
Emerging Role of Hippo-YAP (Yes-Associated Protein)/TAZ (Transcriptional Coactivator with PDZ-Binding Motif) Pathway Dysregulation in Renal Cell Carcinoma Progression

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
Renal Cell Carcinoma Prevalence, Histological Subtypes, and Clinical Challenges
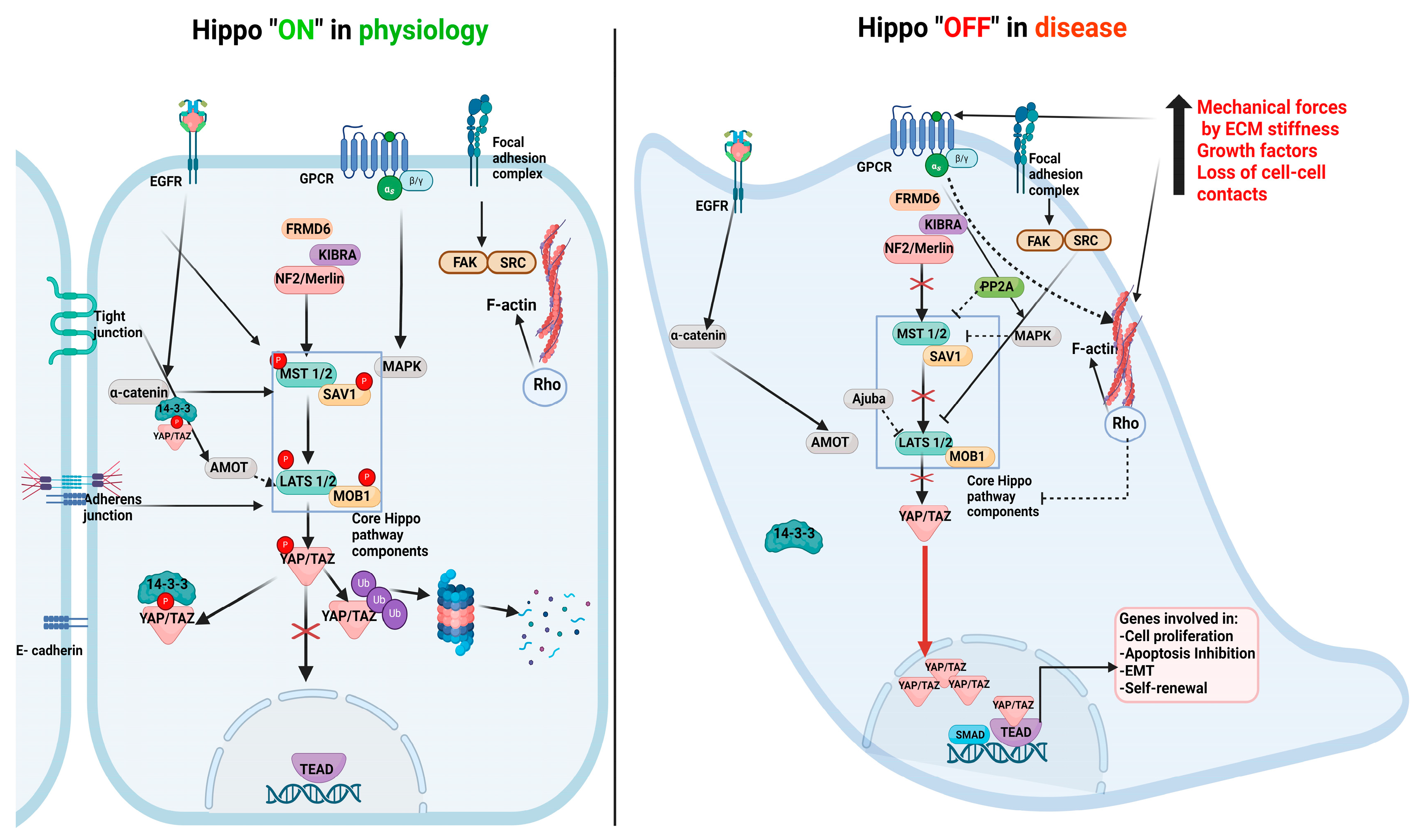
2. Hippo Pathway Components and Their Role in Physiology
3. Hippo Pathway Dysregulation in Renal Disease Progression
Direct Evidence for Core Hippo Pathway Inactivation in Human Kidney Cancer and Mouse Models of RCC Progression
4. Downstream Consequences of YAP Hyperactivation in RCC Progression
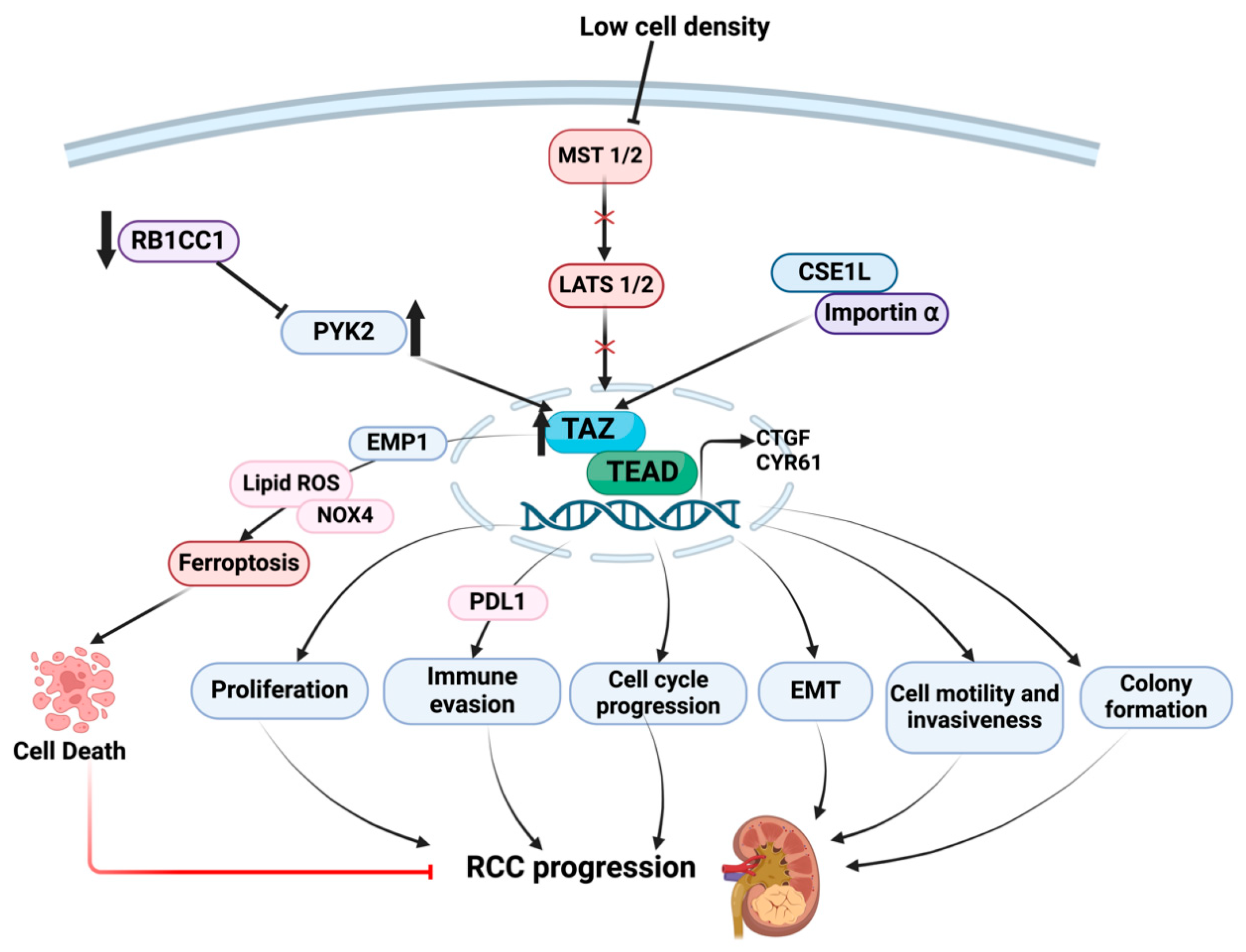
5. Downstream Consequences of TAZ Dysregulation in RCC Progression
6. Upstream Controls on YAP/TAZ Activation in RCC Progression
6.1. Neurofibromin 2 (NF2)/Merlin
6.2. Salvador Homolog 1 (SAV-1)
6.3. Hypoxia
6.4. Proteasome Activator (REGγ)
6.5. MER Proto-Oncogene Tyrosine Kinase (MERTK)
6.6. Syntaxin-Binding Protein 4 (STXBP4)
6.7. SH3-Binding Glutamate-Rich Protein-like 2 (SH3BGRL2)
6.8. Claudin-2
7. Conclusions, Therapeutic Relevance, and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Amot | Angiomotin |
| Ajuba | protein encoded by JUB gene |
| ccRCC | Clear cell Renal cell Carcinoma, |
| CTGF | CCN2 or Connective Tissue Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| ECM | Extracellular Matrix |
| EMT | Epithelial-Mesenchymal Transition |
| FMR1 | Fragile X Messenger Ribonucleoprotein 1 |
| GPCR | G-protein Coupled Receptors |
| KIBRA | Kidney and Brain Expressed Protein |
| HIF1α | Hypoxia Inducible Factor α |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| HK2 | Human Kidney 2 |
| Lats1/2 | Large tumor suppressor kinase 1 |
| MET | Mesenchymal Epithelial Transition |
| MOB1 | Mps One Binder 1 |
| MERTK | MER proto-oncogene tyrosine kinase |
| MST 1/2 | Serine Threonine like kinases |
| mTOR | Mammalian Target of Rapamycin |
| PYK2 | Protein Tyrosine Kinase 2 |
| RASSF | Ras association domain family |
| RB1CC1 | RB1-inducible coiled-coil protein 1 |
| RHO | Ras Homologous |
| ROS | Reactive Oxygen Species |
| SAV1 | Salvador Homolog 1 |
| STXBP4 | Syntaxin-Binding Protein 4 |
| SH3BGRL2 | SH3-Binding Glutamate-Rich Protein Like 2 |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TCGA | The Cancer Genome Atlas |
| TEAD | Transcriptional Enhancer Associated Domain |
| TGF-β | Transforming Growth Factor β |
| VEGF | Vascular Endothelial Growth Factor |
| VHL | Von Hippel–Lindau |
| Wnt | Wingless/Integrated |
| YAP | Yes-associated protein |
| ZO-2 | Zonula Occludens 2 |
References
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Miller, J.D.; Li, J.Z.; Russell, M.W.; Charbonneau, C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): A literature review. Cancer Treat. Rev. 2008, 34, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Decastro, G.J.; McKiernan, J.M. Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol. Clin. N. Am. 2008, 35, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kaelin, W.G. Targeting HIF2 in Clear Cell Renal Cell Carcinoma. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.A.; James, B.R.; Guan, Y.; Torry, D.S.; Wilber, A.; Griffith, T.S. Exploiting natural anti-tumor immunity for metastatic renal cell carcinoma. Hum. Vaccin. Immunother. 2015, 11, 1612–1620. [Google Scholar] [CrossRef]
- Allory, Y.; Culine, S.; de la Taille, A. Kidney cancer pathology in the new context of targeted therapy. Pathobiology 2011, 78, 90–98. [Google Scholar]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed]
- Genevet, A.; Tapon, N. The Hippo pathway and apico-basal cell polarity. Biochem. J. 2011, 436, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: Modulation, function and therapeutic approaches. Nat. Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, L.; Zhang, Y.; Li, W.; Li, J.; Wang, Y.; Meng, C.; Qin, J.; Zheng, Z.H.; Lan, H.Y.; et al. Tubule-Specific Mst1/2 Deficiency Induces CKD via YAP and Non-YAP Mechanisms. J. Am. Soc. Nephrol. 2020, 31, 946–961. [Google Scholar] [CrossRef]
- Happe, H.; van der Wal, A.M.; Leonhard, W.N.; Kunnen, S.J.; Breuning, M.H.; de Heer, E.; Peters, D.J. Altered Hippo signalling in polycystic kidney disease. J. Pathol. 2011, 224, 133–142. [Google Scholar] [CrossRef]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.S.; Meliambro, K.; Ray, J.; Campbell, K.N. Hippo signaling in the kidney: The good and the bad. Am. J. Physiol. Renal Physiol. 2016, 311, F241–F248. [Google Scholar] [CrossRef] [PubMed]
- Duong, N.X.; Le, M.K.; Kondo, T.; Mitsui, T. Heterogeneity of Hippo signalling activity in different histopathologic subtypes of renal cell carcinoma. J. Cell. Mol. Med. 2023, 27, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Rybarczyk, A.; Klacz, J.; Wronska, A.; Matuszewski, M.; Kmiec, Z.; Wierzbicki, P.M. Overexpression of the YAP1 oncogene in clear cell renal cell carcinoma is associated with poor outcome. Oncol. Rep. 2017, 38, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Schnell, U.; Chaney, C.; Tong, B.; Pan, X.; Ye, J.; Mernaugh, G.; Cotton, J.L.; Margulis, V.; Mao, J.; et al. Deletion of Lats1/2 in adult kidney epithelia leads to renal cell carcinoma. J. Clin. Investig. 2021, 131, e144108. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Bratslavsky, G.; Linehan, W.M.; Srinivasan, R. Sarcomatoid renal cell carcinoma: A comprehensive review of the biology and current treatment strategies. Oncologist 2012, 17, 46–54. [Google Scholar] [CrossRef]
- Godlewski, J.; Kiezun, J.; Krazinski, B.E.; Kozielec, Z.; Wierzbicki, P.M.; Kmiec, Z. The Immunoexpression of YAP1 and LATS1 Proteins in Clear Cell Renal Cell Carcinoma: Impact on Patients’ Survival. Biomed. Res. Int. 2018, 2018, 2653623. [Google Scholar] [CrossRef]
- Li, Z.; Su, P.; Yu, M.; Zhang, X.; Xu, Y.; Jia, T.; Yang, P.; Zhang, C.; Sun, Y.; Li, X.; et al. YAP represses the TEAD-NF-κB complex and inhibits the growth of clear cell renal cell carcinoma. Sci. Signal. 2024, 17, eadk0231. [Google Scholar] [CrossRef] [PubMed]
- Schutte, U.; Bisht, S.; Heukamp, L.C.; Kebschull, M.; Florin, A.; Haarmann, J.; Hoffmann, P.; Bendas, G.; Buettner, R.; Brossart, P.; et al. Hippo signaling mediates proliferation, invasiveness, and metastatic potential of clear cell renal cell carcinoma. Transl. Oncol. 2014, 7, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-J.; Zhao, X.-M.; Wang, D.-L.; Chen, K.-H.; Sheng, X.; Li, W.-B.; Li, M.-C.; Liu, W.-J.; He, J. YAP is overexpressed in clear cell renal cell carcinoma and its knockdown reduces cell proliferation and induces cell cycle arrest and apoptosis. Oncol. Rep. 2014, 32, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Li, S.; Luo, C.; Zhang, X.; Shen, Y.; Sui, Y.X.; Wang, F.; Wang, X.; Yang, J.; Liu, P.; et al. Angiomotin promotes renal epithelial and carcinoma cell proliferation by retaining the nuclear YAP. Oncotarget 2016, 7, 12393–12403. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.; Jiang, Y.; Zhang, X.; Liu, M.; Wang, S.; Liu, S.; Liang, H.; Liu, C. YAP1 activation promotes epithelial-mesenchymal transition and cell survival of renal cell carcinoma cells under shear stress. Carcinogenesis 2022, 43, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, H.; Chong, Y.; Guan, B.; Guo, P. YAP Promotes VEGFA Expression and Tumor Angiogenesis Though Gli2 in Human Renal Cell Carcinoma. Arch. Med. Res. 2019, 50, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Duan, Y.; Lu, X.; Chen, L.; Zhang, W.; Wang, H.; Hu, R.; Liu, S. RB1CC1 functions as a tumor-suppressing gene in renal cell carcinoma via suppression of PYK2 activity and disruption of TAZ-mediated PDL1 transcription activation. Cancer Immunol. Immunother. 2021, 70, 3261–3275. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Bao, L.; Song, Z.; Wang, K.; Cao, Q.; Tong, J.; Cheng, G.; Xu, T.; Chen, X.; Liu, D.; et al. High expression of TAZ serves as a novel prognostic biomarker and drives cancer progression in renal cancer. Exp. Cell Res. 2019, 376, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef]
- Nagashima, S.; Maruyama, J.; Honda, K.; Kondoh, Y.; Osada, H.; Nawa, M.; Nakahama, K.I.; Ishigami-Yuasa, M.; Kagechika, H.; Sugimura, H.; et al. CSE1L promotes nuclear accumulation of transcriptional coactivator TAZ and enhances invasiveness of human cancer cells. J. Biol. Chem. 2021, 297, 100803. [Google Scholar] [CrossRef]
- Philips, G.K.; Atkins, M.B. New agents and new targets for renal cell carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e222–e227. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.B.; Xu, J.; Skanderup, A.J.; Dong, Y.; Brannon, A.R.; Wang, L.; Won, H.H.; Wang, P.I.; Nanjangud, G.J.; Jungbluth, A.A.; et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat. Commun. 2016, 7, 13131. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Reuter, V.E.; Eble, J.N.; Vlatkovic, L.; Yaskiv, O.; Swanson, D.; Dickson, B.C.; Antonescu, C.R.; Matoso, A.; Gagan, J.; et al. Biphasic Hyalinizing Psammomatous Renal Cell Carcinoma (BHP RCC): A Distinctive Neoplasm Associated with Somatic NF2 Mutations. Am. J. Surg. Pathol. 2020, 44, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bai, H.; David, K.K.; Dong, J.; Zheng, Y.; Cai, J.; Giovannini, M.; Liu, P.; Anders, R.A.; Pan, D. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 2010, 19, 27–38. [Google Scholar] [CrossRef]
- Morris, Z.S.; McClatchey, A.I. Aberrant epithelial morphology and persistent epidermal growth factor receptor signaling in a mouse model of renal carcinoma. Proc. Natl. Acad. Sci. USA 2009, 106, 9767–9772. [Google Scholar] [CrossRef]
- White, S.M.; Avantaggiati, M.L.; Nemazanyy, I.; Di Poto, C.; Yang, Y.; Pende, M.; Gibney, G.T.; Ressom, H.W.; Field, J.; Atkins, M.B.; et al. YAP/TAZ Inhibition Induces Metabolic and Signaling Rewiring Resulting in Targetable Vulnerabilities in NF2-Deficient Tumor Cells. Dev. Cell 2019, 49, 425–443.e9. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Vats, P.; Cieslik, M.; Cao, X.; Su, F.; Shukla, S.; Udager, A.M.; Wang, R.; Pan, J.; Kasaian, K.; et al. Biallelic Alteration and Dysregulation of the Hippo Pathway in Mucinous Tubular and Spindle Cell Carcinoma of the Kidney. Cancer Discov. 2016, 6, 1258–1266. [Google Scholar] [CrossRef]
- Harvey, K.; Tapon, N. The Salvador-Warts-Hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef]
- Matsuura, K.; Nakada, C.; Mashio, M.; Narimatsu, T.; Yoshimoto, T.; Tanigawa, M.; Tsukamoto, Y.; Hijiya, N.; Takeuchi, I.; Nomura, T.; et al. Downregulation of SAV1 plays a role in pathogenesis of high-grade clear cell renal cell carcinoma. BMC Cancer 2011, 11, 523. [Google Scholar] [CrossRef] [PubMed]
- Bora-Singhal, N.; Saha, B.; Mohankumar, D.; Padmanabhan, J.; Coppola, D.; Chellappan, S. A Novel PHD2/VHL-mediated Regulation of YAP1 Contributes to VEGF Expression and Angiogenesis. Cancer Res. Commun. 2022, 2, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Q.; Wang, L.; Chen, H.; Gao, X.; Gong, D.; Ma, J.; Kubra, S.; Yao, X.; Li, X.; et al. REGgamma deficiency suppresses tumor progression via stabilizing CK1epsilon in renal cell carcinoma. Cell Death Dis. 2018, 9, 627. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, Y.; Leung, J.Y.; Fan, C.; Popov, K.I.; Su, S.; Qian, J.; Wang, X.; Holtzhausen, A.; Ubil, E.; et al. MERTK mediated novel site Akt phosphorylation alleviates SAV1 suppression. Nat. Commun. 2019, 10, 1515. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.E.; Duong, V.T.; Han, H.; Ta, A.P.; Chen, Y.; Zhao, S.; Yang, B.; Seo, G.; Chuc, K.; Oh, S.; et al. Elucidation of WW domain ligand binding specificities in the Hippo pathway reveals STXBP4 as YAP inhibitor. EMBO J. 2020, 39, e102406. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Li, W.; Xu, A.; Shi, H.; Wang, K.; Yang, H.; Wang, R.; Peng, B. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. eBioMedicine 2020, 51, 102596. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Ahmad, R.; Giannico, G.A.; Zent, R.; Talmon, G.A.; Harris, R.C.; Clark, P.E.; Lokeshwar, V.; Dhawan, P.; Singh, A.B. Claudin-2 inhibits renal clear cell carcinoma progression by inhibiting YAP-activation. J. Exp. Clin. Cancer Res. 2021, 40, 77. [Google Scholar] [CrossRef] [PubMed]
- Nyimanu, D.; Behm, C.; Choudhury, S.; Yu, A.S.L. The role of claudin-2 in kidney function and dysfunction. Biochem. Soc. Trans. 2023, 51, 1437–1445. [Google Scholar] [CrossRef]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Sancisi, V. YAP and TAZ Are Not Identical Twins. Trends Biochem. Sci. 2021, 46, 154–168. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondal, V.; Higgins, P.J.; Samarakoon, R. Emerging Role of Hippo-YAP (Yes-Associated Protein)/TAZ (Transcriptional Coactivator with PDZ-Binding Motif) Pathway Dysregulation in Renal Cell Carcinoma Progression. Cancers 2024, 16, 2758. https://doi.org/10.3390/cancers16152758
Mondal V, Higgins PJ, Samarakoon R. Emerging Role of Hippo-YAP (Yes-Associated Protein)/TAZ (Transcriptional Coactivator with PDZ-Binding Motif) Pathway Dysregulation in Renal Cell Carcinoma Progression. Cancers. 2024; 16(15):2758. https://doi.org/10.3390/cancers16152758
Chicago/Turabian StyleMondal, Varsha, Paul J. Higgins, and Rohan Samarakoon. 2024. "Emerging Role of Hippo-YAP (Yes-Associated Protein)/TAZ (Transcriptional Coactivator with PDZ-Binding Motif) Pathway Dysregulation in Renal Cell Carcinoma Progression" Cancers 16, no. 15: 2758. https://doi.org/10.3390/cancers16152758