

Ectopic PU.1 Expression Provides Chimeric Antigen Receptor (CAR) T Cells with Innate Cell Capacities Including IFN-β Release


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. CAR T Cell Generation
2.3. Western Blot Analysis
2.4. Flow Cytometry
2.5. Cytokine Secretion
2.6. Cytotoxicity Assay
2.7. Repetitive Stimulation Assay
2.8. Statistical Analysis
3. Results
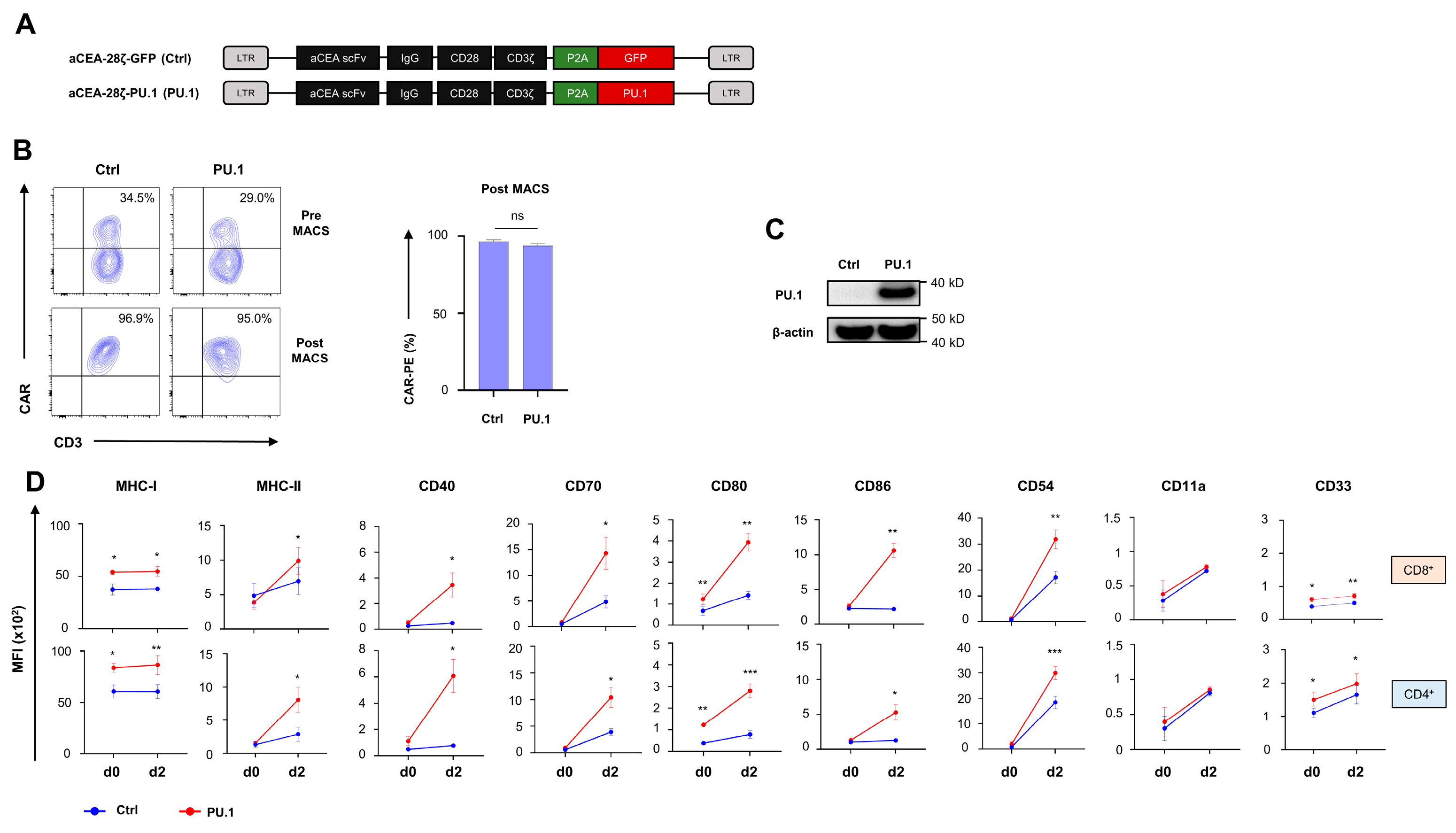
3.1. Ectopic PU.1 Expression Upregulates Costimulatory Receptors in CAR T Cells
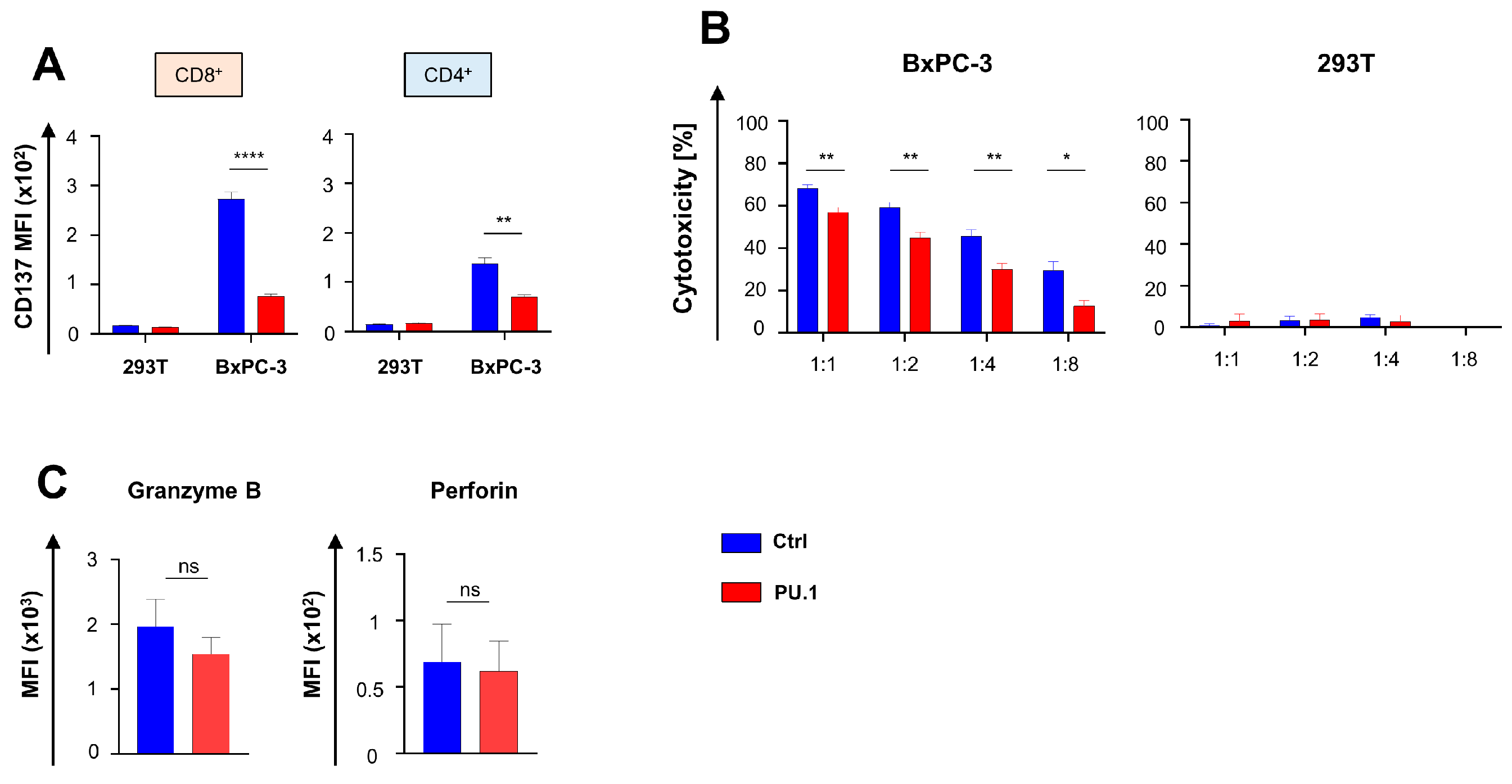
3.2. Ectopic PU.1 Expression Is Accompanied by Decrease in IL-2 Secretion and CAR T Cell Proliferation
3.3. Ectopic PU.1 Expression Diminishes the Cytotoxic Capacities of CAR T Cells
3.4. Ectopic PU.1 Reduces Functional Persistence of CAR T Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holzinger, A.; Abken, H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology 2022, 107, 446–463. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Achmad, H.; Bokov, D.; Abdelbasset, W.K.; Alsadoon, Z.; Chupradit, S.; Suksatan, W.; Shariatzadeh, S.; Hasanpoor, Z.; Yazdanifar, M.; et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res. Ther. 2022, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Dobrin, A.; Lindenbergh, P.L.; Shi, Y.; Perica, K.; Xie, H.; Jain, N.; Chow, A.; Wolchok, J.D.; Merghoub, T.; Sadelain, M.; et al. Synthetic dual co-stimulation increases the potency of HIT and TCR-targeted cell therapies. Nat. Cancer 2024, 5, 760–773. [Google Scholar] [CrossRef]
- Katsarou, A.; Sjöstrand, M.; Naik, J.; Mansilla-Soto, J.; Kefala, D.; Kladis, G.; Nianias, A.; Ruiter, R.; Poels, R.; Sarkar, I.; et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci. Transl. Med. 2021, 13, eabh1962. [Google Scholar] [CrossRef] [PubMed]
- Mansilla-Soto, J.; Eyquem, J.; Haubner, S.; Hamieh, M.; Feucht, J.; Paillon, N.; Zucchetti, A.E.; Li, Z.; Sjöstrand, M.; Lindenbergh, P.L.; et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med. 2022, 28, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; van der Stegen Sjoukje, J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef]
- Chen, H.; Wei, F.; Yin, M.; Zhao, Q.; Liu, Z.; Yu, B.; Huang, Z. CD27 enhances the killing effect of CAR T cells targeting trophoblast cell surface antigen 2 in the treatment of solid tumors. Cancer Immunol. Immunother. 2021, CII 70, 2059–2071. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e6. [Google Scholar] [CrossRef] [PubMed]
- Blokon-Kogan, D.; Levi-Mann, M.; Malka-Levy, L.; Itzhaki, O.; Besser, M.J.; Shiftan, Y.; Szöőr, A.; Vereb, G.; Gross, G.; Abken, H.; et al. Membrane anchored IL-18 linked to constitutively active TLR4 and CD40 improves human T cell anti-tumor capacities for adoptive cell therapy. J. Immunother. Cancer 2022, 10, e001544. [Google Scholar] [CrossRef]
- Chopin, M.; Lun, A.T.; Zhan, Y.; Schreuder, J.; Coughlan, H.; D′Amico, A.; Mielke, L.A.; Almeida, F.F.; Kueh, A.J.; Dickins, R.A.; et al. Transcription Factor PU.1 Promotes Conventional Dendritic Cell Identity and Function via Induction of Transcriptional Regulator DC-SCRIPT. Immunity 2019, 50, 77–90.e5. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V.; Hosokawa, H.; Ungerbäck, J. Mechanisms of Action of Hematopoietic Transcription Factor PU.1 in Initiation of T-Cell Development. Front. Immunol. 2019, 10, 228. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Desbordes, S.C.; Xie, H.; Tillo, E.S.; Pixley, F.; Stanley, E.R.; Graf, T.P.U. 1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6057–6062. [Google Scholar] [CrossRef] [PubMed]
- Montecino-Rodriguez, E.; Casero, D.; Fice, M.; Le, J.; Dorshkind, K. Differential Expression of PU.1 and Key T Lineage Transcription Factors Distinguishes Fetal and Adult T Cell Development. J. Immunol. 2018, 200, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, T.; Nakano, S.; Nomura, K.; Uchida, Y.; Kasakura, K.; Nishiyama, C. A transcription factor PU.1 is critical for Ccl22 gene expression in dendritic cells and macrophages. Sci. Rep. 2019, 9, 1161. [Google Scholar] [CrossRef] [PubMed]
- Turkistany, S.A.; DeKoter, R.P. The transcription factor PU.1 is a critical regulator of cellular communication in the immune system. Arch. Immunol. Ther. Exp. 2011, 59, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Schenkel, C.; Bezler, V.; Kaljanac, M.; Hartley, J.; Barden, M.; Pan, H.; Holzinger, A.; Herr, W.; Abken, H. CAR Triggered Release of Type-1 Interferon Limits CAR T-Cell Activities by an Artificial Negative Autocrine Loop. Cells 2022, 11, 3839. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Rappl, G.; Abken, H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T Cell Response against CD30− Tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Casey, N.P.; Persiconi, I.; Moharrami, N.N.; Sike, A.; Jin, Y.; Kyte, J.A. Development of a TGFβ-IL-2/15 Switch Receptor for Use in Adoptive Cell Therapy. Biomedicines 2023, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Ortner, E.; Stenger, D.; Mahdawi, J.; Apfelbeck, A.; Habjan, N.; Weißer, T.; Kaeuferle, T.; Willier, S.; Kobold, S.; et al. Design and Evaluation of TIM-3-CD28 Checkpoint Fusion Proteins to Improve Anti-CD19 CAR T-Cell Function. Front. Immunol. 2022, 13, 845499. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, T.; Cadilha, B.L.; Obeck, H.; Benmebarek, M.-R.; Märkl, F.; Michaelides, S.; Strzalkowski, T.; Briukhovetska, D.; Müller, P.J.; Nandi, S.; et al. Rational design of PD-1-CD28 immunostimulatory fusion proteins for CAR T cell therapy. Br. J. Cancer 2023, 129, 696–705. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.; Lofgren, M.; Watt, A.; Horton, H.; Kieffer-Kwon, P.; Ding, J.; Kobold, S.; Baeuerle, P.A.; Hofmeister, R.; Gutierrez, D.A.; et al. Functional enhancement of mesothelin-targeted TRuC-T cells by a PD1-CD28 chimeric switch receptor. Cancer Immunol. Immunother. 2023, CII 72, 4195–4207. [Google Scholar] [CrossRef]
- Muto, M.; Baghdadi, M.; Maekawa, R.; Wada, H.; Seino, K.-I. Myeloid molecular characteristics of human γδ T cells support their acquisition of tumor antigen-presenting capacity. Cancer Immunol. Immunother. 2015, CII 64, 941–949. [Google Scholar] [CrossRef]
- Murira, A.; Lamarre, A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Bocarsly, P.; Dai, J.; Singh, S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008, 19, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J.; Thomas, C.; Garcia, K.C.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Huff, A.L.; Wongthida, P.; Thompson, J.; Kottke, T.; Tonne, J.; Schuelke, M.; Ayasoufi, K.; Driscoll, C.B.; Shim, K.G.; et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020, 11, 3187. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Eder, M.; Barden, M.; Pan, H.; Herr, W.; Abken, H. Ectopic PU.1 Expression Provides Chimeric Antigen Receptor (CAR) T Cells with Innate Cell Capacities Including IFN-β Release. Cancers 2024, 16, 2737. https://doi.org/10.3390/cancers16152737
Harrer DC, Eder M, Barden M, Pan H, Herr W, Abken H. Ectopic PU.1 Expression Provides Chimeric Antigen Receptor (CAR) T Cells with Innate Cell Capacities Including IFN-β Release. Cancers. 2024; 16(15):2737. https://doi.org/10.3390/cancers16152737
Chicago/Turabian StyleHarrer, Dennis Christoph, Matthias Eder, Markus Barden, Hong Pan, Wolfgang Herr, and Hinrich Abken. 2024. "Ectopic PU.1 Expression Provides Chimeric Antigen Receptor (CAR) T Cells with Innate Cell Capacities Including IFN-β Release" Cancers 16, no. 15: 2737. https://doi.org/10.3390/cancers16152737
APA StyleHarrer, D. C., Eder, M., Barden, M., Pan, H., Herr, W., & Abken, H. (2024). Ectopic PU.1 Expression Provides Chimeric Antigen Receptor (CAR) T Cells with Innate Cell Capacities Including IFN-β Release. Cancers, 16(15), 2737. https://doi.org/10.3390/cancers16152737