Simple Summary
Cancer is a difficult-to-cure disease with high worldwide incidence and mortality. Among the many changes observed in cancer cells and patient samples is altered glycosylation, a commonly observed modification of biomolecules such as proteins. These glycan structures can dictate protein function, and dysregulation of glycosylation can contribute to tumor migration and metastasis. Thus, manipulation of glycosylation states may be a novel approach to cancer treatment. One target of the well-known tumor suppressor p53 is FUCA1, encoding alpha-L-fucosidase, which plays a role in glycosylation, although the exact mechanism linking FUCA1 to cancer is unclear. Investigation into these glycosylation processes and the mechanisms linking the p53-FUCA1 axis to cancer development may provide new insights into this disease and suggest new drug targets for cancer therapies.
Abstract
Cancer is a difficult-to-cure disease with high worldwide incidence and mortality, in large part due to drug resistance and disease relapse. Glycosylation, which is a common modification of cellular biomolecules, was discovered decades ago and has been of interest in cancer research due to its ability to influence cellular function and to promote carcinogenesis. A variety of glycosylation types and structures regulate the function of biomolecules and are potential targets for investigating and treating cancer. The link between glycosylation and carcinogenesis has been more recently revealed by the role of p53 in energy metabolism, including the p53 target gene alpha-L-fucosidase 1 (FUCA1), which plays an essential role in fucosylation. In this review, we summarize roles of glycan structures and glycosylation-related enzymes to cancer development. The interplay between glycosylation and tumor microenvironmental factors is also discussed, together with involvement of glycosylation in well-characterized cancer-promoting mechanisms, such as the epidermal growth factor receptor (EGFR), phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) and p53-mediated pathways. Glycan structures also modulate cell–matrix interactions, cell–cell adhesion as well as cell migration and settlement, dysfunction of which can contribute to cancer. Thus, further investigation of the mechanistic relationships among glycosylation, related enzymes and cancer progression may provide insights into potential novel cancer treatments.
1. Introduction
In recent years, glycosylation has attracted a lot of research interest because of its importance in cancer development and diagnosis, as well as potential cancer therapies. Glycosylation is the enzyme-catalyzed addition of saccharides to biomolecules such as proteins, sphingolipids and free-circulating polypeptides [1], and it plays a major role in modulating cellular functions. Glycosylation can be classified as O- or N-linked, depending on its target [2]. The covalent linking of sugar molecules requires them to be in a positive energetic state, most commonly provided by sugar nucleotides. Examples of such sugar nucleotides include uridine diphosphate N-acetylgalactosamine (UDP-GalNAc) (Figure 1a), uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) (Figure 1a), guanosine diphosphate mannose (GDP-mannose) (Figure 1b) and guanosine diphosphate fucose (GDP-fucose) [2,3,4,5] (Figure 1c).
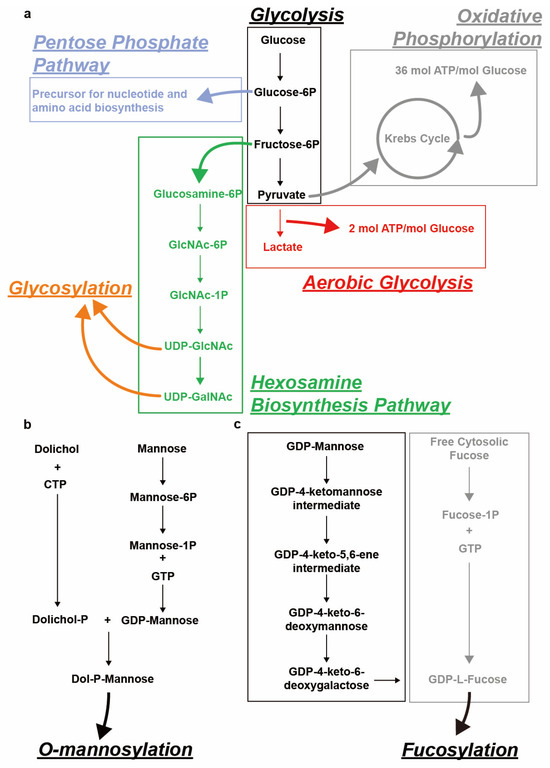
Figure 1.
Schematical representation of sugar nucleotide biosynthesis for various types of glycosylation. (a) Uridine diphosphate N-acetylgalactosamine (UDP-GalNAc), UDP-N-acetylglucosamine (UDP-GlcNAc) biosynthesis from glucose via the glycolysis-related hexosamine biosynthesis pathway (HBP). Other glycolysis-related pathways including the pentose phosphate pathway, aerobic glycolysis and oxidative phosphorylation are also depicted. (b) Dolichol-phosphate-mannose (Dol-P-Man) biosynthesis from Dolichol and mannose. (c) Guanosine diphosphate fucose (GDP-fucose) biosynthesis from guanosine diphosphate mannose (GDP-mannose) (de novo synthesis) and free cytosolic fucose (salvage pathway).
The main cellular compartments where glycosylation takes place include the endoplasmic reticulum (ER) and the Golgi apparatus, although some glycosylation can occur in the cytosol [1]. These glycosyl modifications can take many diverse forms through the attachment of various types of saccharides, different sugar–sugar and sugar–target linkage types and the generation of complex branching structures, each of which can potentially modulate biological function [2]. The tumor suppressor p53 has been shown to inhibit aerobic glycolysis and protein glycosylation [6]. One target of p53 is FUCA1, which encodes alpha-L-fucosidase 1, a hydrolase that plays a role in degrading glycan structures [7]. These observations suggest that characterization of glycan structures and their regulation may provide novel insights into cancer development.
This review describes the various glycosylation processes, the enzymes involved, and their relationship with cancer development. We further discuss the roles of glycosylation in tumorigenesis, progression and metastasis, together with the role of several well-known cancer-associated cell signaling pathways such as epidermal growth factor receptor (EGFR), phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) and p53 pathways affected by glycosylation.
2. Cancer and Glycosylation
Glycosylation refers to the process of adding carbohydrates (monosaccharides or glycans) to a noncarbohydrate molecule, referred to as an aglycone. Depending on the aglycone type, the resulting glycosylated molecule can be classified as glycoprotein, glycolipid or proteoglycan. The addition of various carbohydrates can modulate the structure and conformation of the target and regulate intermolecular interactions such as cell–cell and cell–ECM (extracellular matrix) interactions. Accordingly, aberrant glycosylation can result in the dysregulation of these interactions and potential pathology [1].
Protein glycosylation occurs post-translationally, mostly in the ER and the Golgi apparatus and sometimes in the cytoplasm or nucleus. Glycoproteins can be further divided into several subtypes, depending on the linkage site. The two most common subtypes are N-linked to asparagine (Asn) or arginine (Arg), and O-linked to serine (Ser) or threonine (Thr) [2].
Depending on the conjugated monosaccharide, O-linked glycans can be of several types, as follows: O-GalNAcylated mucin (UDP-GalNAc), O-GlcNAcylated glycan (UDP-GlcNAc) or O-mannosylated glycan (GDP-mannose) (Figure 2a). High expression of O-glycans has been observed in lung cancer patients and are a consequence of signal transducer and activator of transcription 3 (STAT3) hyper-activation induced by the PI3K/Akt pathway, which has been implicated in oncogenesis [8,9]. The generation of truncated O-glycans, resulting from premature terminal formation, is also associated with cancer progression [1,2]. Sialylation, a process of covalently adding sialic acids—derivatives of neuraminic acids—to elongated glycans, is a common step in generating terminal structures of carbohydrate chains [10]. These antigens are targets for antigen-binding adhesin which mediates cell–cell interactions [2]. Premature O-glycan truncation often leads to heavy sialylation, which causes an increase in net negative charge and consequent steric hinderance of cell–cell interaction, and thereby may lead to metastasis [11,12]. Furthermore, O-GlcNAc transferase (OGT), the enzyme responsible for the initial generation of O-GlcNAcylated glycans (Table 1), has been linked to carcinogenesis. The translocation of nuclear factor kappa B (NF-κB) to the nucleus, and consequent transcriptional activity, is modulated by O-GlcNAcylation [13] (Figure 3). OGT also modulates the function of RNA polymerase II (Pol II), as O-GlcNAcylation can compete with phosphorylation of regulatory sites in the enzyme complex [14,15] (Figure 3). The observed elevation of O-GlcNAcylation in cancer cells may be due to the preference of OGT for unstructured and flexible protein regions, which have a higher abundance in cancer compared to normal cells [16,17,18,19,20].
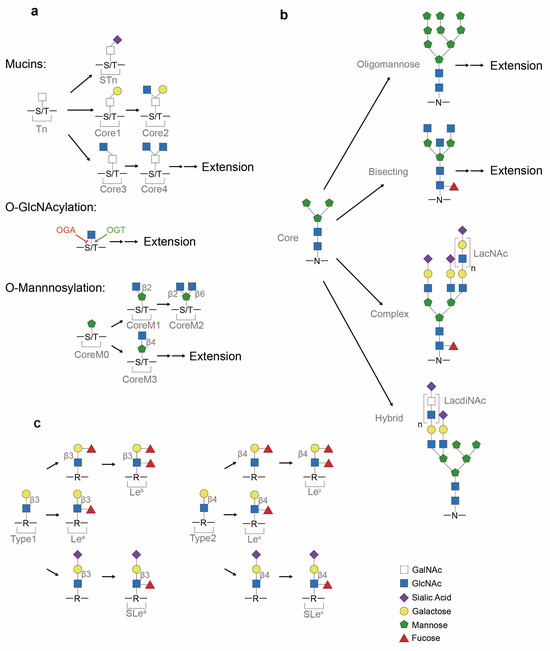
Figure 2.
Representative glycans common in protein glycosylation. (a) O-linked (mucins, O-GlcNAcylated and O-mannosylated) glycosylation. Core structures of O-GalNAcylated mucins consist of Thomsen-nouveau (Tn) antigen, Thomsen–Freidenreich antigen (TF antigen or T antigen or core 1), core 2, core 3, core 4 and sialyl-Tn (STn). After adding the first GalNAc to serine/threonine (S/T), this monosaccharide-linked structure is termed the Tn antigen. If galactose is added to the Tn antigen, it will result in the formation of core 1. If a sialic acid is attached to Tn, a terminal structure STn is formed. O-GlcNAcylation of proteins is balanced between O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). O-mannosylation contains the major structures of coreM0, coreM1, coreM2 and coreM3. Linkage between GlcNAc and mannose differs CoreM1 (β1-2 attachment) and CoreM3 (β1-4 attachment) structures. (b) Asparagine (N)-linked (N-GlcNAcylated) glycosylation. Mature N-glycan precursor is formed from the Man3GlcNAc2 core into oligomannose, complex and hybrid glycans. Extension by the Galβ1-4GlcNAc building block (LacNAc sequence) or GalNAcβ1-4GlcNAc building block (LacdiNAc sequence) is commonly observed in glycans. (c) Terminal glycan structures (Lewis antigens). Common precursors of Type 1 and Type 2 chains are synthesized by a β1,3-GlcNActransferase which catalyzes the attachment of β-GlcNAc block onto glycans and glycolipids (represented by R). Attachment of the outermost galactose defers Type 1 (β1-3 attachment) and Type 2 (β1-4 attachment) glycans. From Type 1 and Type 2 backbones, Lewis antigens, assisted by fucosyltransferases (FUTs) and sialyltransferases, are differentiated into Leb, Lea, SLea, Ley, Lex and SLex.
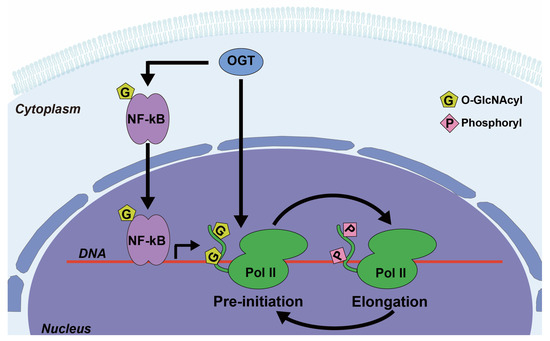
Figure 3.
Examples of O-GlcNAc transferase (OGT) involvement in the regulation of various signaling pathways. OGT catalyzes the addition of O-GlcNAc to nuclear factor kappa B (NF-κB), which enhances its nuclear localization and transcriptional activity. OGT also adds O-GlcNAc to the C-terminal domain (CTD) of RNA polymerase II (Pol II), which competes with phosphorylation at the same sites and can switch Pol II between the pre-initiation and the elongation states.
N-glycosylation serves to localize and direct proteins to secretory pathways, to track proteins within cells, and to regulate cell signaling [21,22,23,24]. A diversity of N-glycan glycoforms (Figure 2b) are involved in cellular and disease processes [25,26]. These processes can be influenced by N-contained side chain exposure, changes in protein conformation and the availability of glycosidase and glycosyltransferases inside the respective subcompartments [24]. N-glycosylation is frequently observed on receptors involved in cellular signaling pathways such as the EGFR pathway, and the biological outcome of these pathways is significantly influenced by the extent of N-glycosylation [23], including an association of N-glycosylation with carcinogenesis.
Glycan extension can also be terminated by special structures such as Lewis antigens (Lea, Leb, SLea, Lex, Ley and SLex) (Figure 2c). Overexpression of Lewis antigens and the resulting loss of cellular adhesion have been observed in cancer samples. For instance, SLea expression has been used to monitor patient responses to cancer therapy, as detection of SLea by assay is a clinically approved cancer-associated marker [2]. SLex, on the other hand, has been implicated in mediating cell–cell adhesion as ligands for lectins, and can contribute to tumor cell migration and metastasis when dysregulated [2].
Glycosylated proteins play a role in various cellular processes predominantly as secreted and cell-surface proteins. These aberrant glycan structures can disturb receptor–ligand binding and cell–cell adhesion and thereby skew cellular signaling pathways toward conditions favoring carcinogenesis, invasion and metastasis [27,28,29,30,31,32,33,34,35,36]. As such, cancer-associated glycosylation patterns and expression of glycosylation-related enzymes could serve as potential biomarkers for cancer detection and as potential drug targets for cancer therapeutics.

Table 1.
Summary of major glycosylation-related enzymes together with their enzymatic functions, implication in glycosylation process and cancer indication.
Table 1.
Summary of major glycosylation-related enzymes together with their enzymatic functions, implication in glycosylation process and cancer indication.
| Gene | Enzyme | Mechanism of Action | Glycosylation Process | Cancer Indication | Reference |
|---|---|---|---|---|---|
| OGT | O-N-acetylglucosamine (O-GlcNAc) transferase | Addition of GlcNAc to serine/threonine (Ser/Thr) | O-GlcNAcylation initiation | Transcription; cancer epigenetics; cell signaling; carcinogenesis | [37] |
| OGA | O-GlcNAcase | Removal of GlcNAc from Ser/Thr | DeGlcNAcylation of O-glycan | Transcription; cancer epigenetics; cell signaling; carcinogenesis | [37] |
| FUT1 | Fucosyltransferase 1 | α1,2-Fucosyltrasferase | Lewis antigen Leb/y synthesis | Cell proliferation; metastasis; invasion; angiogenesis | [38,39] |
| FUT2 | Fucosyltransferase 2 | α1,2-Fucosyltrasferase | Leb/y antigen synthesis | Cell migration; invasion; cancer progression | [38,39] |
| FUT3 | Fucosyltransferase 3 | α1,3/4-Fucosyltrasferase | Lea/b/x/y, SLea/x antigen synthesis | Cancer progression; poor prognosis; Epithelial-to-mesenchymal transition (EMT) | [38,39] |
| FUT4 | Fucosyltransferase 4 | α1,3-Fucosyltrasferase | Lex, SLex antigen synthesis | Cell proliferation; anti-apoptosis; multidrug resistance (MDR) | [38,39] |
| FUT5 | Fucosyltransferase 5 | α1,3-Fucosyltrasferase | SLex antigen synthesis | Cell proliferation; metastasis | [38,39] |
| FUT6 | Fucosyltransferase 6 | α1,3-Fucosyltrasferase | SLex antigen synthesis | Cell proliferation; metastasis; MDR | [38,39] |
| FUT7 | Fucosyltransferase 7 | α1,3-Fucosyltrasferase | SLex antigen synthesis | Cell proliferation; anti-apoptosis | [38,39] |
| FUT8 | Fucosyltransferase 8 | α1,6-Fucosyltrasferase | Core fucosylation of N-glycans | Cell proliferation; metastasis; MDR; poor prognosis | [38,39] |
| FUT9 | Fucosyltransferase 9 | α1,3-Fucosyltrasferase | Lex antigen synthesis | Cancer stemness; cell proliferation; MDR | [38,39,40] |
| FUT10 | Fucosyltransferase 10 | α1,3-Fucosyltrasferase | Core fucosylation of N-glycans | Not yet observed in human | [39,41] |
| FUT11 | Fucosyltransferase 11 | α1,3-Fucosyltrasferase | Core fucosylation of N-glycans | Not yet observed in human | [39,41] |
| POFUT1/FUT12 | Protein O-fucosyltransferase 1 | Transfer fucose to Ser/Thr | O-Fucosylation | High expression in cancer samples; invasion; differentiation | [39,42] |
| POFUT2/FUT13 | Protein O-fucosyltransferase 2 | Transfer fucose to Ser/Thr | O-Fucosylation | High expression in cancer samples; poor prognosis; invasion; differentiation | [39,42,43] |
| FUCA1 | alpha-L-fucosidase 1 | Removal of attached fucose | Defucosylation | Cell proliferation; patient survival | [7,39] |
| FUCA2 | alpha-L-fucosidase 2 | Removal of attached fucose | Defucosylation | High expression in cancer samples; poor prognosis | [39] |
| ST6GALNACs | α-N-acetylgalactosaminide (GalNAc) α-2,6-sialyltransferases | α6-Sialylation of O-GalNAc | Terminal sialylation | Sialyl-Thomsen-nouveau (STn) overexpression; cancer prognosis marker; cell proliferation; migration; cell adhesion | [2,44] |
| ST3GALs | ST3 β-galactoside α-2,3-sialyltransferases | α3-Sialylation of galactose | Terminal sialylation | O-glycan truncation; metastasis; invasion; cell proliferation; cancer prognosis marker | [2,44] |
| GMDS | GDP-mannose-4,6-dehydratase | GDP-mannose-4,6-Dehydratase | GDP-fucose de novo synthesis | High expression in cancer samples; anti-apoptosis; EMT | [5,45,46] |
| GFUS | Guanosine diphosphate fucose (GDP-L-fucose) synthase | Synthesis of GDP-fucose | GDP-fucose de novo synthesis | Cell–selectin binding; cell differentiation; cell proliferation; extravasation | [47,48] |
3. Glycosylation and the Tumor Microenvironment
Not only do cancer cells adapt to grow and survive, but they also alter interactions within the local microenvironment to promote tumor maintenance and metastasis. Glycosylation plays an important role in these interactions. The tumor microenvironment (TME) is a complex set of cellular, physical and soluble mediators surrounding the tumor tissue. These include blood and lymph vessels, endothelial cells and immune cells [49,50] as well as acellular components such as cytokines and the ECM [50,51,52]. These cell–stromal interactions can skew the physical and biochemical properties of the TME toward conditions that favor tumor growth and survival.
Dysregulated cell growth subjects cells within tumor tissue to constant stress, including hypoxia (Table 2). The arrangement of blood and lymph vessel surrounding tumors tends to be disorganized, resulting in poor oxygen penetration into the tumor center [53]. As a result, tumor cells may shift from oxidative phosphorylation to aerobic glycolysis [54,55] (Figure 1a). This shift is called the Warburg effect, and it is an adaptation to the limited oxygen availability that serves to maintain the energy required for cell growth [54]. One response induced by hypoxia is the activation of hypoxia-inducible transcription factors (HIFs) and their signaling pathways [56,57,58,59]. These HIFs transcriptionally regulate glycolysis and glycosylation, although exactly how they alter glycosylation is unclear at present [60]. Hypoxia causes a shift toward the biosynthesis of glycans with low α2,6-sialylation and high β1,6-branching, as well as elongation by poly-N-acetyllactosamine (poly-LacNAc) [61] (Figure 2b). This biased generation of glycans leads to weaker cell adhesion, which could potentially facilitate metastasis.
Acidification of the TME is another adaptive strategy to promote the growth of tumor versus normal cells by creating a hostile, low pH extracellular microenvironment [62,63,64,65] (Table 2). TME acidification is attributed to increased aerobic glycolysis and lactate production due to the Warburg effect [55]. Additionally, the disorganization of lymphatic vessels responsible for the degradation of lactic acids also contributes to the lower pH of the TME [63,66]. Moreover, the accumulation of lactic acids contributes to the synthesis of hypoxanthine (a potential oxygen free radical generator) [67], as well as the expression of the cell-surface CD44 antigen [68]. CD44 antigens, which are themselves glycoproteins, function as receptors of proteoglycans (one subtype of glycan) including hyaluronic acid [2,60,69,70] and chondroitin sulfates [71,72], and these interactions are known to be tumor-associated [2]. Thus, TME acidification and the consequent alteration in CD44-mediated cellular interactions provide another example linking glycosylation with cancer progression and tumor metastasis.
In addition, biological molecules involved in cell–cell and cell–matrix adhesion may be altered in the TME, influencing endothelial adhesion, cell mobility and metastasis [60] (Table 3) Altered glycosylation of intercellular adhesion molecule 1 (ICAM1) [73,74], vascular cell adhesion protein 1 (VCAM1) [75] or platelet endothelial cell adhesion molecule (PECAM1) [76] can influence monocyte rolling and cell adhesion (Table 3). Altered glycosylation of glycoproteins, collagen, glycosaminoglycans (GAGs) and proteoglycans (multi-GAG chains) residing in the ECM are also potential mediators of cell–matrix interactions [2].
In summary, a hostile TME favors the survival of tumor cells over normal. Glycosylation plays various essential roles in the establishment of the TME, the adaptation of tumors to this environment and the downstream consequences of these processes. This involves altered communication between cells and the extracellular environment, mediated by glycosylated cellular surface receptors and transmembrane proteins. As such, dysregulation in glycosylation can lead to the loss of cell–cell and cell–matrix contact, which, in turn, can facilitate cancer cell migration from the primary tumor to distant sites [52].
Table 2.
Summary of glycosylation alterations that are induced during tumor microenvironment establishment and maintenance.
Table 2.
Summary of glycosylation alterations that are induced during tumor microenvironment establishment and maintenance.
| Tumor Microenvironment Properties | Causation | Cancer-Promoting Functions | Glycosylation Alteration |
|---|---|---|---|
| Hypoxia |
|
|
|
| Low pH |
|
|
|
Table 3.
Summary of glycosylated and glycosylation-related molecules that are implicated in tumor microenvironment establishment and maintenance.
Table 3.
Summary of glycosylated and glycosylation-related molecules that are implicated in tumor microenvironment establishment and maintenance.
| Function | Glycosylated & Glycosylation-Related Molecules | Cancer Indication |
|---|---|---|
| Cell adhesion |
| Cell rolling, migration and adhesion [73,74]; cancer progression [27,28,29,30,31,32,33]. |
| Trans-Endothelial Migration |
| Loss of cell adhesion [60]; cell intravasation, rolling and extravasation [60]; tumor metastasis [82,83,84,85]; cell migration [86]. |
4. p53 and Glycosylation
The gene TP53 codes for the tumor protein p53—the well characterized “guardian of the genome”—which was originally characterized as a tumor suppressor that activates the transcription of target genes responsible for repairing DNA damage (Figure 4a). However, subsequent studies revealed that p53 function is not limited to the DNA damage response and repair pathways but also extends to the maintenance of energy metabolism [6]. A high rate of intracellular glycolysis and the acidification of the TME due to lactate accumulation induce p53 expression and activation, which functions to protect cells from glycolytic stress [87]. During tumor development, the loss of p53 in cells surrounding the tumors can further acidify the TME and promote carcinogenesis. Moreover, reactive oxygen species (ROS) that are produced during mitochondrial respiration via oxidative phosphorylation are also harmful to cells (Figure 1a) [88,89], and wild-type p53 is upregulated in response to protect cells from high levels of toxic ROS. However, oncogenic mutation of p53 (mt p53) in cancer cells abrogates this protective response against ROS, leading to increased oxidative damage to cells [90].
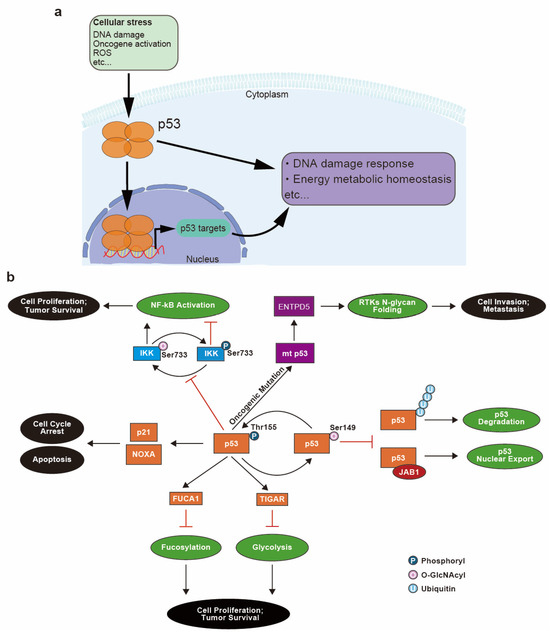
Figure 4.
Involvement of p53 in cell stress responses and glycosylation-related pathways and the relationship between p53 glycosylation and cancer progression. (a) Upon cellular stress, p53 is upregulated and translocated into the nucleus to transcriptionally regulate genes that are implicated in stress response pathways including the DNA damage response and energy metabolism homeostasis. (b) During the response to DNA damage, wild-type p53 is phosphorylated and activated, which leads to cell cycle arrest and apoptosis via transcription of p21 and NOXA. p53 also transcriptionally regulates expression of alpha-L-Fucosidase 1 (FUCA1) and TP53-induced glycolysis and apoptosis regulator (TIGAR) protein to modulate fucosylation and glycolysis pathways. Hyperactivation of these two pathways is associated with cell proliferation and tumor survival. p53 inhibits pro-proliferative NF-κB activation by favoring phosphorylation (Ser733) of Inhibitor of nuclear factor-κB (IκB) kinase (IKK), which competes with O-GlcNAcylation (Ser733) of IKK. O-GlcNAcylation of p53 (Ser149) competes with phosphorylation (Thr155), which attenuates ubiquitination-mediated p53 degradation as well as p53 nuclear export mediated by Jun activation domain-binding protein 1 (JAB1). Oncogenic mutant of p53 (mt p53) also promotes cancer by inducing N-glycan folding of receptor tyrosine kinases (RTKs) via the transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5) protein.
The inhibition of glycolysis and shift to aerobic metabolism, known as the Warburg effect, are also known to depend on p53 activity [91]. Thus, the loss of p53 function in various cancers further promotes aerobic glycolysis [6], a hallmark of cancer cell metabolism [92]. Increased glucose uptake and lactate production from aerobic glycolysis is considered to be an adaptation of cancer cells to the hypoxia microenvironment created by tumor growth [55,62]. High energy demand, TME acidification and an increase in ROS production can induce the expression and activation of normal p53 [87,88,89]. p53 in turn regulates energy metabolism by modulating the glycolysis pathways [91]. A main effector of this regulation is the TP53-induced glycolysis regulatory phosphatase protein (TIGAR), whose expression is induced by activated p53 (Figure 4b) and which functions to reduce levels of fructose-1,6-biphosphate, an intermediate of glycolysis [93]. Limiting the abundance of this substrate results in a reduction in glycolysis, which in turn prevents extensive production of harmful ROS. In contrast, loss of functional p53 is a profound enhancer of aerobic glycolysis [94].
Another link between p53 and cellular energy metabolism is seen in the regulation of NF-κB-dependent transcriptional activity by p53. Mutation of p53 causes enhanced O-GlcNAcylation of IKK, inhibitor of nuclear factor-κB (IκB) kinase, thereby upregulating IKK activity and upregulating NF-κB activity (Figure 4b). IKK is composed of the subunits IKKα and IKKβ, and O-GlcNAcylation of IKKβ occurs at Ser733, which is also the site of inactivating phosphorylation. O-GlcNAc interferes with phosphorylation at this site, enhancing IKK activity and promoting the ubiquitination and removal of IκB. This leads to enhanced NF-κB activation and increased cell survival [95] (Figure 4b). Upregulation of NF-κB also can promote aerobic glycolysis [96] and drive oncogenesis by enhancing expression of solute carrier family 2 member 3 (SLC2A3) [97]. O-GlcNAcylation of IKKβ provides another example of a competition between glycosylation and phosphorylation that can dictate protein function, similar to the example of Pol II regulation mentioned earlier.
Furthermore, p53 itself can be O-GlcNAcylated (Figure 4b). Studies have demonstrated that O-GlcNAcylation of p53 occurs at Ser149, and this modification inhibits ubiquitin-based degradation of p53. Ser149 is proximal to a phosphorylation site at Thr155 and when O-GlcNAcylated can potentially suppress phosphorylation at Thr155 and thereby decrease susceptibility to ubiquitination [98] (Figure 4b). Moreover, Thr155 phosphorylation is necessary for p53 nuclear export mediated by Jun activation domain-binding protein 1 (JAB1) (Figure 4b). Thus, O-GlcNAcylation prevents phosphorylation at Thr155, inhibits p53 nuclear export, inhibits ubiquitination and consequently increases p53 half-life in cells [99,100]. Thus, the p53 protein, which previously has been characterized as a DNA damage response factor, is also implicated in glycosylation. p53 not only modulates glycosylation on other proteins via its effect on glycolysis, but is itself glycosylated in a manner that can affect its function.
Mutated p53 in cancer cells has also been implicated in the aberrant folding of N-glycosylated proteins via its transcriptional regulation of the ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5) gene [101,102]. ENTPD5 protein is a component of the calnexin/calreticulin chaperon system that facilitates N-glycoprotein folding [103,104]. Receptor tyrosine kinases (RTKs), including transforming growth factor beta (TGFβ) receptor and EGFR [105], are highly modified by N-glycans, and their oncogenic mutations are associated with the calnexin/calreticulin chaperon system [103,106]. Moreover, ENTPD5 expression level is highly associated with the presence of mt p53 at both the transcript and protein levels, and knockdown of either ENTPD5 or mt p53 attenuates the invasiveness and metastatic potential of cancer cells [102]. These observations indicate that mt p53 can modulate the folding of N-glycoproteins essential for tumorigenesis and cancer progression via ENTPD5 and the calnexin/calreticulin chaperon system in cancer cells (Figure 4b).
Taken together, p53 is involved in the modulation of energy metabolism and generation of protein glycoforms, including modulation of its own activity by glycosylation, illustrating the potential contribution of p53 to cancer development from the energy homeostasis perspective [98].
5. Fucosylation in Cancer
Another type of protein glycosylation is fucosylation, which has been extensively studied in human metabolism and has been the subject of much interest due to its apparent involvement in oncogenesis. Fucosylation is classified into terminal (α1-2 or α1-3/4 linked) and core fucosylation (α1-6 linked), depending on the sites of attachment [2]. Unlike GlcNAcylation or GalNAcylation, which are direct linkages to the target, fucosylation decorates previously synthesized glycans with fucose.
5.1. Fucose Nucleotide Biosynthesis
GDP-fucose is the sugar donor used for fucosylation. This sugar nucleotide is formed in the cytosol via the following two possible routes: de novo synthesis and salvage pathways. 90% of GDP-fucose comes from the de novo pathway, during which cytosolic GDP-mannose is converted step-wise into GDP-fucose via catalysis by GDP-mannose-4,6-dehydratase (GMDS) [5] and GDP-L-fucose synthase (GFUS or protein FX) [47] (Figure 1c) (Table 1). In contrast, the salvage pathway utilizes free cytosolic fucose to synthesize GDP-fucose via catalysis by fucose kinases and fucose-1-phosphate guanylyltransferase (FPGT) [107] (Figure 1c). GDP-fucose is thereafter transported into the Golgi apparatus and serves as the sugar donor for fucosylation, catalyzed by FUTs [108].
5.2. Fucosyltransferases (FUTs) and Fucosidases
The human genome encodes 13 FUTs that are responsible for terminal and core fucosylation with different α-linkages (Table 1). FUTs are mostly located in the following two subcellular compartments: the N-glycan targeting FUTs reside in the Golgi apparatus while O-fucosyltransferases reside in the ER [39]. Two fucosidases (FUCA1 and alpha-L-Fucosidase 2 (FUCA2)) were found to carry out the defucosylation of glycans in humans [39]. The distinct functions of each FUT and fucosidase is listed in Table 1. Fucosylation is enhanced in cancer development [109,110,111,112] and cancer aggressiveness is apparently inhibited when fucosylation is reduced [113] Thus, FUTs and fucosidases may be novel candidate drug targets for cancer therapeutics.
5.3. Fucosylation and Cancer
Aberrant fucosylation is frequently observed in cancer samples. Lewis antigen Lex (Figure 2c) is used as a biomarker for glioblastoma multiforme (GBM) due to the correlation between its high expression and the tumorigenic potential of cells [109,110]. FUT8, which attenuates cancer aggressiveness when knocked down [113], is overexpressed at both the mRNA and protein levels in lung and colorectal cancer (CRC) samples and its expression is associated with poor prognosis [111]. In addition, the sialylated Lewis antigens SLex and SLea (Figure 2c) are globally elevated in cancer specimens [2] and are implicated in carcinogenesis and tumor metastasis via their ability to enhance cancer cell mobility across the endothelium [114,115]. General upregulation of fucosylation is also observed in lung cancer adenocarcinoma, often accompanied by increased GMDS expression [45]. FUCA1 mRNA expression is generally lower in cancer samples compared to normal counterparts, and this is associated with poor patient prognosis [7,116]. In contrast, FUCA2 is proposed to be a cancer prognostic marker due to its high mRNA expression in pan-cancer patients and association with poor survival rates [117]. The correlation between FUCA2 high expression and cancer has also been confirmed at the protein level in lung carcinoma and uterine corpus endometrial carcinoma [117].
Current studies on the contribution of fucosylation to carcinogenesis have revealed its effects on malignant cell proliferation, invasion, metastasis, immune surveillance escape and multidrug resistance (MDR) [39].
5.4. Fucosylation in Cancer Cell Proliferation
Fucosylation levels are positively associated with cancer cell proliferation. Maintaining higher fucosylation in cancer cells requires securing higher amounts of its sugar nucleotide donor GDP-fucose. Inhibition of GDP-fucose synthesis leads to decreased fucosylation of receptors implicated in the EGFR signaling pathway and, thus, reduces cancer cell proliferation [118]. In addition, knocking down GMDS induces cell cycle arrest and activates apoptosis [45]. FUTs have been shown to promote cell cycle progression [119,120,121,122]. In addition, alterations in FUTs’ mRNA expressions result in changes in the levels of Lewis antigen abundance, which interfere with various well-known cancer-associated signaling pathways including the PI3K/Akt [123] and EGFR/mitogen-activated protein kinase (MAPK) pathways [124]. Multiple lines of evidence have also supported the notion that fucosidases are involved in cancer progression and metastasis, as follows: (1) ectopic expression of FUCA1 in samples having low endogenous levels of FUCA1 significantly attenuates the invasiveness of cancer cells; (2) transient FUCA1 knockdown results in the loss of cell–cell contact; and (3) prolonged FUCA1 knockdown leads to increased cancer cell proliferation, invasion and migration [125]. Moreover, fucosylation can contribute to the hyperactivation of the EGFR, a well-known driver of carcinogenesis, and this can be reversed by FUCA1 overexpression, which reduces fucosylation [7]. These observations indicate the tumor suppressor potential of FUCA1. In contrast, FUCA2 expression is commonly elevated in cancer [117,126]. Anti-FUCA2 antibodies inhibit the proliferation of breast cancer cells, and this is reversed by ectopic expression of FUCA2, suggesting that this fucosidase has oncogenic potential [127].
5.5. Fucosylation in Cancer Stem Cells
Similarities have been observed between cancer development and embryonic development, and thus important biological pathways for embryogenesis are expected to demonstrate similar significance in cancer progression. Although the exact role of fucosylation in human embryo development has not been fully elucidated, resultant lethal phenotypes in mouse embryos arising from defects in fucosylation hints at the essentiality of this process for proper cell growth and proliferation [128]. This is further supported by fucosidosis, a rare neurodegenerative autosomal recessive disorder, being mediated by FUCA1 mutation [129].
Cancer stem cells (CSCs), as the postulated origin of most human tumors, have also been proposed to contribute to cancer development. Research in oral squamous cell carcinoma has demonstrated that inhibiting fucosylation negatively affected tumor initiation and CSCs invasion, and fucosylated SLex antigen is implicated in metastasis associated with CSCs [130]. These observations could be explained by the fucosylation of integrins—cellular surface molecules that function to guide movement and localization of cells through intercellular interactions with glycan-binding proteins (GBPs) presented on other cells. This implication is not limited to cancerous cells. For example, modulating the fucosylation status of integrins is a potential strategy for the delivery of noncancerous multipotent stromal cells to target tissues for tissue repair [131]. Moreover, enhanced fucosylation of cytotoxic T lymphocytes improves their homing to tumor tissue and thus is proposed to be an effective strategy for cancer treatment [132]. These results indicate that fucosylation is also implicated in cancer progression from the aspect of stem cells potentially through its role in guiding cell migration.
5.6. Fucosylation in the Epithelial-to-Mesenchymal Transition (EMT)
Fucosylation has been shown to be involved in the EMT. EMT refers to a biological process consisting of the following: (1) the transition of epithelial tumor cells bearing epithelial cellular markers to cells that mimic the mesenchymal cell phenotype and exhibit mesenchymal cellular markers; and (2) the degradation of basement membrane, enhancing the capability of tumor cells to migrate to distant sites [133]. Similar to other types of glycosylation, fucosylation may also affect cell–cell adhesion by modulating the function of EMT factors. Cancer-associated upregulation of GMDS and FUT8 leads to an enhancement in core fucosylation followed by appearance of mesenchymal markers [46], indicating enhanced EMT. Furthermore, transcription of EMT factors is induced by fucosylation of translocation-facilitating proteins, which in turn promote the nuclear translocation of transcription coactivators that skew cells toward the mesenchymal cell phenotype [134]. Additionally, upregulation of fucosylation promotes an elevation in matrix metalloproteinases (MMPs) that degrade the ECM [135], clearing boundaries around tumor cells and facilitating metastasis. EMT can also be induced by signaling pathways such as the TGFβ-dependent cascade, which is regulated by both FUT8 and FUCA1 [136,137]. Expression of FUCA1 decreases during TGFβ-induced EMT, resulting in the generation of highly fucosylated N-glycans associated with cancer development [136].
5.7. Fucosylation and Tumor Cell Trans-Endothelial Migration
Translocation of leukocytes across tissue barriers during the inflammatory response involves glycoprotein-mediated alterations in cell adhesion, and tumors utilize this mechanism to allow trans-endothelial migration and metastasis [71,138,139]. Glycosylated epitopes on tumor cells facilitate cell rolling along the endothelium by binding to GBPs exposed on endothelial cells [71,80] (Figure 5). These interactions between GBPs exposed on the endothelial cellular surface and the glycan structures of circulating biomolecules such as leukocytes and tumor cells are modulated by enzymes such as glucosyltransferases and FUTs [80,140] (Table 3).
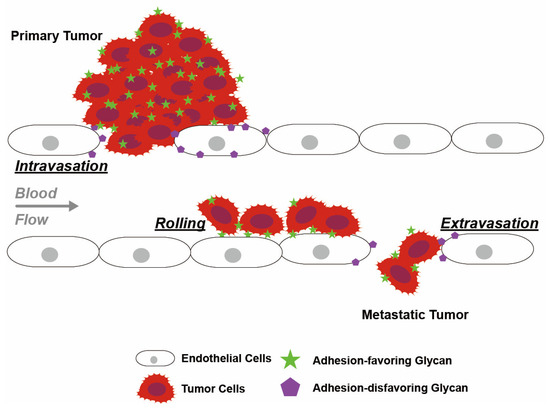
Figure 5.
Glycan-mediated intravasation, rolling and extravasation of tumor cells along the endothelium. Adhesion-disfavoring glycans attenuate adhesion and reduce cell–cell interactions with the endothelium and enhance endothelial permeability, thereby facilitating tumor intravasation and extravasation. Glycans on tumor cells bind to lectin receptors on endothelial cells with low affinity, which contributes to tumor cell rolling along the endothelium. Glycan structures involved in this process include Lewis antigens and lectin receptors.
Fucosylation reduces cell adhesion, thereby allowing intravasation and extravasation of tumor cells across the endothelium [60]. Specifically, these cellular interactions are modulated by terminal fucosylated glycans binding to lectins. Fucosylation alters the interactions of glycans with lectin, allowing enhanced cell rolling mobility [79]. Similarly, fucosylation allows tumor cells to mimic leukocyte transportation and travel from the primary site to colonize distant parts of the body [27,28,29,30,31,32,33,71,138,139] (Figure 5). This argument is consistent with the observed increase in the abundance of fucosylated SLex motifs on migrating tumor cells [27,28,29,30,31,32,33].
Various glycosylation-related enzymes contribute to the synthesis of fucosylated glycans. One example of a sialylation-related enzyme implicated in cancer development is ST3 beta-galactoside alpha-2,3-sialyltransferase 6 (ST3GAL6), which is essential for sialylated Lewis antigen synthesis. Knockdown of ST3GAL6 was observed to attenuate cell trans-endothelial migration ability, while elevation of ST3GAL6 expression was found to be associated with low overall survival in multiple myeloma patients [81] (Table 1). Reduction in enzymes involved in fucosylation decreases the synthesis of the lectin ligands SLea and SLex, leading to attenuated binding of tumor cells to lectins [48].
These observations indicate that cancer-associated changes in fucosylation-related enzymes affect the adhesion of cells essential for endothelium integrity, contributing to tumor cell trans-endothelial migration and metastasis.
5.8. Fucosylation in Metastasis
The function of fucosylation in metastasis is not limited to facilitating EMT and trans-endothelial migration of tumor cells across blood vessels. The amount of circulating tumor cells (CTCs) shed from primary tumors as well as their rolling velocities along blood vessels are also dependent on fucosylation levels. For instance, in a simulated model, knockdown of FUT3 significantly reduced the number of flowing cells under shear stress. High rolling velocity that disfavors settlement of flowing cells was also observed in cells with FUT3 knockdown [141]. Moreover, knockdown of FUT3 lowered expression of the lectin ligand SLex, hence weakening interactions between CTCs and GBPs present on other cells [141,142]. As proper binding of CTCs toward GBPs is required for the attachment of tumor cells to secondary sites [142], the attenuated binding affinity to GBPs caused by FUT3 knockdown is one example of the metastatic regulation of CTCs by fucosylation alteration.
In addition, fucosylation affects information transmission between tumor cells and other cells within TME. This behavior, primarily regulated by tumor-derived exosomes, also facilitates metastasis. Exosomes are integrin-coated extracellular vesicles carrying miRNAs, mRNAs, long noncoding RNAs (lncRNAs), DNAs and lipids, and are primarily involved intercellular communication [143]. Although the mechanisms behind regulation of exosomes by fucosylation remain underexplored, evidence suggests an association between these two biological features. Compared to those from normal cells, exosomes derived from tumors display analogous features to their parent cells such as aberrant fucosylation. For instance, evidence has shown that integrins were over-expressed and heavily fucosylated in tumor-derived exosomes [144]. Meanwhile, others have demonstrated that fucosylated exosomes promote cancer growth while soluble fucosylated glycans had no effect on cell proliferation [145]. Moreover, exosomes derived from tumor cells could be used to relieve cellular stress to maintain cell growth and survival. Evidence suggests that tumor cells sequester miRNAs that disfavor tumor growth into fucosylated exosomes to mitigate the suppressive effects of these miRNAs [146].
Taken together, fucosylation status significantly affects metastasis in almost every step of the process including EMT, detachment of tumor cells from primary tumor, intravasation, CTCs rolling along blood vessels, extravasation, and settlement to secondary sites. The most well-characterized mechanism of fucosylation contribution to metastasis is through intercellular interactions between fucosylated surface molecules and tumor cells or tumor-derived exosomes with GBPs presented on the surface of other cells. Despite this, studies have been limited to the effects of FUTs on fucosylation-dependent metastasis promotion, while the function and relevance of fucosidases remain underexplored.
5.9. Fucosylation in Immune Surveillance
Fucosylation also participates in the escape of tumor cells from immune surveillance. The innate immune response involves binding of receptors on immune cells to antigens presented on target cells. The binding and function of either the immune receptors or target cell antigens can be modulated by glycosylation, including fucosylation. Often tumor cells utilize glycosylation to mask their foreignness and escape from immune surveillance [39]. For example, fucosylation of tumor cell antigens promotes their recognition by Natural Killer (NK) cells [147]. Accordingly, some cancer cells have reduced GDP-fucose production due to a mutation in GMDS [148,149], leading to a reduction in the abundance of Lewis antigen. This attenuates NK cell responses that depend on recognition of antigen, including activation of tumor necrosis factor (TNF) receptor superfamily member 6 (CD95) and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis pathways, thus protecting tumor cells from immune cell attack [148,150].
These results demonstrate that fucosylation is essential for antigen presentation on the cell surface, and when compromised can enable tumor cells to evade the immune response.
5.10. Fucosylation in Multidrug Resistance (MDR)
MDR is a major obstacle to cancer treatment, as it results in resistance to chemotherapy [39]. Fucosylation has been linked to the acquisition of MDR in tumor cells, suggesting that it could be targeted as an adjunct to chemotherapy. Cells that have acquired MDR often display N-linked glycans with high levels of core fucosylation [151], attributed to altered FUT8 and FUCA1 activities [7,126,137,152,153]. Furthermore, enhanced expression of FUTs observed in cancers can contribute to MDR development by activating the PI3K/Akt and extracellular signal-regulated kinase (ERK)/MAPK pathways, which facilitate the survival of cells and compromise the efficacy of anticancer therapies [154,155]. In addition, MDR-associated protein 1 (MRP1), which promotes MDR, has been reported to be regulated by FUTs [154].
In summary, fucosylation can modulate the functional properties of proteins and has been linked to carcinogenesis and cancer progression, similar to other glycosylation processes. One major effect is the regulation of Lewis antigen synthesis, affecting their interaction with lectin receptors on immune cells to escape from immune surveillance and on endothelial cells to promote tumor migration [147,148,149]. The strong correlation between fucosylation levels and cancer progression suggests that these fucosylation-related enzymes could be potential therapeutic targets for new cancer treatments. This may include inhibition of FUTs to decrease fucosylation [156], or enhancement of FUCA1 activity, which has been speculated to possess tumor-suppressive potential.
6. Regulation of Fucosylation by the p53-FUCA1 Axis
Investigations of the tumor-suppressive potential of FUCA1 led to the discovery that its gene is a transcriptional target of p53. Thus, p53, in addition to its other reported functions, appears to promote cancer by regulating fucosylation [7].
6.1. Transcriptional Regulation of FUCA1 by p53
The idea that FUCA1 is regulated by p53 was first proposed based on the observed co-expression of p53 protein and FUCA1 mRNA [7,157]. This was confirmed by the observation that p53 protein binds to the FUCA1 gene [7] and that proper binding of p53 to its responsive elements on FUCA1 is necessary for the transcription activation of FUCA1 [157]. Thus, FUCA1 was deemed a direct transcriptional target of p53. Moreover, chemotherapies that induce p53-dependent pathways upregulate FUCA1 mRNA expression in a p53-dependent manner [157]. However, FUCA1 does not respond to regulation by p73, a p53 family member that shares many transcription targets with p53 [157]. These results indicate that transcriptional control of FUCA1 by p53 is innate and specific to endogenous p53.
Confirmation of the transcription regulation of FUCA1 by p53 is not limited to in vitro but also extends to in vivo studies. In specimens collected from breast cancer patients bearing wild-type p53, FUCA1 expression is high, whereas specimens containing mutant p53 show low levels of FUCA1 [7]. The same trend was observed in thyroid cancer patients, as follows: FUCA1 RNA expression is high in papillary thyroid cancers (PTCs) which mostly carry wild-type p53, while low levels of FUCA1 RNA were detected in anaplastic thyroid cancers (ATCs) where p53 is frequently mutated. These observations indicate that regulation of FUCA1 transcription is dependent on p53 status (wild-type or mutated). In highly aggressive cancer types, common phenotypes include the loss of wild-type p53, which could explain the observed correlation between cancer aggressiveness and the difference in FUCA1 expression in ATCs that are derived from more differentiated PTCs [158].
6.2. Regulation of Fucosylation by the p53-FUCA1 Axis
FUCA1 fucosidase activity is also positively corelated with the extent of p53 induction [157]. FUCA1 fucosidase activity can be upregulated by DNA damage and overall fucosylation levels are decreased in cells having wild-type p53 [7]. Moreover, fucosidase activity can be induced by exogenous p53 overexpression, and this is further potentiated by the additional induction of DNA damage [7]. Similarly, increases in the levels of FUCA1 proteins was observed following p53 induction, whereas knocking down FUCA1 led to a loss of overall fucosidase activity in the cell [157]. Taken together, these results indicate that FUCA1 fucosidase activity as well as cellular fucosylation levels are both regulated by p53.
6.3. Tumor Suppression by the p53-FUCA1 Axis
This relationship with p53 suggests that FUCA1 could function to suppress tumors. This is supported by the observation that p53 exhibits a greater impact on tumor suppression compared to p73 [157], consistent with FUCA1 being a specific target of p53 but not p73.
The p53-FUCA1 axis have been shown to play a role in the induction of apoptosis [7]. Overexpression of FUCA1 inhibits cell proliferation by increasing apoptosis in breast cancer, CRC, GBM, and lung cancer cells, independent of p53 status [7]. FUCA1 was shown to reduce fucosylation of EGFR by catalyzing α1,6-defucosylation (Figure 6). The resulting decrease in EGFR fucosylation in turn represses phosphorylation of both EGFR and Akt [7] (Figure 6), a downstream effector of the EGFR pathway, indicating attenuated activity of both EGFR and Akt. The observation that expression of FUCA1 inhibits the EGFR pathway, implicated in promoting cancer, again supports the potential role of FUCA1 as a tumor suppressor.
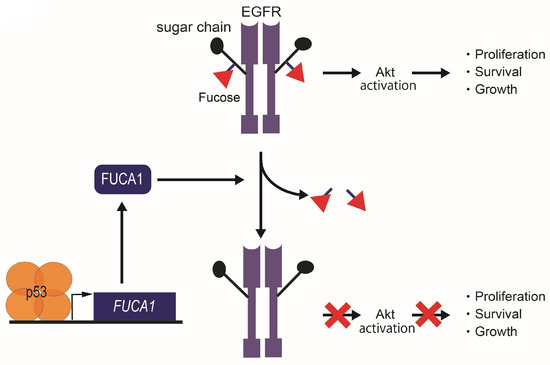
Figure 6.
Tumor suppression function in cancer cells by the p53-FUCA1 axis. FUCA1 is one of the p53 target genes. FUCA1 reduces α1,6-fucosylation of EGFR and inhibits the Akt signaling pathway in cancer cells.
Another study has shown that FUCA1 fucosidase plays a role in p53-mediated apoptosis in an osteosarcoma cell line previously subjected to chemotherapy [157]. This study showed that while overexpression of FUCA1 does not affect cancer cell viability, knockdown of FUCA1 impairs the ability of p53 to induce activation of effector caspases that would otherwise cause apoptosis and cell death [157]. On the other hand, other studies have demonstrated that overexpression of FUCA1 does not affect the clonogenic potential of thyroid cancer or normal cells [158].
These results demonstrate the tumor suppressing potential of FUCA1 is at least partially dependent on its regulation by p53. On the other hand, the inconsistent effects of FUCA1 on cancer cell viability show that the mechanisms linking FUCA1 and cancer progression are as yet not fully understood. As such, further investigation on FUCA1, fucosylation and p53 would improve our understanding of cancer development from the aspect of protein glycosylation and could provide novel targets for cancer therapies.
7. Conclusions and Future Directions
Glycoconjugates are present in every cellular compartment and can play a critical role in determining the function of biomolecules. The enzymatic regulation of glycosylation and the generation of multiple glycoforms play an important role in many cellular mechanisms, including the development of cancer [1]. Glycosylation patterns often change when cells are subjected to stress [2]. Understanding these changes and their mechanistic consequences may explain the relationship between glycosylation and cancer development. These mechanisms include receptor–ligand binding, cell–cell adhesion, extracellular interaction and communication within the tumor microenvironment. There are a number of unexplored areas here. For instance, although a correlation between the expression of the p53 target FUCA1 and cancer has been reported, the precise mechanism that underlies this correlation is not well understood [7,116,125,126]. Understanding the mechanisms by which altered glycosylation can contribute to tumorigenesis, cell migration, metastasis, immune escape and the drug resistance of cancer cells may inform the development of cancer therapeutics that modulate glycosylation. In addition, investigation into cancer-promoting glycan structures and the responsible enzymes could enhance our understanding of the relationship between cancer development and glycosylation [1,2,24,37,39,52,60,159]. Moreover, utilizing bioinformatic strategies such as metabolomics, glycomics and glycoproteomics could unravel more biomarkers for cancer diagnosis and prognosis, as well as identify novel candidates for cancer drug development.
Author Contributions
D.H. wrote this manuscript. N.K. created figures and edited the paper. R.O. conceptualized and edited this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partly supported by a Grant-in-Aid for Scientific Research (B, #20H03523, C, #22K07181, Challenging Exploratory Research, #23K18382 to R.O.) from MEXT of Japan; P-Direct, P-Create and P-Promote from AMED of Japan (R.O.); research grants of the Okinaka Memorial Institute for Medical Research (R.O.), the Life Science Foundation of Japan (R.O.), Princess Takamatsu Cancer Research Fund (R.O.).
Acknowledgments
We thank Marc Lamphier for critical reading of the manuscript.
Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- Lemjabbar-Alaoui, H.; McKinney, A.; Yang, Y.W.; Tran, V.M.; Phillips, J.J. Glycosylation alterations in lung and brain cancer. Adv. Cancer Res. 2015, 126, 305–344. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Ten Hagen, K.G.; Fritz, T.A.; Tabak, L.A. All in the family: The UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2003, 13, 1R–16R. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.O.; Halmo, S.M.; Wells, L. Recent advancements in understanding mammalian O-mannosylation. Glycobiology 2017, 27, 806–819. [Google Scholar] [CrossRef]
- Webb, N.A.; Mulichak, A.M.; Lam, J.S.; Rocchetta, H.L.; Garavito, R.M. Crystal structure of a tetrameric GDP-D-mannose 4,6-dehydratase from a bacterial GDP-D-rhamnose biosynthetic pathway. Protein Sci. 2004, 13, 529–539. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. Glycolysis links p53 function with NF-κB signaling: Impact on cancer and aging process. J. Cell. Physiol. 2010, 224, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ezawa, I.; Sawai, Y.; Kawase, T.; Okabe, A.; Tsutsumi, S.; Ichikawa, H.; Kobayashi, Y.; Tashiro, F.; Namiki, H.; Kondo, T.; et al. Novel p53 target gene FUCA1 encodes a fucosidase and regulates growth and survival of cancer cells. Cancer Sci. 2016, 107, 734–745. [Google Scholar] [CrossRef]
- Gao, J.; McConnell, M.J.; Yu, B.; Li, J.; Balko, J.M.; Black, E.P.; Johnson, J.O.; Lloyd, M.C.; Altiok, S.; Haura, E.B. MUC1 is a downstream target of STAT3 and regulates lung cancer cell survival and invasion. Int. J. Oncol. 2009, 35, 337–345. [Google Scholar] [PubMed]
- Raina, D.; Kosugi, M.; Ahmad, R.; Panchamoorthy, G.; Rajabi, H.; Alam, M.; Shimamura, T.; Shapiro, G.I.; Supko, J.; Kharbanda, S.; et al. Dependence on the MUC1-C oncoprotein in non-small cell lung cancer cells. Mol. Cancer Ther. 2011, 10, 806–816. [Google Scholar] [CrossRef]
- Ghosh, S. Sialic acids and sialoglycans in endocrinal disorders. In Sialic Acids and Sialoglycoconjugates in the Biology of Life, Health and Disease; Ghosh, S., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 247–268. [Google Scholar]
- Kudelka, M.R.; Ju, T.; Heimburg-Molinaro, J.; Cummings, R.D. Simple sugars to complex disease—Mucin-type O-glycans in cancer. Adv. Cancer Res. 2015, 126, 53–135. [Google Scholar] [CrossRef]
- Cao, Y.; Blohm, D.; Ghadimi, B.M.; Stosiek, P.; Xing, P.X.; Karsten, U. Mucins (MUC1 and MUC3) of gastrointestinal and breast epithelia reveal different and heterogeneous tumor-associated aberrations in glycosylation. J. Histochem. Cytochem. 1997, 45, 1547–1557. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.G.; Dahmus, M.E.; Hart, G.W. RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J. Biol. Chem. 1993, 268, 10416–10424. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.A.; Burlingame, A.L.; Myers, S.A. Human RNA Polymerase II Promoter Recruitment in Vitro Is Regulated by O-Linked N-Acetylglucosaminyltransferase (OGT). J. Biol. Chem. 2016, 291, 14056–14061. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kohler, J.J. Glycosylation of the nuclear pore. Traffic 2014, 15, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, T.W.; Cecioni, S.; Eskandari, R.; Zandberg, W.F.; Vocadlo, D.J. O-GlcNAc occurs cotranslationally to stabilize nascent polypeptide chains. Nat. Chem. Biol. 2015, 11, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Fardini, Y.; Perez-Cervera, Y.; Camoin, L.; Pagesy, P.; Lefebvre, T.; Issad, T. Regulatory O-GlcNAcylation sites on FoxO1 are yet to be identified. Biochem. Biophys. Res. Commun. 2015, 462, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell. Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Takahashi, M.; Gu, J.G.; Miyoshi, E.; Matsumoto, A.; Kitazume, S.; Taniguchi, N. Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 2008, 99, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Moremen, K.W.; Lewis, N.E.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2022; pp. 103–116. [Google Scholar]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Gout, S.; Morin, C.; Houle, F.; Huot, J. Death receptor-3, a new E-Selectin counter-receptor that confers migration and survival advantages to colon carcinoma cells by triggering p38 and ERK MAPK activation. Cancer Res. 2006, 66, 9117–9124. [Google Scholar] [CrossRef] [PubMed]
- Barthel, S.R.; Gavino, J.D.; Wiese, G.K.; Jaynes, J.M.; Siddiqui, J.; Dimitroff, C.J. Analysis of glycosyltransferase expression in metastatic prostate cancer cells capable of rolling activity on microvascular endothelial (E)-selectin. Glycobiology 2008, 18, 806–817. [Google Scholar] [CrossRef]
- Barthel, S.R.; Hays, D.L.; Yazawa, E.M.; Opperman, M.; Walley, K.C.; Nimrichter, L.; Burdick, M.M.; Gillard, B.M.; Moser, M.T.; Pantel, K.; et al. Definition of molecular determinants of prostate cancer cell bone extravasation. Cancer Res. 2013, 73, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Barthel, S.R.; Wiese, G.K.; Cho, J.; Opperman, M.J.; Hays, D.L.; Siddiqui, J.; Pienta, K.J.; Furie, B.; Dimitroff, C.J. Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc. Natl. Acad. Sci. USA 2009, 106, 19491–19496. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, S.; Kameyama, M.; Imaoka, S.; Furukawa, H.; Ishikawa, O.; Sasaki, Y.; Izumi, Y.; Irimura, T. Involvement of carbohydrate antigen sialyl Lewisx in colorectal cancer metastasis. Dis. Colon Rectum 1997, 40, 420–431. [Google Scholar] [CrossRef]
- St. Hill, C.A. Interactions between endothelial selectins and cancer cells regulate metastasis. Front. Biosci. Landmark 2011, 16, 3233–3251. [Google Scholar] [CrossRef]
- Witz, I.P. Tumor-microenvironment interactions: The selectin-selectin ligand axis in tumor-endothelium cross talk. Cancer Treat. Res. 2006, 130, 125–140. [Google Scholar] [PubMed]
- Hakomori, S. Glycosylation defining cancer malignancy: New wine in an old bottle. Proc. Natl. Acad. Sci. USA 2002, 99, 10231–10233. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.W.; Laferte, S.; Waghorne, C.; Breitman, M.L.; Kerbel, R.S. β1-6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science 1987, 236, 582–585. [Google Scholar] [CrossRef] [PubMed]
- West, C.M.; Hart, G.W. Nucleocytoplasmic Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 223–238. [Google Scholar]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell. Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Blanas, A.; Sahasrabudhe, N.M.; Rodriguez, E.; van Kooyk, Y.; van Vliet, S.J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 2018, 8, 39. [Google Scholar] [CrossRef]
- Shan, M.; Yang, D.; Dou, H.; Zhang, L. Fucosylation in cancer biology and its clinical applications. Prog. Mol. Biol. Transl. Sci. 2019, 162, 93–119. [Google Scholar] [CrossRef]
- Blanas, A.; Zaal, A.; van der Haar Avila, I.; Kempers, M.; Kruijssen, L.; de Kok, M.; Popovic, M.A.; van der Horst, J.C.; van Vliet, S.J. FUT9-Driven Programming of Colon Cancer Cells towards a Stem Cell-Like State. Cancers 2020, 12, 2580. [Google Scholar] [CrossRef]
- Mollicone, R.; Oriol, R. Fucosyltransferases 10, 11. GDP-Fucose N-Glycan Core α1,3-Fucosyltransferases (FUT10, FUT11). In Handbook of Glycosyltransferases and Related Genes, 2nd ed.; Taniguchi, N., Honke, K., Fukuda, M., Narimatsu, H., Yamaguchi, Y., Angata, T., Eds.; Springer: Tokyo, Japan, 2014; pp. 605–622. [Google Scholar]
- Wang, P.; Liu, X.; Yu, J.; Meng, Z.; Lv, Z.; Shang, C.; Geng, Q.; Wang, D.; Xue, D.; Li, L. Fucosyltransferases Regulated by Fusobacterium Nucleatum and Act as Novel Biomarkers in Colon Adenocarcinoma. J. Inflamm. Res. 2023, 16, 747–768. [Google Scholar] [CrossRef]
- Chen, C.I.; Keusch, J.J.; Klein, D.; Hess, D.; Hofsteenge, J.; Gut, H. Structure of human POFUT2: Insights into thrombospondin type 1 repeat fold and O-fucosylation. EMBO J. 2012, 31, 3183–3197. [Google Scholar] [CrossRef]
- Dobie, C.; Skropeta, D. Insights into the role of sialylation in cancer progression and metastasis. Br. J. Cancer 2021, 124, 76–90. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, K.; Qin, H.; Zhu, J.; Qin, Q.; Yu, Y.; Wang, H. GMDS knockdown impairs cell proliferation and survival in human lung adenocarcinoma. BMC Cancer 2018, 18, 600. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Comunale, M.A.; Rawat, S.; Casciano, J.C.; Lamontagne, J.; Herrera, H.; Ramanathan, A.; Betesh, L.; Wang, M.; Norton, P.; et al. Intrinsic hepatocyte dedifferentiation is accompanied by upregulation of mesenchymal markers, protein sialylation and core alpha 1,6 linked fucosylation. Sci. Rep. 2016, 6, 27965. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.; Sturla, L.; Bisso, A.; Benatti, U.; De Flora, A. Synthesis of GDP-L-fucose by the human FX protein. J. Biol. Chem. 1996, 271, 27274–27279. [Google Scholar] [CrossRef] [PubMed]
- Zipin, A.; Israeli-Amit, M.; Meshel, T.; Sagi-Assif, O.; Yron, I.; Lifshitz, V.; Bacharach, E.; Smorodinsky, N.I.; Many, A.; Czernilofsky, P.A.; et al. Tumor-microenvironment interactions: The fucose-generating FX enzyme controls adhesive properties of colorectal cancer cells. Cancer Res. 2004, 64, 6571–6578. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Dal Collo, G.; Bazzoni, R.; Krampera, M. Role of mesenchymal stromal cell-derived extracellular vesicles in tumour microenvironment. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Fong, G.H. Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension 2007, 49, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Ferrer, L.; Legler, K.; Milde-Langosch, K. Role of protein glycosylation in cancer metastasis. Semin. Cancer Biol. 2017, 44, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.B.; Costello, C.E.; Rahimi, N. Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis. Cells 2019, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Mendez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; Garcia-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Riemann, A.; Schneider, B.; Gundel, D.; Stock, C.; Gekle, M.; Thews, O. Acidosis Promotes Metastasis Formation by Enhancing Tumor Cell Motility. Adv. Exp. Med. Biol. 2016, 876, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim-Hashim, A.; Estrella, V. Acidosis and cancer: From mechanism to neutralization. Cancer Metastasis Rev. 2019, 38, 149–155. [Google Scholar] [CrossRef]
- Sanchez-Illana, A.; Pineiro-Ramos, J.D.; Ramos-Garcia, V.; Ten-Domenech, I.; Vento, M.; Kuligowski, J. Oxidative stress biomarkers in the preterm infant. Adv. Clin. Chem. 2021, 102, 127–189. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Shuster, S.; Neudecker, B.A.; Formby, B. Lactate stimulates fibroblast expression of hyaluronan and CD44: The Warburg effect revisited. Exp. Cell Res. 2002, 276, 24–31. [Google Scholar] [CrossRef]
- Salathia, S.; Gigliobianco, M.R.; Casadidio, C.; Di Martino, P.; Censi, R. Hyaluronic Acid-Based Nanosystems for CD44 Mediated Anti-Inflammatory and Antinociceptive Activity. Int. J. Mol. Sci. 2023, 24, 7286. [Google Scholar] [CrossRef]
- Graca, M.F.P.; Miguel, S.P.; Cabral, C.S.D.; Correia, I.J. Hyaluronic acid-Based wound dressings: A review. Carbohydr. Polym. 2020, 241, 116364. [Google Scholar] [CrossRef]
- Kawashima, H.; Hirose, M.; Hirose, J.; Nagakubo, D.; Plaas, A.H.; Miyasaka, M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 2000, 275, 35448–35456. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Kitagawa, H. Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta 2013, 1830, 4719–4733. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Scott, D.W.; Dunn, T.S.; Ballestas, M.E.; Litovsky, S.H.; Patel, R.P. Identification of a high-mannose ICAM-1 glycoform: Effects of ICAM-1 hypoglycosylation on monocyte adhesion and outside in signaling. Am. J. Physiol. Cell Physiol. 2013, 305, C228–C237. [Google Scholar] [CrossRef]
- Abe, Y.; Smith, C.W.; Katkin, J.P.; Thurmon, L.M.; Xu, X.; Mendoza, L.H.; Ballantyne, C.M. Endothelial α2,6-linked sialic acid inhibits VCAM-1-dependent adhesion under flow conditions. J. Immunol. 1999, 163, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Imamaki, R.; Kurimoto, A.; Ogawa, K.; Kato, M.; Yamaguchi, Y.; Tanaka, K.; Ishida, H.; Ando, H.; Kiso, M.; et al. Interaction of platelet endothelial cell adhesion molecule (PECAM) with α2,6-sialylated glycan regulates its cell surface residency and anti-apoptotic role. J. Biol. Chem. 2014, 289, 27604–27613. [Google Scholar] [CrossRef] [PubMed]
- Tiemeyer, M.; Swiedler, S.J.; Ishihara, M.; Moreland, M.; Schweingruber, H.; Hirtzer, P.; Brandley, B.K. Carbohydrate ligands for endothelial-leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1991, 88, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.L.; Nudelman, E.; Gaeta, F.C.; Perez, M.; Singhal, A.K.; Hakomori, S.; Paulson, J.C. ELAM-1 mediates cell adhesion by recognition of a carbohydrate ligand, sialyl-Lex. Science 1990, 250, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Mehta-D’souza, P.; Klopocki, A.G.; Oganesyan, V.; Terzyan, S.; Mather, T.; Li, Z.; Panicker, S.R.; Zhu, C.; McEver, R.P. Glycan Bound to the Selectin Low Affinity State Engages Glu-88 to Stabilize the High Affinity State under Force. J. Biol. Chem. 2017, 292, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Truty, J.; Lawrence, R.; Johns, S.C.; Srinivasan, R.S.; Handel, T.M.; Fuster, M.M. A critical role for lymphatic endothelial heparan sulfate in lymph node metastasis. Mol. Cancer 2010, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Glavey, S.V.; Manier, S.; Natoni, A.; Sacco, A.; Moschetta, M.; Reagan, M.R.; Murillo, L.S.; Sahin, I.; Wu, P.; Mishima, Y.; et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 2014, 124, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, V.V.; Glinsky, G.V.; Rittenhouse-Olson, K.; Huflejt, M.E.; Glinskii, O.V.; Deutscher, S.L.; Quinn, T.P. The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res. 2001, 61, 4851–4857. [Google Scholar] [PubMed]
- Glinsky, V.V.; Huflejt, M.E.; Glinsky, G.V.; Deutscher, S.L.; Quinn, T.P. Effects of Thomsen-Friedenreich antigen-specific peptide P-30 on β-galactoside-mediated homotypic aggregation and adhesion to the endothelium of MDA-MB-435 human breast carcinoma cells. Cancer Res. 2000, 60, 2584–2588. [Google Scholar]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Tu, X.; Dong, H.; Boughey, J.C.; Goetz, M.P.; Weinshilboum, R.; Lou, Z.; et al. Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res. 2020, 30, 590–601. [Google Scholar] [CrossRef]
- Yu, L.G.; Andrews, N.; Zhao, Q.; McKean, D.; Williams, J.F.; Connor, L.J.; Gerasimenko, O.V.; Hilkens, J.; Hirabayashi, J.; Kasai, K.; et al. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. J. Biol. Chem. 2007, 282, 773–781. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, X.; Nash, G.B.; Stone, P.C.; Hilkens, J.; Rhodes, J.M.; Yu, L.G. Circulating galectin-3 promotes metastasis by modifying MUC1 localization on cancer cell surface. Cancer Res. 2009, 69, 6799–6806. [Google Scholar] [CrossRef]
- Birts, C.N.; Banerjee, A.; Darley, M.; Dunlop, C.R.; Nelson, S.; Nijjar, S.K.; Parker, R.; West, J.; Tavassoli, A.; Rose-Zerilli, M.J.J.; et al. p53 is regulated by aerobic glycolysis in cancer cells by the CtBP family of NADH-dependent transcriptional regulators. Sci. Signal. 2020, 13, eaau9529. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.H.; et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine 2016, 3, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Madan, E.; Gogna, R.; Bhatt, M.; Pati, U.; Kuppusamy, P.; Mahdi, A.A. Regulation of glucose metabolism by p53: Emerging new roles for the tumor suppressor. Oncotarget 2011, 2, 948–957. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of p53 enhances catalytic activity of IKKβ through O-linked β-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Hu, Z.; Ji, S.; Jin, F.; Jiang, K.; Li, C.; Zhao, P.; Tu, Z.; Chen, X.; Di, L.; et al. NFκB up-regulation of glucose transporter 3 is essential for hyperactive mammalian target of rapamycin-induced aerobic glycolysis and tumor growth. Cancer Lett. 2015, 359, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. Onco. Targets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Kim, J.E.; Nam, H.W.; Ju, J.W.; Kim, H.S.; Kim, Y.S.; Cho, J.W. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. 2006, 8, 1074–1083. [Google Scholar] [CrossRef]
- Lee, E.W.; Oh, W.; Song, H.P.; Kim, W.K. Phosphorylation of p53 at threonine 155 is required for Jab1-mediated nuclear export of p53. BMB Rep. 2017, 50, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.W.; Oh, W.; Song, J. Jab1 as a mediator of nuclear export and cytoplasmic degradation of p53. Mol. Cells 2006, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Schneikert, J.; Stiewe, T. Pro-metastatic p53 mutants control folding of N-glycoproteins. Cell Cycle 2017, 16, 591–592. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010, 143, 711–724. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.J.; Lowe, J.B. Fucose: Biosynthesis and biological function in mammals. Glycobiology 2003, 13, 41R–53R. [Google Scholar] [CrossRef]
- Luhn, K.; Wild, M.K.; Eckhardt, M.; Gerardy-Schahn, R.; Vestweber, D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat. Genet. 2001, 28, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Veillon, L.; Fakih, C.; Abou-El-Hassan, H.; Kobeissy, F.; Mechref, Y. Glycosylation Changes in Brain Cancer. ACS Chem. Neurosci. 2018, 9, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Noda, M.; Okayama, H.; Kofunato, Y.; Chida, S.; Saito, K.; Tada, T.; Ashizawa, M.; Nakajima, T.; Aoto, K.; Kikuchi, T.; et al. Prognostic role of FUT8 expression in relation to p53 status in stage II and III colorectal cancer. PLoS ONE 2018, 13, e0200315. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, E.; Moriwaki, K.; Nakagawa, T. Biological function of fucosylation in cancer biology. J. Biochem. 2008, 143, 725–729. [Google Scholar] [CrossRef]
- Chen, C.Y.; Jan, Y.H.; Juan, Y.H.; Yang, C.J.; Huang, M.S.; Yu, C.J.; Yang, P.C.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 630–635. [Google Scholar] [CrossRef]
- Nakagoe, T.; Sawai, T.; Tsuji, T.; Jibiki, M.; Nanashima, A.; Yamaguchi, H.; Kurosaki, N.; Yasutake, T.; Ayabe, H. Circulating sialyl Lewis(x), sialyl Lewis(a), and sialyl Tn antigens in colorectal cancer patients: Multivariate analysis of predictive factors for serum antigen levels. J. Gastroenterol. 2001, 36, 166–172. [Google Scholar] [CrossRef]
- Konno, A.; Hoshino, Y.; Terashima, S.; Motoki, R.; Kawaguchi, T. Carbohydrate expression profile of colorectal cancer cells is relevant to metastatic pattern and prognosis. Clin. Exp. Metastasis 2002, 19, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Otero-Estevez, O.; Martinez-Fernandez, M.; Vazquez-Iglesias, L.; Paez de la Cadena, M.; Rodriguez-Berrocal, F.J.; Martinez-Zorzano, V.S. Decreased expression of alpha-L-fucosidase gene FUCA1 in human colorectal tumors. Int. J. Mol. Sci. 2013, 14, 16986–16998. [Google Scholar] [CrossRef]
- Zhong, A.; Chen, T.; Xing, Y.; Pan, X.; Shi, M. FUCA2 Is a Prognostic Biomarker and Correlated With an Immunosuppressive Microenvironment in Pan-Cancer. Front. Immunol. 2021, 12, 758648. [Google Scholar] [CrossRef]
- Zhou, Y.; Fukuda, T.; Hang, Q.; Hou, S.; Isaji, T.; Kameyama, A.; Gu, J. Inhibition of fucosylation by 2-fluorofucose suppresses human liver cancer HepG2 cell proliferation and migration as well as tumor formation. Sci. Rep. 2017, 7, 11563. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.S.; Liu, S.; Liu, Y.J.; Liu, J.W.; Liu, T.J.; Wang, X.Q.; Yan, Q. Overexpression of fucosyltransferase IV promotes A431 cell proliferation through activating MAPK and PI3K/Akt signaling pathways. J. Cell. Physiol. 2010, 225, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.X.; Gao, W.; Cai, L. Fucosyltransferase VII promotes proliferation via the EGFR/AKT/mTOR pathway in A549 cells. Onco Targets Ther. 2017, 10, 3971–3978. [Google Scholar] [CrossRef]
- Liang, L.; Gao, C.; Li, Y.; Sun, M.; Xu, J.; Li, H.; Jia, L.; Zhao, Y. miR-125a-3p/FUT5-FUT6 axis mediates colorectal cancer cell proliferation, migration, invasion and pathological angiogenesis via PI3K-Akt pathway. Cell Death Dis. 2017, 8, e2968. [Google Scholar] [CrossRef]
- Guo, Q.; Guo, B.; Wang, Y.; Wu, J.; Jiang, W.; Zhao, S.; Qiao, S.; Wu, Y. Functional analysis of α1,3/4-fucosyltransferase VI in human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 417, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, B.; Hao, Y.; Qi, Y.; Zhu, L.; Li, F.; Liu, D.; Cong, J.; Zhang, S.; Iwamori, M. Lewis y antigen promotes the proliferation of ovarian carcinoma-derived RMG-I cells through the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer Res. 2009, 28, 154. [Google Scholar] [CrossRef]
- Liu, J.J.; Lin, B.; Hao, Y.Y.; Li, F.F.; Liu, D.W.; Qi, Y.; Zhu, L.C.; Zhang, S.L.; Iwamori, M. Lewis(y) antigen stimulates the growth of ovarian cancer cells via regulation of the epidermal growth factor receptor pathway. Oncol. Rep. 2010, 23, 833–841. [Google Scholar]
- Cheng, T.C.; Tu, S.H.; Chen, L.C.; Chen, M.Y.; Chen, W.Y.; Lin, Y.K.; Ho, C.T.; Lin, S.Y.; Wu, C.H.; Ho, Y.S. Down-regulation of α-L-fucosidase 1 expression confers inferior survival for triple-negative breast cancer patients by modulating the glycosylation status of the tumor cell surface. Oncotarget 2015, 6, 21283–21300. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Guo, Q.; Feng, Y.; Cheng, P.; Wu, A. Dual role of fucosidase in cancers and its clinical potential. J. Cancer 2022, 13, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Dong, H.T.; Zhao, T.; Yao, F.; Xu, Y.; Chen, B.; Wu, Y.; Jin, F.; Xing, P. Cancer-associated adipocytes release FUCA2 to promote aggressiveness in TNBC. Endocr. Relat. Cancer 2022, 29, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Stowell, S.R.; Hoffmeister, K.M.; Freeze, H.H.; Varki, A. Glycans in Systemic Physiology. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2022; pp. 549–554. [Google Scholar]
- Domin, A.; Zabek, T.; Kwiatkowska, A.; Szmatola, T.; Deregowska, A.; Lewinska, A.; Mazur, A.; Wnuk, M. The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns. Genes 2021, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, V.; Papagerakis, P.; Tirino, V.; Zheng, L.; Matossian, M.; Prince, M.E.; Paino, F.; Mele, L.; Papaccio, F.; Montella, R.; et al. Increased fucosylation has a pivotal role in invasive and metastatic properties of head and neck cancer stem cells. Oncotarget 2015, 6, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Awan, B.; Turkov, D.; Schumacher, C.; Jacobo, A.; McEnerney, A.; Ramsey, A.; Xu, G.; Park, D.; Kalomoiris, S.; Yao, W.; et al. FGF2 Induces Migration of Human Bone Marrow Stromal Cells by Increasing Core Fucosylations on N-Glycans of Integrins. Stem Cell Rep. 2018, 11, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, G.; Qiao, N.; Zhang, M.; Zope, M.; Perakis, A.A.; Sukhumalchandra, P.; Philips, A.V.; Garber, H.R.; Kerros, C.; St John, L.S.; et al. Fucosylation Enhances the Efficacy of Adoptively Transferred Antigen-Specific Cytotoxic T Lymphocytes. Clin. Cancer Res. 2019, 25, 2610–2620. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.B.; Jung, S.H.; Choi, H.J.; et al. β-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci. Rep. 2019, 9, 18440. [Google Scholar] [CrossRef]
- Yan, L.; Lin, B.; Gao, L.; Gao, S.; Liu, C.; Wang, C.; Wang, Y.; Zhang, S.; Iwamori, M. Lewis (y) antigen overexpression increases the expression of MMP-2 and MMP-9 and invasion of human ovarian cancer cells. Int. J. Mol. Sci. 2010, 11, 4441–4452. [Google Scholar] [CrossRef]
- Guo, J.; Li, X.; Tan, Z.; Lu, W.; Yang, G.; Guan, F. Alteration of N-glycans and expression of their related glycogenes in the epithelial-mesenchymal transition of HCV29 bladder epithelial cells. Molecules 2014, 19, 20073–20090. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.F.; Wu, M.Y.; Lin, Y.C.; Kannagi, R.; Yang, R.B. FUT8 promotes breast cancer cell invasiveness by remodeling TGF-β receptor core fucosylation. Breast Cancer Res. 2017, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Wagnerova, J.; Cervenakova, L.; Balabanov, R.; Zitron, I.; Dore-Duffy, P. Cytokine regulation of E-selectin in rat CNS microvascular endothelial cells: Differential response of CNS and non-CNS vessels. J. Neurol. Sci. 2002, 195, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Otero, D.C.; Takahashi, A.A.; Bradley, L.M. PSGL-1: A New Player in the Immune Checkpoint Landscape. Trends Immunol. 2017, 38, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Renkonen, J.; Tynninen, O.; Hayry, P.; Paavonen, T.; Renkonen, R. Glycosylation might provide endothelial zip codes for organ-specific leukocyte traffic into inflammatory sites. Am. J. Pathol. 2002, 161, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Rana, K.; Ponmudi, V.; King, M.R. Knockdown of fucosyltransferase III disrupts the adhesion of circulating cancer cells to E-selectin without affecting hematopoietic cell adhesion. Carbohydr. Res. 2010, 345, 2334–2342. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento, J.C.; Ferreira Sde, A.; Vasconcelos, J.L.; da Silva-Filho, J.L.; Barbosa, B.T.; Bezerra, M.F.; Rocha, C.R.; Beltrao, E.I. Fut3 role in breast invasive ductal carcinoma: Investigating its gene promoter and protein expression. Exp. Mol. Pathol. 2015, 99, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ma, X.; Yu, J. Exosomes and organ-specific metastasis. Mol. Ther. Methods Clin. Dev. 2021, 22, 133–147. [Google Scholar] [CrossRef]
- Islam, M.K.; Dhondt, B.; Syed, P.; Khan, M.; Gidwani, K.; Webber, J.; Hendrix, A.; Jenster, G.; Lamminen, T.; Bostrom, P.J.; et al. Integrins are enriched on aberrantly fucosylated tumour-derived urinary extracellular vesicles. J. Extracell. Biol. 2022, 1, e64. [Google Scholar] [CrossRef]
- Choi, H.; Ju, S.; Kang, K.; Seo, M.H.; Kim, J.M.; Miyoshi, E.; Yeo, M.K.; Park, S.Y. Terminal fucosylation of haptoglobin in cancer-derived exosomes during cholangiocarcinoma progression. Front. Oncol. 2023, 13, 1183442. [Google Scholar] [CrossRef]
- Zhuang, W.; Liu, C.; Hong, Y.; Zheng, Y.; Huang, M.; Tang, H.; Zhao, L.; Huang, Z.; Tu, M.; Yu, L.; et al. Tumor-suppressive miR-4732-3p is sorted into fucosylated exosome by hnRNPK to avoid the inhibition of lung cancer progression. J. Exp. Clin. Cancer Res. 2024, 43, 123. [Google Scholar] [CrossRef]
- Higai, K.; Ichikawa, A.; Matsumoto, K. Binding of sialyl Lewis X antigen to lectin-like receptors on NK cells induces cytotoxicity and tyrosine phosphorylation of a 17-kDa protein. Biochim. Biophys. Acta 2006, 1760, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Noda, K.; Furukawa, Y.; Ohshima, K.; Uchiyama, A.; Nakagawa, T.; Taniguchi, N.; Daigo, Y.; Nakamura, Y.; Hayashi, N.; et al. Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology 2009, 137, 188–198.e2. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Shinzaki, S.; Miyoshi, E. GDP-mannose-4,6-dehydratase (GMDS) deficiency renders colon cancer cells resistant to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor- and CD95-mediated apoptosis by inhibiting complex II formation. J. Biol. Chem. 2011, 286, 43123–43133. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S.; Igarashi, Y. Glycosphingolipids and sphingolipids closely associated with or causing apoptosis. Acta Histochem. Cytochem. 1995, 28, 72–88. [Google Scholar] [CrossRef][Green Version]
- Wu, J.; Qin, H.; Li, T.; Cheng, K.; Dong, J.; Tian, M.; Chai, N.; Guo, H.; Li, J.; You, X.; et al. Characterization of site-specific glycosylation of secreted proteins associated with multi-drug resistance of gastric cancer. Oncotarget 2016, 7, 25315–25327. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, E.; Noda, K.; Ko, J.H.; Ekuni, A.; Kitada, T.; Uozumi, N.; Ikeda, Y.; Matsuura, N.; Sasaki, Y.; Hayashi, N.; et al. Overexpression of α1-6 fucosyltransferase in hepatoma cells suppresses intrahepatic metastasis after splenic injection in athymic mice. Cancer Res. 1999, 59, 2237–2243. [Google Scholar]
- Agrawal, P.; Fontanals-Cirera, B.; Sokolova, E.; Jacob, S.; Vaiana, C.A.; Argibay, D.; Davalos, V.; McDermott, M.; Nayak, S.; Darvishian, F.; et al. A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer Cell 2017, 31, 804–819.e7. [Google Scholar] [CrossRef]
- Cheng, L.; Luo, S.; Jin, C.; Ma, H.; Zhou, H.; Jia, L. FUT family mediates the multidrug resistance of human hepatocellular carcinoma via the PI3K/Akt signaling pathway. Cell Death Dis. 2013, 4, e923. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Liu, J.; Wang, X.; Yan, Q. Cyclophosphamide-induced apoptosis in A431 cells is inhibited by fucosyltransferase IV. J. Cell. Biochem. 2011, 112, 1376–1383. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Z.; Chen, M.; Lv, Y.; Tian, S.; Meng, F.; Zhang, Y.; Guo, X.; Chen, Y.; Yang, M.; et al. FDW028, a novel FUT8 inhibitor, impels lysosomal proteolysis of B7-H3 via chaperone-mediated autophagy pathway and exhibits potent efficacy against metastatic colorectal cancer. Cell Death Dis. 2023, 14, 495. [Google Scholar] [CrossRef] [PubMed]
- Baudot, A.D.; Crighton, D.; O’Prey, J.; Somers, J.; Sierra Gonzalez, P.; Ryan, K.M. p53 directly regulates the glycosidase FUCA1 to promote chemotherapy-induced cell death. Cell Cycle 2016, 15, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, N.; Ikeda, M.A.; Iotashino, U.; Grieco, M.; Vecchio, G. FUCA1 is induced by wild-type p53 and expressed at different levels in thyroid cancers depending on p53 status. Int. J. Oncol. 2017, 50, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.; Wang, J.; Lu, M.; Xue, Y.; Qiao, J.; Tao, L. Protein O-mannosylation across kingdoms and related diseases: From glycobiology to glycopathology. Biomed. Pharmacother. 2022, 148, 112685. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).