukemia. Mol. Ther. 2021, 29, 3410–3421. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Yilmaz, M.; Ravandi, F. The potential role of Bi-specific antibodies in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2020, 33, 101218. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [PubMed]
- Warlick, E.D.; Weisdorf, D.J.; Vallera, D.A.; Wangen, R.; Lewis, D.; Knox, J.; Schroeder, M.; Felices, M.; Miller, J.S. GTB-3550 TriKE™ for the Treatment of High-Risk Myelodysplastic Syndromes (MDS) and Refractory/Relapsed Acute Myeloid Leukemia (AML) Safely Drives Natural Killer (NK) Cell Proliferation At Initial Dose Cohorts. Blood 2020, 136 (Suppl. S1), 7–8. [Google Scholar] [CrossRef]
- Stein, A.S.; Jongen-Lavrencic, M.; Garciaz, S.; Huls, G.A.; Maiti, A.; Boissel, N.; Botton, S.D.; Fleming, S.; Zwaan, C.M.; Leeuw, D.C.d.; et al. A first-in-human study of CD123 NK cell engager SAR443579 in relapsed or refractory acute myeloid leukemia, B-cell acute lymphoblastic leukemia, or high-risk myelodysplasia. J. Clin. Oncol. 2023, 41 (Suppl. S16), 7005. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| CART Target Antigen | Trial Name | Number |
|---|---|---|
| CD38 | CART-38 in Adult AML and MM Patients | NCT0544280 |
| CLL1 | Anti-CLL1 CART-cell Therapy in CLL1 Positive Relapsed/Refractory AML | NCT04884984 NCT04923919 |
| CD19 | CART-19 T Cell in CD19 Positive in R/R AML | NCT03896854 |
| CD33 | CD33KO-HSPC-Infusion Followed by CART-33 Infusion(s) for R/R AML | NCT05945849 |
| FLT3 | Anti-FLT3 CAR T-cell Therapy in FLT3 Positive R/R AML | NCT05023707 |
| CD7 | Dose-Escalation and Dose-Expansion Study to Evaluate the Safety and Tolerability of Anti-CD7 Allogeneic CAR T-Cell (WU-CART-007) in CD7+ Hematological Malignancies | NCT05377827 |
| CD33, CD38 CD56, CD117, CD123, CD34, and Muc1 | CAR-T Cells Combined with Peptide Specific WT1 Dendritic Cell in Relapsed/Refractory Leukemia/MDS | NCT03291444 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, D.J.; Alber, B.; Saultz, J.N. The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies. Cancers 2024, 16, 2615. https://doi.org/10.3390/cancers16152615
Chandra DJ, Alber B, Saultz JN. The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies. Cancers. 2024; 16(15):2615. https://doi.org/10.3390/cancers16152615
Chicago/Turabian StyleChandra, Daniel J., Bernhard Alber, and Jennifer N. Saultz. 2024. "The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies" Cancers 16, no. 15: 2615. https://doi.org/10.3390/cancers16152615
APA StyleChandra, D. J., Alber, B., & Saultz, J. N. (2024). The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies. Cancers, 16(15), 2615. https://doi.org/10.3390/cancers16152615