
Insulin Resistance in Women Correlates with Chromatin Histone Lysine Acetylation, Inflammatory Signaling, and Accelerated Aging
 , , , , , , , , , add
Show full author list
, , , , , , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Human PBMC Isolation
2.3. ChIP-Rx
2.4. ChIP-Seq Analyses
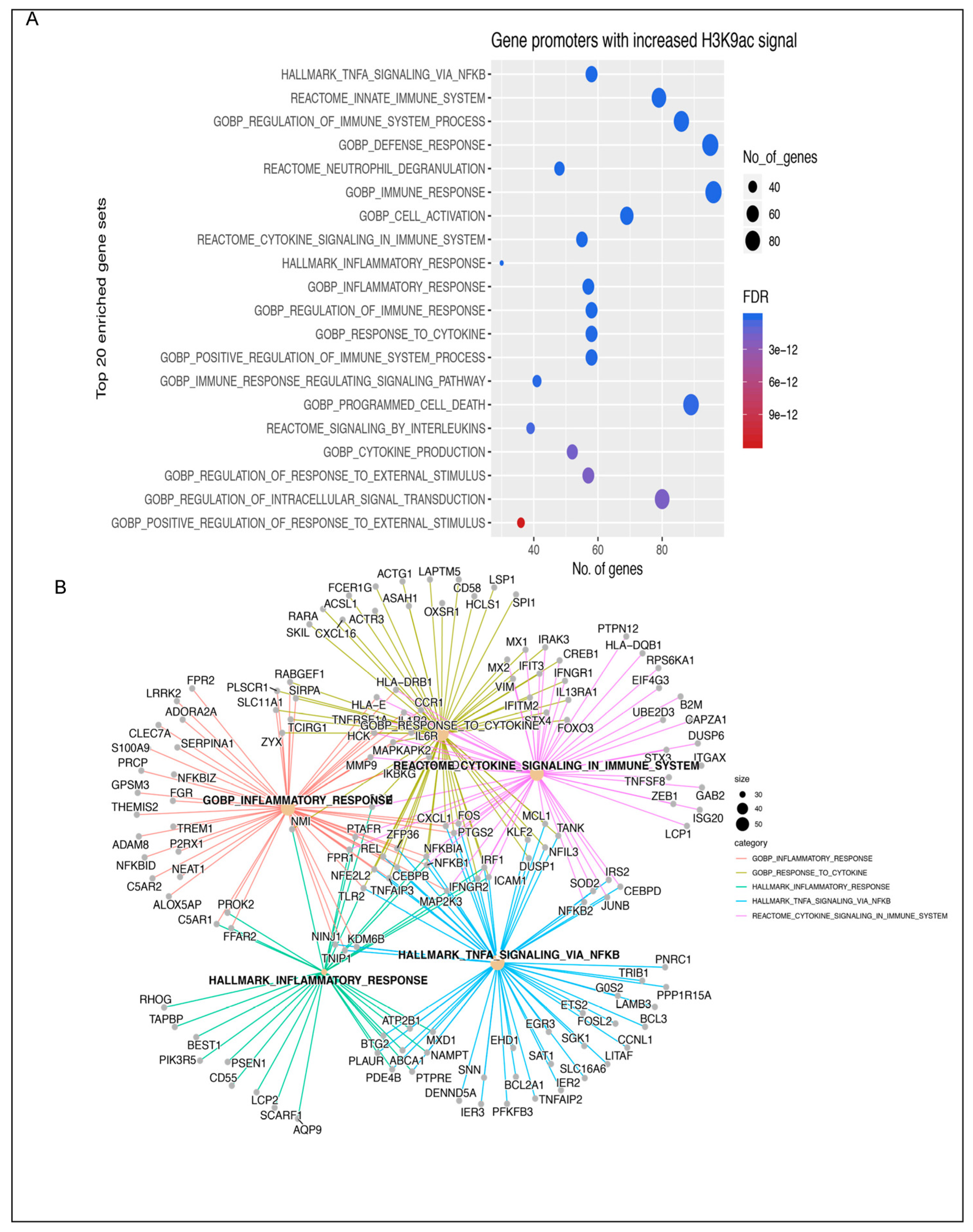
2.5. Gene Set Enrichment
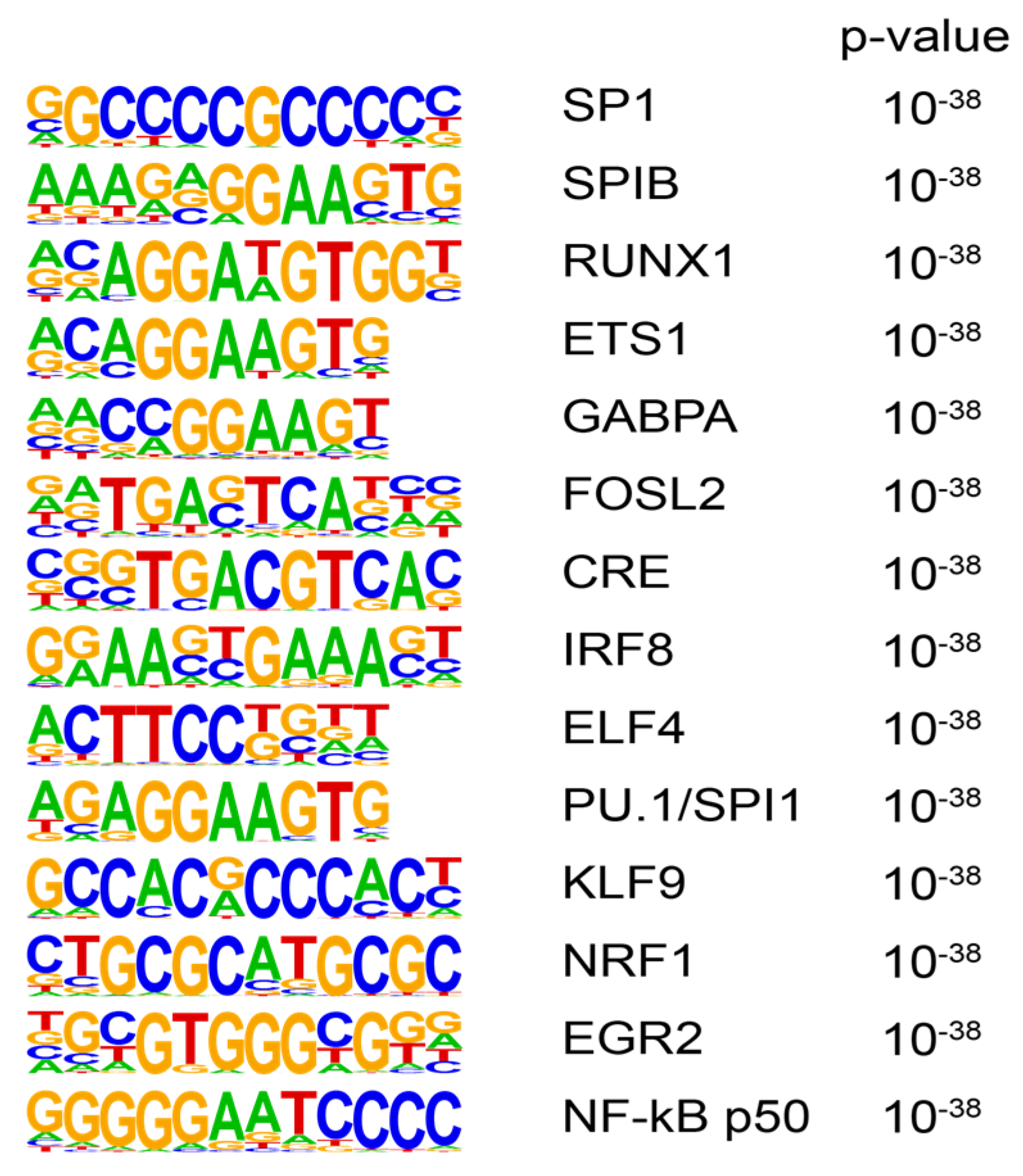
2.6. Motif Enrichment
2.7. Cytokine Arrays
2.8. Senescence Assay in Circulating Lymphocytes Using Flow Cytometry
2.9. DNA Methylation DNAme Profiling and Data Preprocessing
2.10. Assignment of Disparities Index
2.11. Statistics
2.12. DNAme Age Analysis
3. Results
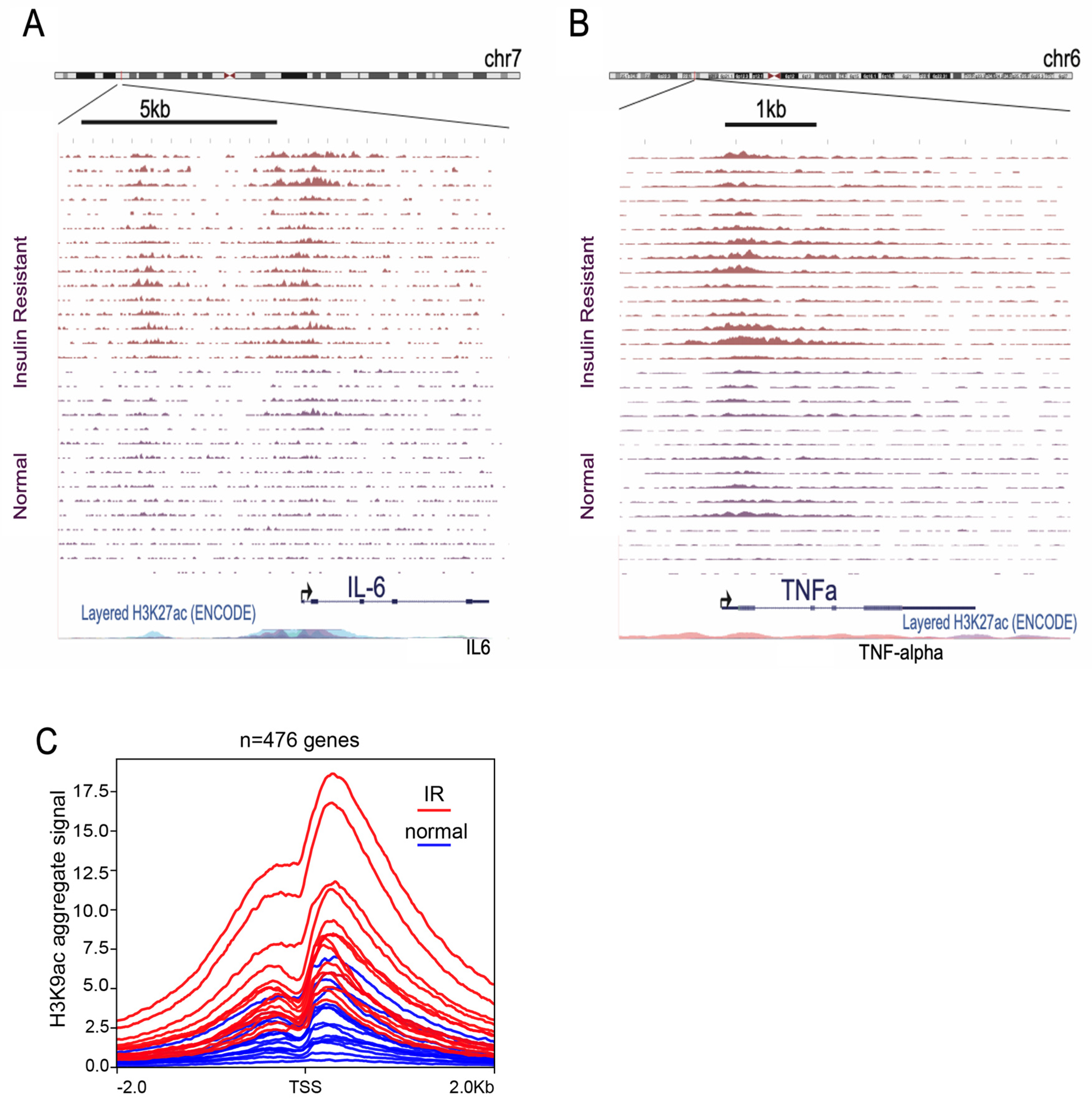
3.1. Insulin Induces H3K9 Acetylation on Gene Promoter Regions Involved in Inflammatory Signaling
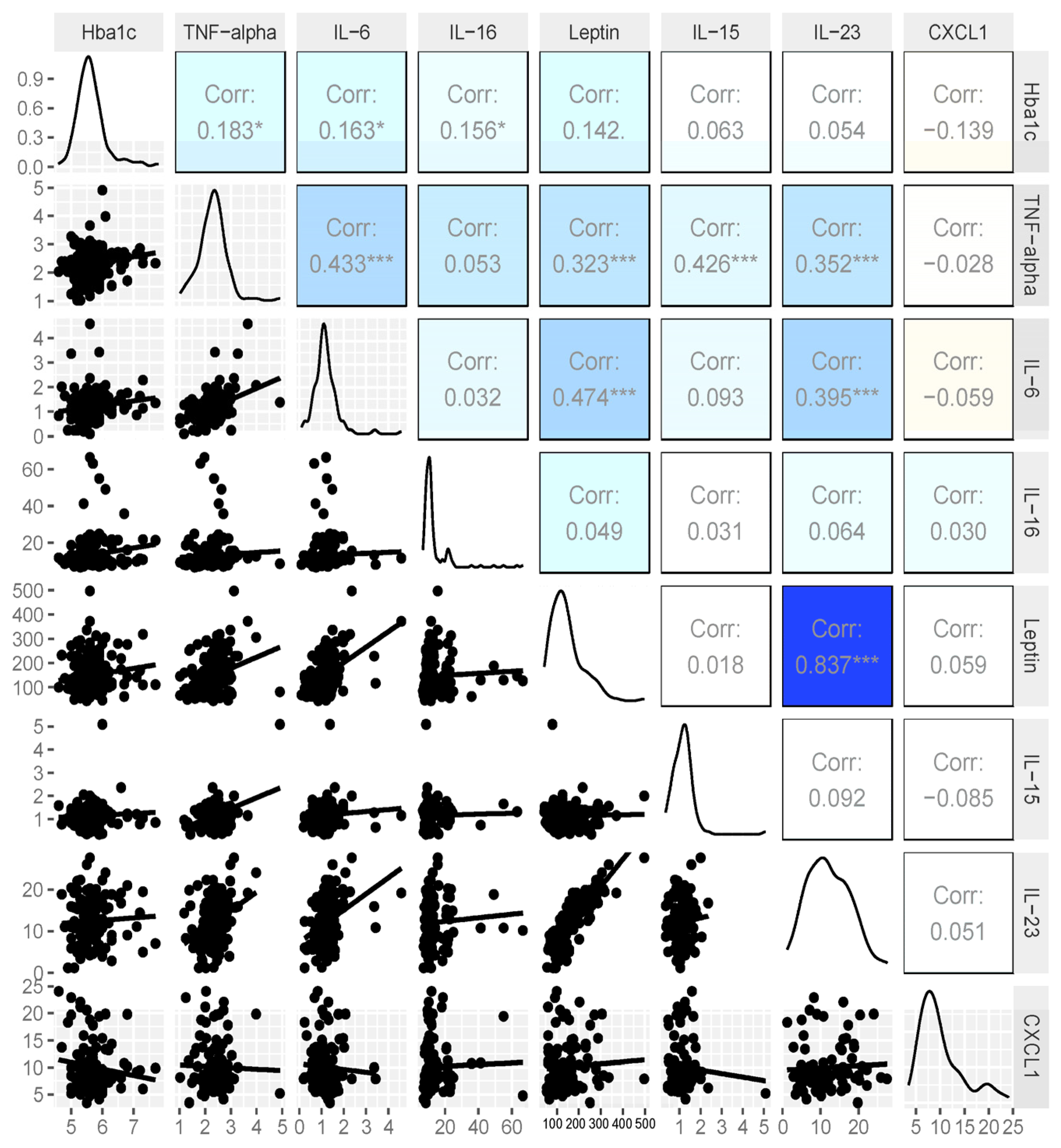
3.2. Cytokine Analysis
3.3. Transcription Factor Motif Analysis
3.4. Expanded Analysis of HbA1c and Cytokines
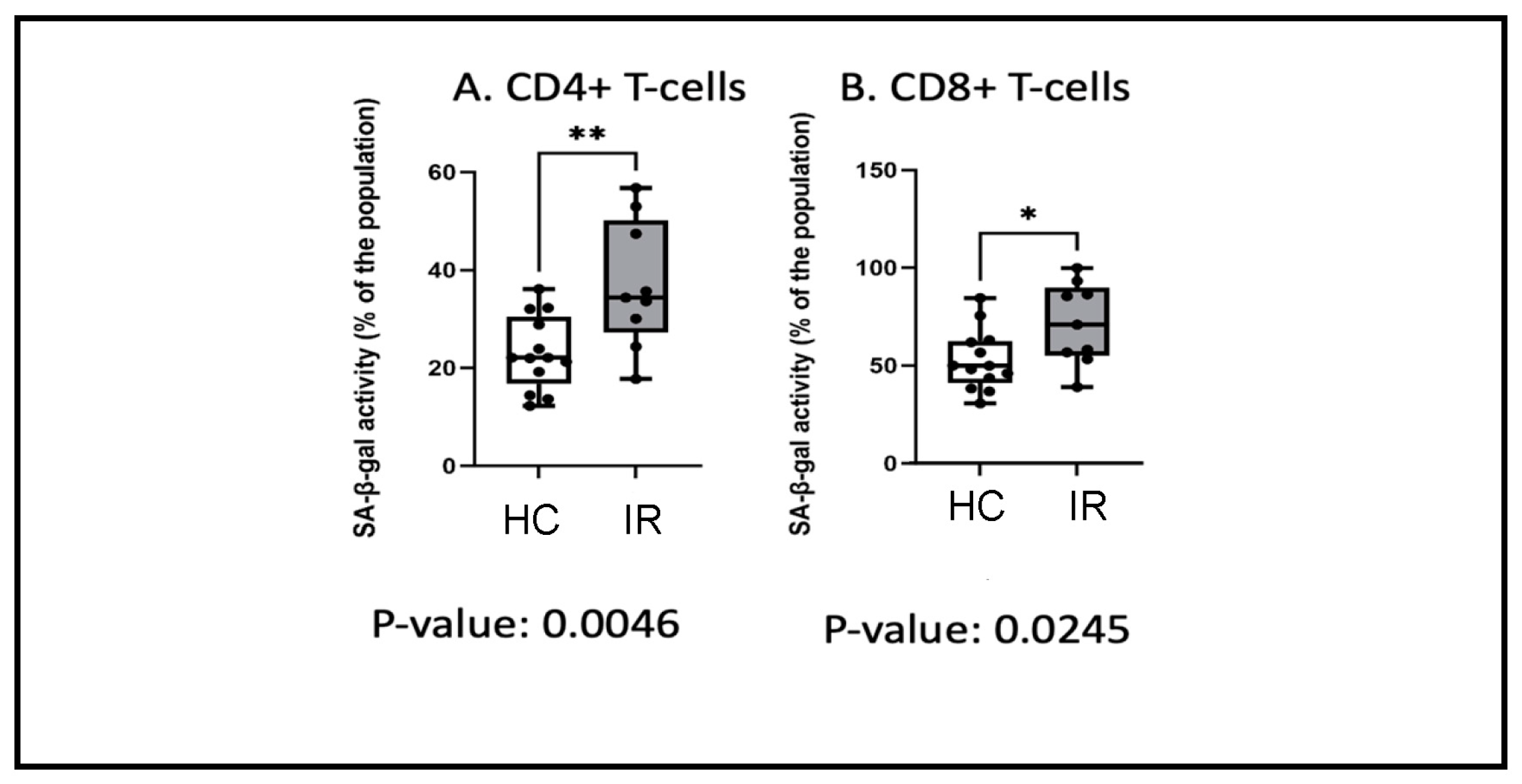
3.5. Insulin Resistance and Senescence
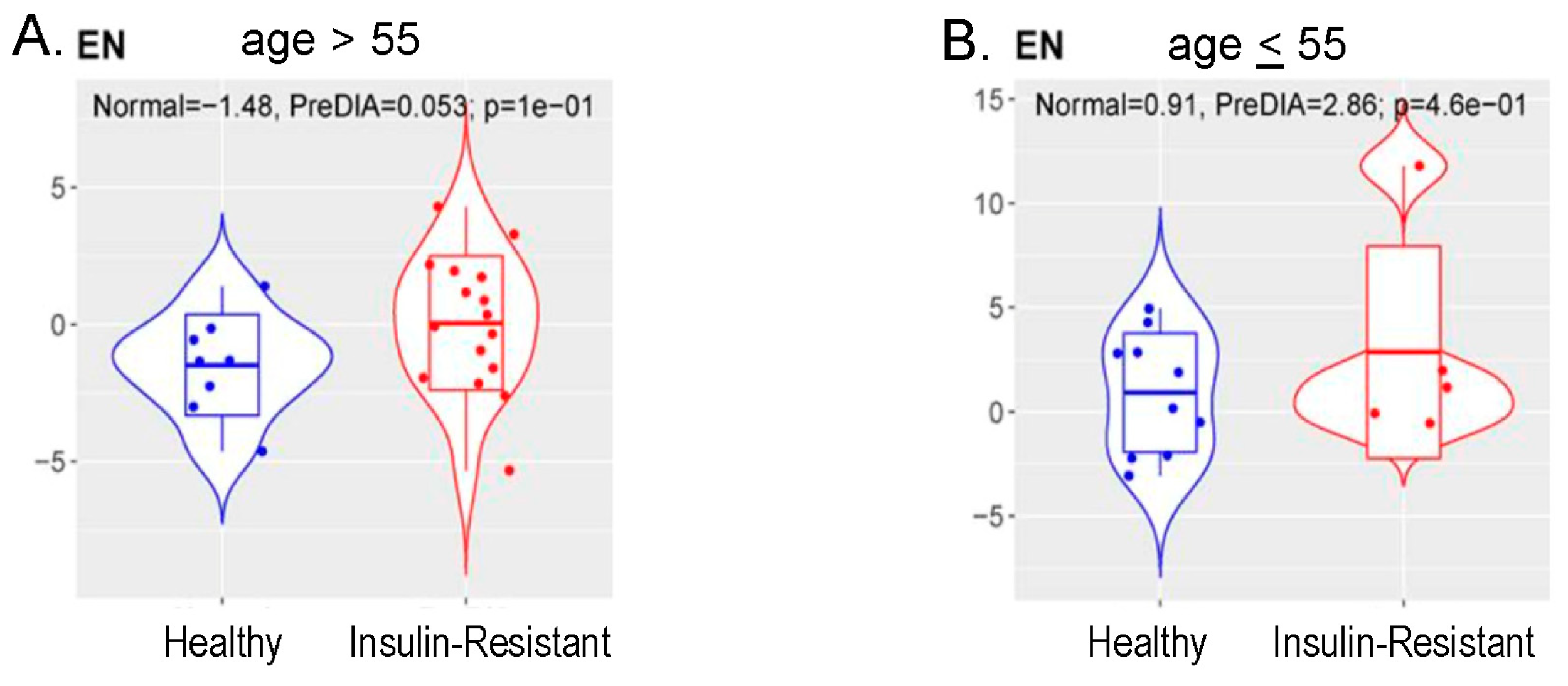
3.6. Methylation Measures of Aging
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raghubeer, S. The influence of epigenetics and inflammation on cardiometabolic risks. Semin. Cell Dev. Biol. 2024, 154, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; Peters, J.L.; Cooper, C.; Lowe, J.; Knight, B.A.; Powell, R.J.; Jones, A.; Hyde, C.J.; Hattersley, A.T. Can clinical features be used to differentiate type 1 from type 2 diabetes? A systematic review of the literature. BMJ Open 2015, 5, e009088. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Chavez, T.A.; Seewaldt, V.L. Obesity and Triple-Negative Breast Cancer: Disparities, Controversies, and Biology. Am. J. Pathol. 2018, 188, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.niddk.nih.gov/health-information/diabetes/overview/what-is-diabetes/prediabetes-insulin-resistance (accessed on 27 June 2024).
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.D.; Mortimer, J.E.; Natarajan, R.; Dietze, E.C.; Seewaldt, V.L. Metabolic Health, Insulin, and Breast Cancer: Why Oncologists Should Care About Insulin. Front Endocrinol 2020, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Senapati, P.; Kato, H.; Lee, M.; Leung, A.; Thai, C.; Sanchez, A.; Gallagher, E.J.; LeRoith, D.; Seewaldt, V.L.; Ann, D.K.; et al. Hyperinsulinemia promotes aberrant histone acetylation in triple-negative breast cancer. Epigenet. Chromatin 2019, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Senapati, P.; Miyano, M.; Sayaman, R.W.; Basam, M.; Leung, A.; LaBarge, M.A.; Schones, D.E. Loss of epigenetic suppression of retrotransposons with oncogenic potential in aging mammary luminal epithelial cells. Genome Res. 2023, 33, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D. Bubble Bubble, Senescent Cells Are a Cauldron of Tumor Trouble. Cancer Res. 2020, 80, 3193–3194. [Google Scholar] [CrossRef]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Iwasaki, K.; Abarca, C.; Aguayo-Mazzucato, C. Regulation of Cellular Senescence in Type 2 Diabetes Mellitus: From Mechanisms to Clinical Applications. Diabetes Metab. J. 2023, 47, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, S.F.; Miyano, M.; Sayaman, R.W.; Lopez, J.C.; Jokela, T.A.; Todhunter, M.E.; Hinz, S.; Garbe, J.C.; Stampfer, M.R.; Kessenbrock, K.; et al. Evidence for accelerated aging in mammary epithelia of women carrying germline BRCA1 or BRCA2 mutations. Nat. Aging 2021, 1, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 27 June 2024).
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Orlando, D.A.; Chen, M.W.; Brown, V.E.; Solanki, S.; Choi, Y.J.; Olson, E.R.; Fritz, C.C.; Bradner, J.E.; Guenther, M.G. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014, 9, 1163–1170. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef]
- Touleimat, N.; Tost, J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef]
- Fortin, J.P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef]
- Kind, A.J.H.; Buckingham, W.R. Making Neighborhood-Disadvantage Metrics Accessible—The Neighborhood Atlas. N. Engl. J. Med. 2018, 378, 2456–2458. [Google Scholar] [CrossRef]
- Available online: https://www.neighborhoodatlas.medicine.wisc.edu(accessed on 27 June 2024).
- Pelegi-Siso, D.; de Prado, P.; Ronkainen, J.; Bustamante, M.; Gonzalez, J.R. methylclock: A Bioconductor package to estimate DNA methylation age. Bioinformatics 2021, 37, 1759–1760. [Google Scholar] [CrossRef]
- Sistrunk, C.; Tolbert, N.; Sanchez-Pino, M.D.; Erhunmwunsee, L.; Wright, N.; Jones, V.; Hyslop, T.; Miranda-Carboni, G.; Dietze, E.C.; Martinez, E.; et al. Impact of Federal, State, and Local Housing Policies on Disparities in Cardiovascular Disease in Black/African American Men and Women: From Policy to Pathways to Biology. Front. Cardiovasc. Med. 2022, 9, 756734. [Google Scholar] [CrossRef]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HbA1c | Age | Race | Ethnicity | BMI (kg/m2) |
|---|---|---|---|---|
| Metabolically Healthy | ||||
| 5.3 | 54 | Asian | 24 | |
| 5.4 | 59 | Asian | 20 | |
| 5.2 | 64 | Black/Indigenous | 24 | |
| 5.4 | 60 | not specified | Latina | 31 |
| 5.5 | 60 | White | 29 | |
| 5.5 | 59 | White | 26 | |
| 5.6 | 61 | White | Latina | 26 |
| 5.1 | 65 | White | Latina | 24 |
| 5.6 | 54 | Asian | 25 | |
| 5.6 | 66 | White | 27 | |
| 5.6 | 31 | White | 23 | |
| 5.2 | 64 | White | Latina | 31 |
| 5.1 | 45 | White | 20 | |
| 5.3 | 54 | Asian | 24 | |
| 5.4 | 59 | Asian | 20 | |
| Insulin Resistant | ||||
| 5.8 | 59 | White | Latina | 33 |
| 5.9 | 66 | White | 29 | |
| 5.8 | 56 | White | 24 | |
| 5.7 | 58 | Black | 24 | |
| 5.7 | 73 | White | 34 | |
| 5.8 | 48 | Asian | 24 | |
| 6.1 | 61 | Black | 31 | |
| 6.0 | 58 | Black | 19 | |
| 6.0 | 66 | White | 26 | |
| 5.8 | 50 | Asian | 21 | |
| 5.7 | 64 | White | 26 | |
| 5.7 | 74 | White | 28 | |
| 6.1 | 62 | Black | 32 | |
| Race | N (%) |
|---|---|
| Asian | 26 (10.6) |
| Black | 11 (4.5) |
| Native American | 4 (1.6) |
| White | 186 (75.9) |
| Unknown | 18 (7.3) |
| Ethnicity | |
| Hispanic | 52 (21.2) |
| Non-Hispanic | 179 (73.1) |
| Unknown | 14 (5.7) |
| Family History of Diabetes | |
| Yes | 126 (51.4) |
| No | 119 (48.6) |
| US Born | |
| Yes | 48 (19.6) |
| No | 197 (80.4) |
| Health Status | |
| Poor | 2 (0.8) |
| Fair | 19 (7.8) |
| Average | 70 (28.6) |
| Good | 119 (48.6) |
| Excellent | 35 (14.3) |
| HbA1c | Age | Race | Ethnicity |
|---|---|---|---|
| Metabolically Healthy | |||
| 4.7 | 37 | White | |
| 4.9 | 53 | White | |
| 5.0 | 34 | White | |
| 5.0 | 53 | White | Latina |
| 5.2 | 52 | White | |
| 5.2 | 48 | White | |
| 5.3 | 51 | White | Latina |
| 5.4 | 47 | Asian | |
| 5.4 | 55 | White | |
| 5.5 | 48 | White | |
| 5.5 | 56 | White | |
| 5.5 | 43 | Native American | |
| 5.6 | 55 | White | Latina |
| Insulin Resistant | |||
| 5.7 | 52 | White | Latina |
| 5.7 | 62 | White | |
| 5.7 | 61 | Asian | |
| 5.9 | 52 | Black | |
| 5.9 | 51 | Black | |
| 5.8 | 53 | White | |
| 6.3 | 45 | Asian | |
| 6.0 | 53 | White | |
| 6.3 | 51 | White | Latina |
| HbA1c | Age | Race | Ethnicity | Deprivation Index | BMI | Methylation Score |
|---|---|---|---|---|---|---|
| Metabolically Healthy | ||||||
| 4.7 | 37 | White | 2 | 22.9 | 33.4 | |
| 4.9 | 53 | White | 3 | 29.7 | 48.4 | |
| 5 | 53 | White | Latina | 4 | 30.5 | 43.1 |
| 5.2 | 48 | White | 2 | 25.9 | 38.9 | |
| 5.3 | 55 | Asian | Filipina | 4 | 23.8 | 47.2 |
| 5.3 | 54 | White | Latina | 9 | 31.6 | 43.4 |
| 5.3 | 48 | White | 2 | 22.1 | 40.0 | |
| 5.3 | 51 | White | Latina | 9 | 37.1 | 48.3 |
| 5.3 | 40 | Asian | 1 | 20.1 | 30.1 | |
| 5.4 | 51 | White | 1 | 20.4 | 44.9 | |
| 5.4 | 56 | White | 6 | 31 | 36.0 | |
| 5.4 | 47 | Asian | 6 | 33.7 | 43.2 | |
| 5.4 | 55 | White | 3 | 22 | 46.4 | |
| 5.5 | 66 | White | 3 | 29.4 | 53.2 | |
| 5.5 | 54 | White | 3 | 26.2 | 45.3 | |
| 5.5 | 53 | White | 5 | 21.1 | 45.9 | |
| 5.5 | 48 | White | 4 | 32.1 | 38.3 | |
| 5.5 | 54 | White | 1 | 34.3 | 50.8 | |
| 5.5 | 56 | White | 4 | 28.8 | 46.6 | |
| 5.5 | 64 | White | 1 | 19 | 54.5 | |
| 5.5 | 31 | White | Latina | 4 | 33.6 | 22.5 |
| 5.6 | 57 | White | 3 | 23.5 | 45.1 | |
| 5.6 | 76 | White | 1 | 40.5 | 63.7 | |
| 5.6 | 62 | White | 1 | 26.6 | 48.6 | |
| 5.62 | 60 | Black | 3 | 20.3 | 44.8 | |
| Insulin Resistant | ||||||
| 5.7 | 51 | Asian | Filipina | 3 | 23.7 | 38.5 |
| 5.7 | 62 | White | 10 | 37.1 | 50.4 | |
| 5.7 | 61 | Asian | 4 | 21 | 50.0 | |
| 5.7 | 70 | White | 4 | 27.8 | 63.5 | |
| 5.7 | 59 | Black | 8 | 24 | 43.7 | |
| 5.8 | 56 | White | Latina | 4 | 32.9 | 50.9 |
| 5.8 | 74 | White | 5 | 31.3 | 56.7 | |
| 5.8 | 47 | Asian | 4 | 21.8 | 37.5 | |
| 5.8 | 49 | White | Latina | 9 | 32.8 | 32.7 |
| 5.8 | 46 | White | 6 | 23.6 | 38.5 | |
| 5.8 | 50 | White | 5 | 24 | 59.1 | |
| 5.8 | 69 | White | 9 | 33.8 | 51.5 | |
| 5.8 | 53 | White | 3 | 35.9 | 43.1 | |
| 5.8 | 83 | White | 7 | 21.6 | 58.3 | |
| 5.8 | 61 | White | 10 | 33.3 | 52.7 | |
| 5.9 | 63 | White | 2 | 29.2 | 53.9 | |
| 5.9 | 59 | White | Latina | 7 | 29.2 | 45.4 |
| 5.9 | 49 | White | 8 | 23 | 42.5 | |
| 5.9 | 52 | Black | 9 | 31.6 | 34.5 | |
| 5.9 | 51 | Black | 2 | 38.2 | 36.6 | |
| 5.9 | 56 | White | 3 | 30.8 | 48.5 | |
| 6 | 74 | White | 4 | 36.6 | 60.6 | |
| 6 | 54 | White | 5 | 35.3 | 38.3 | |
| 6 | 72 | Asian | 2 | 19.6 | 59.7 | |
| 6.1 | 57 | Black | 3 | 30.7 | 40.4 | |
| 6.2 | 48 | White | Latina | 5 | 32.4 | 38.1 |
| 6.3 | 51 | White | Latina | 4 | 27.1 | 41.9 |
| 6.3 | 73 | Asian | 3 | 25.1 | 67.2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal, C.M.; Alva-Ornelas, J.A.; Chen, N.Z.; Senapati, P.; Tomsic, J.; Robles, V.M.; Resto, C.; Sanchez, N.; Sanchez, A.; Hyslop, T.; et al. Insulin Resistance in Women Correlates with Chromatin Histone Lysine Acetylation, Inflammatory Signaling, and Accelerated Aging. Cancers 2024, 16, 2735. https://doi.org/10.3390/cancers16152735
Vidal CM, Alva-Ornelas JA, Chen NZ, Senapati P, Tomsic J, Robles VM, Resto C, Sanchez N, Sanchez A, Hyslop T, et al. Insulin Resistance in Women Correlates with Chromatin Histone Lysine Acetylation, Inflammatory Signaling, and Accelerated Aging. Cancers. 2024; 16(15):2735. https://doi.org/10.3390/cancers16152735
Chicago/Turabian StyleVidal, Christina M., Jackelyn A. Alva-Ornelas, Nancy Zhuo Chen, Parijat Senapati, Jerneja Tomsic, Vanessa Myriam Robles, Cristal Resto, Nancy Sanchez, Angelica Sanchez, Terry Hyslop, and et al. 2024. "Insulin Resistance in Women Correlates with Chromatin Histone Lysine Acetylation, Inflammatory Signaling, and Accelerated Aging" Cancers 16, no. 15: 2735. https://doi.org/10.3390/cancers16152735