

Sulfamoylated Estradiol Analogs Targeting the Actin and Microtubule Cytoskeletons Demonstrate Anti-Cancer Properties In Vitro and In Ovo

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Methods, and Chemicals
2.2. Microtubule Dynamic Instability Parameters: Time-Lapse Fluorescence Microscopy
2.3. Fluorescence Microscopy: Effect of the Molecules on the Actin Skeleton
2.4. Fluorescence Microscopy: Effect of the Molecules on the Microtubules
2.5. Reverse Phase Protein Array Analyses
2.6. Western Blots
2.7. Wound Healing Assays
2.8. MatrigelTM Invasion Assay
2.9. Anti-Angiogenic Properties of ESE-15-One and ESE-16: Human Umbilical Vein Endothelial Cell-Migration Test
2.10. HUVEC Invasion and Migration Using xCELLigence® Real-Time Cell Analysis
2.11. Anti-Tumor and Anti-Metastatic Properties of the Novel Compounds In Ovo
2.12. Statistical Analysis
3. Results
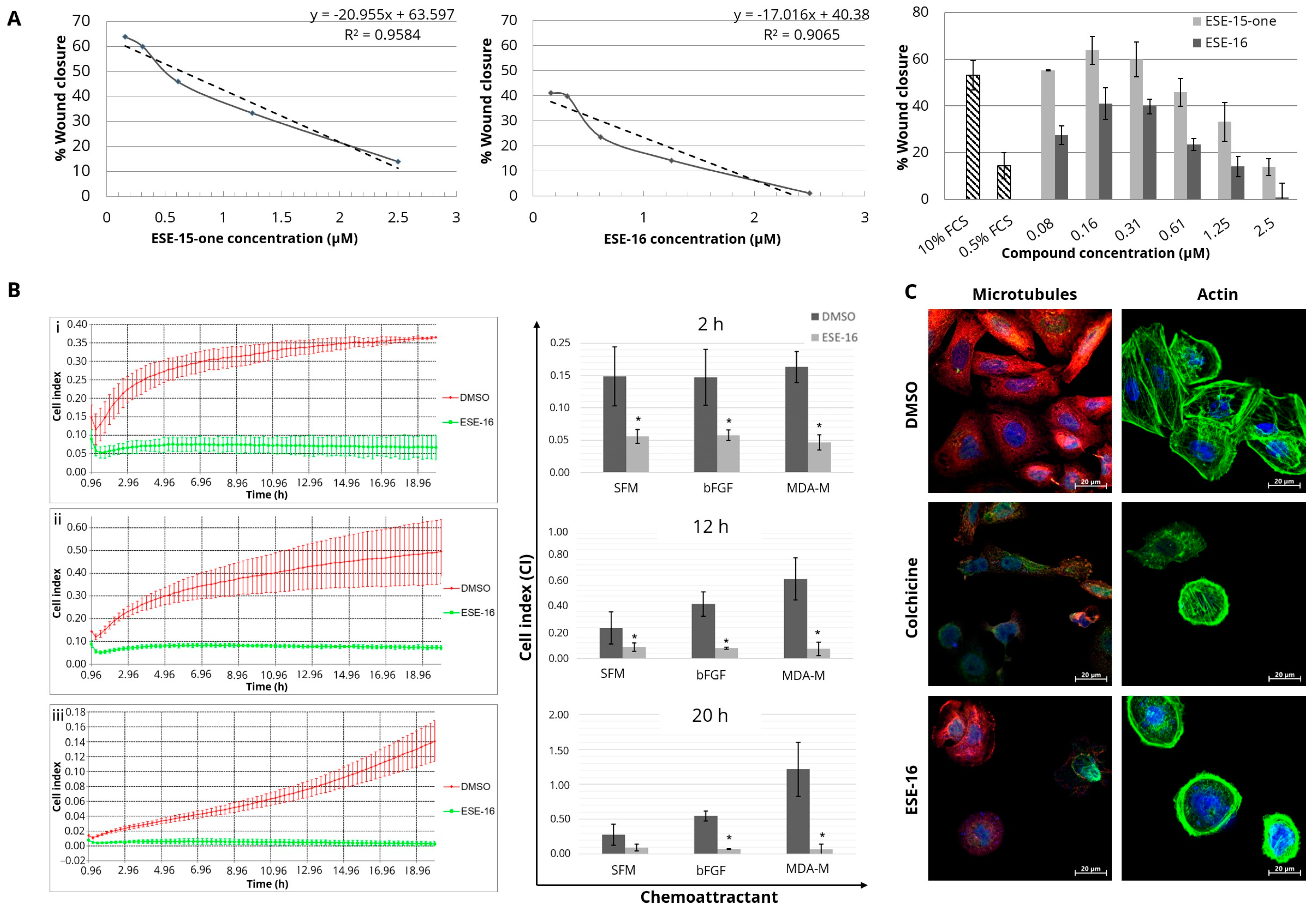
3.1. Temporal Analysis of the Effect of the Compounds on Cell Microtubules and Actin Filament
3.2. The Compounds Slow Down the Migration and the Invasion of Neoplastic Cells
3.3. The Effect of the Compounds on Endothelial Cell Migration
3.4. Together with Cofilin Phosphorylation, P-Ezrin/Radixin/Moesin May Regulate the Actin Response to the Molecules, Which Also Activate Anti-Angiogenic, Extra-Cellular, and Adhesion Pathways
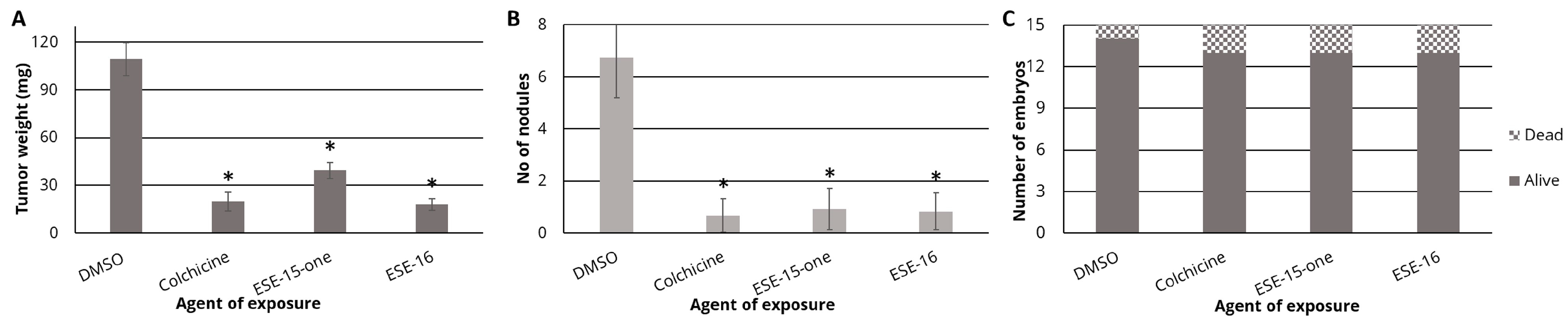
3.5. ESE-15-One and ESE-16 Exhibit Anti-Tumor and Anti-Metastatic Properties In Ovo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tischer, J.; Gergely, F. Anti-mitotic therapies in cancer. J. Cell Biol. 2018, 218, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 methoxyestradiol (Panzem®NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef]
- Mooberry, S.L. Mechanism of action of 2-methoxyestradiol: New developments. Drug Resist. Updates 2003, 6, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Tevaarwerk, A.J.; Holen, K.D.; Alberti, D.B.; Sidor, C.; Arnott, J.; Quon, C.; Wilding, G.; Liu, G. Phase I trial of 2-methoxyestradiol NanoCrystal dispersion in advanced solid malignancies. Clin. Cancer Res. 2009, 15, 1460–1465. [Google Scholar] [CrossRef]
- Van Zijl, C.; Lottering, M.L.; Steffens, F.; Joubert, A. In Vitro effects of 2-methoxyestradiol on MCF-12A and MCF-7 cell growth, morphology and mitotic spindle formation. Cell Biochem. Funct. 2008, 26, 632–642. [Google Scholar] [CrossRef]
- Verenich, S.; Gerk, P.M. Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges. Mol. Pharm. 2010, 7, 2030–2039. [Google Scholar] [CrossRef]
- Sweeney, C.; Liu, G.; Yiannoutsos, C.; Kolesar, J.; Horvath, D.; Staab, M.J.; Fife, K.; Armstrong, V.; Treston, A.; Sidor, C.; et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin. Cancer Res. 2005, 11, 6625–6633. [Google Scholar] [CrossRef]
- Stander, A.; Joubert, F.; Joubert, A. Docking, synthesis, and in vitro evaluation of antimitotic estrone analogs. Chem. Biol. Drug Des. 2011, 77, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Theron, A.; Prudent, R.; Nolte, E.; van den Bout, I.; Punchoo, R.; Marais, S.; du Toit, P.; Hlophe, Y.; van Papendorp, D.; Lafanechère, L.; et al. Novel in silico-designed estradiol analogues are cytotoxic to a multidrug-resistant cell line at nanomolar concentrations. Cancer Chemother. Pharmacol. 2015, 75, 431–437. [Google Scholar] [CrossRef]
- Theron, A.; Nolte, E.; Lafanechere, L.; Joubert, A. Molecular crosstalk between apoptosis and autophagy induced by a novel 2-methoxyestradiol analogue in cervical adenocarcinoma cells. Cancer Cell Int. 2013, 13, 87. [Google Scholar] [CrossRef]
- Mercier, A.E.; Prudent, R.; Pepper, M.S.; De Koning, L.; Nolte, E.; Peronne, L.; Nel, M.; Lafanechère, L.; Joubert, A.M. Characterization of signalling pathways that link apoptosis and autophagy to cell death induced by estrone Analogues which reversibly depolymerize microtubules. Molecules 2021, 26, 706. [Google Scholar] [CrossRef] [PubMed]
- Helena, J.; Joubert, A.; Mabeta, P.; Coetzee, M.; Lakier, R.; Mercier, A. Intracellular Signaling Responses Induced by Radiation within an In Vitro Bone Metastasis Model after Pre-Treatment with an Estrone Analogue. Cells 2021, 10, 2105. [Google Scholar] [CrossRef] [PubMed]
- Nolte, E.; Joubert, A.; Lakier, R.; van Rensburg, A.; Mercier, A. Exposure of breast and lung cancer cells to a novel estrone analog prior to radiation enhances Bcl-2-mediated cell death. Int. J. Mol. Sci. 2018, 19, 2887. [Google Scholar] [CrossRef]
- Nolte, E.M.; Joubert, A.M.; Lafanechère, L.; Mercier, A.E. Radiosensitization of Breast Cancer Cells with a 2-Methoxyestradiol Analogue Affects DNA Damage and Repair Signaling In Vitro. Int. J. Mol. Sci. 2023, 24, 3592. [Google Scholar] [CrossRef] [PubMed]
- Sherbet, G.V. Suppression of angiogenesis and tumour progression by combretastatin and derivatives. Cancer Lett. 2017, 403, 289–295. [Google Scholar] [CrossRef]
- Honore, S.; Braguer, D. Investigating microtubule dynamic instability using microtubule-targeting agents. Methods Mol. Biol. 2011, 777, 245–260. [Google Scholar] [CrossRef]
- Paturle-Lafanechere, L.; Manier, M.; Trigault, N.; Pirollet, F.; Mazarguil, H.; Job, D. Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J. Cell Sci. 1994, 107, 1529–1543. [Google Scholar] [CrossRef]
- Vassal, E.; Barette, C.; Fonrose, X.; Dupont, R.; Sans-Soleilhac, E.; Lafanechère, L. Miniaturization and validation of a sensitive multiparametric cell-based assay for the concomitant detection of microtubule-destabilizing and microtubule-stabilizing agents. J. Biomol. Screen. 2006, 11, 377–389. [Google Scholar] [CrossRef]
- Troncale, S.; Barbet, A.; Coulibaly, L.; Henry, E.; He, B.; Barillot, E.; Dubois, T.; Hupé, P.; de Koning, L. NormaCurve: A SuperCurve-based method that simultaneously quantifies and normalizes reverse phase protein array data. PLoS ONE 2012, 7, e38686. [Google Scholar] [CrossRef]
- Watanabe, N.; Okochi, E.; Mochizuki, M.; Sugimura, T.; Ushijima, T. The presence of single nucleotide instability in human breast cancer cell lines. Cancer Res. 2001, 61, 7739–7742. [Google Scholar] [PubMed]
- American Type Culture Collection (ATCC). Available online: https://www.atcc.org/products/htb-26#detailed-product-information (accessed on 15 February 2024).
- Morelli, M.P.; Tentler, J.J.; Kulikowski, G.N.; Tan, A.-C.; Bradshaw-Pierce, E.L.; Pitts, T.M.; Brown, A.M.; Nallapareddy, S.; Arcaroli, J.J.; Serkova, N.J.; et al. Preclinical activity of the rational combination of selumetinib (AZD6244) in combination with vorinostat in KRAS-mutant colorectal cancer models. Clin. Cancer Res. 2012, 18, 1051–1062. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P.; Pepper, M.S. Altered expression of platelet factor 4 and basic fibroblast growth factor correlates with the inhibition of tumor growth in mice. Biomed. Pharmacother. 2015, 69, 186–190. [Google Scholar] [CrossRef]
- Goodwin, A.M. In vitro assays of angiogenesis for assessment of angiogenic and anti-angiogenic agents. Microvasc. Res. 2007, 74, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Potapova, I.A.; Gaudette, G.R.; Brink, P.R.; Robinson, R.B.; Rosen, M.R.; Cohen, I.S.; Doronin, S.V. Mesenchymal stem cells support migration, extracellular matrix invasion, proliferation, and survival of endothelial cells in vitro. Stem Cells 2007, 25, 1761–1768. [Google Scholar] [CrossRef]
- Faridi, A.; Rudlowski, C.; Biesterfeld, S.; Schuh, S.; Rath, W.; Schröder, W. Long-term follow-up and prognostic significance of angiogenic basic fibroblast growth factor (bFGF) expression in patients with breast cancer. Pathol. Res. Pract. 2002, 198, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Payne, S.; Wang, F.; Claus, P.; Su, Z.; Groth, J.; Geradts, J.; de Ridder, G.; Alvarez, R.; Marcom, P.K.; et al. Nuclear basic fibroblast growth factor regulates triple-negative breast cancer chemo-resistance. Breast Cancer Res. 2015, 17, 91. [Google Scholar] [CrossRef] [PubMed]
- Lokman, N.A.; Elder, A.S.F.; Ricciardelli, C.; Oehler, M.K. Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int. J. Mol. Sci. 2012, 13, 9959–9970. [Google Scholar] [CrossRef]
- Zebda, N.; Bernard, O.; Bailly, M.; Welti, S.; Lawrence, D.S.; Condeelis, J.S. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J. Cell Biol. 2000, 151, 1119–1128. [Google Scholar] [CrossRef]
- Xiang, Y.; Zheng, K.; Ju, H.; Wang, S.; Pei, Y.; Ding, W.; Chen, Z.; Wang, Q.; Qiu, X.; Zhong, M.; et al. Cofilin 1-mediated biphasic F-actin dynamics of neuronal cells affect herpes simplex virus 1 infection and replication. J. Virol. 2012, 86, 8440–8451. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Takai, Y.; Nakamura, T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 1999, 147, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Keezer, S.M.; Ivie, S.E.; Krutzsch, H.C.; Tandle, A.; Libutti, S.K.; Roberts, D.D. Angiogenesis inhibitors target the endothelial cell cytoskeleton through altered regulation of heat shock protein 27 and cofilin. Cancer Res. 2003, 63, 6405–6412. [Google Scholar] [PubMed]
- van Haren, J.; Wittmann, T. Microtubule Plus End Dynamics − Do We Know How Microtubules Grow? Bioessays 2019, 41, 1800194. [Google Scholar] [CrossRef]
- Michaels, T.C.T.; Feng, S.; Liang, H.; Mahadevan, L. Mechanics and kinetics of dynamic instability. eLife 2020, 9, e54077. [Google Scholar] [CrossRef] [PubMed]
- Kamath, K.; Okouneva, T.; Larson, G.; Panda, D.; Wilson, L.; Jordan, M.A. 2-Methoxyestradiol suppresses microtubule dynamics and arrests mitosis without depolymerizing microtubules. Mol. Cancer Ther. 2006, 5, 2225–2233. [Google Scholar] [CrossRef]
- Cramer, L.P.; Siebert, M.; Mitchison, T.J. Identification of Novel Graded Polarity Actin Filament Bundles in Locomoting Heart Fibroblasts: Implications for the Generation of Motile Force. J. Cell Biol. 1997, 136, 1287–1305. [Google Scholar] [CrossRef]
- Matsudalra, P. Modular organization of actin crosslinking proteins. Trends Biochem. Sci. 1991, 16, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, S.; Mellor, H. Actin stress fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef]
- McHardy, L.M.; Sinotte, R.; Troussard, A.; Sheldon, C.; Church, J.; Williams, D.E.; Andersen, R.J.; Dedhar, S.; Roberge, M.; Roskelley, C.D. The tumor invasion inhibitor dihydromotuporamine C activates RHO, remodels stress fibers and focal adhesions, and stimulates sodium–proton exchange. Cancer Res. 2004, 64, 1468–1474. [Google Scholar] [CrossRef]
- Maddox, A.S.; Burridge, K. RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J. Cell Biol. 2003, 160, 255–265. [Google Scholar] [CrossRef]
- Teng, B.; Lukasz, A.; Schiffer, M. The ADF/Cofilin-pathway and actin dynamics in podocyte injury. Int. J. Cell Biol. 2012, 2012, 320531. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Leu, J.-D.; Lee, Y.-J. The actin depolymerizing factor (ADF)/cofilin signaling pathway and DNA damage responses in cancer. Int. J. Mol. Sci. 2015, 16, 4095–4120. [Google Scholar] [CrossRef] [PubMed]
- Kanellos, G.; Zhou, J.; Patel, H.; Ridgway, R.A.; Huels, D.; Gurniak, C.B.; Sandilands, E.; Carragher, N.O.; Sansom, O.J.; Witke, W.; et al. ADF and Cofilin1 Control Actin Stress Fibers, Nuclear Integrity, and Cell Survival. Cell Rep. 2015, 13, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell. Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Shibuya, A.; Nakamura, T. Activation of LIM kinases by myotonic dystrophy kinase-related Cdc42-binding kinase α. J. Biol. Chem. 2001, 276, 23092–23096. [Google Scholar] [CrossRef]
- Nagata-Ohashi, K.; Ohta, Y.; Goto, K.; Chiba, S.; Mori, R.; Nishita, M.; Ohashi, K.; Kousaka, K.; Iwamatsu, A.; Niwa, R.; et al. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J. Cell Biol. 2004, 165, 465–471. [Google Scholar] [CrossRef]
- Akhshi, T.K.; Wernike, D.; Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton 2014, 71, 1–23. [Google Scholar] [CrossRef]
- Roig, J.; Tuazon, P.T.; Zipfel, P.A.; Pendergast, A.M.; Traugh, J.A. Functional interaction between c-Abl and the p21-activated protein kinase γ-PAK. Proc. Natl. Acad. Sci. USA 2000, 97, 14346–14351. [Google Scholar] [CrossRef]
- Valerius, N.H.; Stendahl, O.; Hartwig, J.H.; Stossel, T.P. Distribution of actin-binding protein and myosin in polymorphonuclear leukocytes during locomotion and phagocytosis. Cell 1981, 24, 195–202. [Google Scholar] [CrossRef]
- Rubino, S.; Fighetti, M.; Unger, E.; Cappuccinelli, P. Location of actin, myosin, and microtubular structures during directed locomotion of Dictyostelium amebae. J. Cell Biol. 1984, 98, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers—Assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- McHardy, L.M.; Warabi, K.; Andersen, R.J.; Roskelley, C.D.; Roberge, M. Strongylophorine-26, a Rho-dependent inhibitor of tumor cell invasion that reduces actin stress fibers and induces nonpolarized lamellipodial extensions. Mol. Cancer Ther. 2005, 4, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.S.; Gardel, M.; Ma, X.; Adelstein, R.S.; Waterman, C.M. Local cortical tension by myosin II guides 3D endothelial cell branching. Curr. Biol. 2009, 19, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Prager-Khoutorsky, M.; Lichtenstein, A.; Krishnan, R.; Rajendran, K.; Mayo, A.; Kam, Z.; Geiger, B.; Bershadsky, A.D. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat. Cell Biol. 2011, 13, 1457–1465. [Google Scholar] [CrossRef]
- Jansen, K.A.; Donato, D.M.; Balcioglu, H.E.; Schmidt, T.; Danen, E.H.J.; Koenderink, G.H. A guide to mechanobiology: Where biology and physics meet. BBA Mol. Cell Res. 2015, 1853, 3043–3052. [Google Scholar] [CrossRef]
- Curran, S.; Murray, G.I. Matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 1999, 189, 300–308. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother. Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur. J. Cell Biol. 1997, 74, 111–122. [Google Scholar] [PubMed]
- Dubey, R.K.; Jackson, E.K. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol. Metab. 2009, 20, 374–379. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr. Opin. Oncol. 2003, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- LaVallee, T.M.; Burke, P.A.; Swartz, G.M.; Hamel, E.; Agoston, G.E.; Shah, J.; Suwandi, L.; Hanson, A.D.; Fogler, W.E.; Sidor, C.F.; et al. Significant antitumor activity in vivo following treatment with the microtubule agent ENMD-1198. Mol. Cancer Ther. 2008, 7, 1472–1482. [Google Scholar] [CrossRef]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Katakowski, M.; Chen, X.; Wang, L.; Lu, D.; Lu, M.; Gautam, S.C.; Chopp, M. Intravenous bone marrow stromal cell therapy reduces apoptosis and promotes endogenous cell proliferation after stroke in female rat. J. Neurosci. Res. 2003, 73, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, T.; Stabile, E.; Burnett, M.S.; Lee, C.W.; Barr, S.; Fuchs, S.; Epstein, S.E. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ. Res. 2004, 94, 678–685. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Dark, G.G.; Hill, S.A.; Prise, V.E.; Tozer, G.M.; Pettit, G.R.; Chaplin, D.J. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997, 57, 1829–1834. [Google Scholar] [PubMed]
- Bryan, B.A.; D’Amore, P.A. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell. Mol. Life Sci. 2007, 64, 2053–2065. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nishita, M.; Mishima, T.; Ohashi, K.; Mizuno, K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006, 25, 713–726. [Google Scholar] [CrossRef]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, W. The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front. Endocrinol. 2015, 6, 159. [Google Scholar] [CrossRef]
- Kallergi, G.; Markomanolaki, H.; Giannoukaraki, V.; Papadaki, M.A.; Strati, A.; Lianidou, E.S.; Georgoulias, V.; Mavroudis, D.; Agelaki, S. Hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in circulating tumor cells of breast cancer patients. Breast Cancer Res. 2009, 11, R84. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.W.; Mayo, L.D.; Dunbar, J.D.; Kessler, K.M.; Ozes, O.N.; Warren, R.S.; Donner, D.B. VRAP is an adaptor protein that binds KDR, a receptor for vascular endothelial cell growth factor. J. Biol. Chem. 2000, 275, 6059–6062. [Google Scholar] [CrossRef] [PubMed]
- Le Boeuf, F.; Houle, F.; Huot, J. Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J. Biol. Chem. 2004, 279, 39175–39185. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Houle, F.; Huot, J. Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J. Biol. Chem. 2006, 281, 34009–34020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | DMSO (0.05% v/v) | Colchicine 0.05 µM | ESE-16 0.125 μM (¼ IG50) | ESE-16 0.25 μM (½ IG50) | ESE-16 0.5 μM (IG50) | ESE-15-One 0.045 μM (¼ IG50) | ESE-15-One 0.09 μM (½ IG50) | ESE-15-One 0.186 μM (IG50) |
|---|---|---|---|---|---|---|---|---|
| % Time spent growing | 80.44 | 58.4 * | 62.08 * | 29.18 *** | TD | 58.92 ** | 37.51 *** | 25.46 *** |
| % Time spent in pause | 19.56 | 41.60 * | 37.32 * | 70.82 *** | TD | 41.08 ** | 62.03 *** | 74.54 *** |
| Growth rate (µm/min ± SEM) | 16.18 ± 0.42 | 14.61 ± 0.36 * | 14.04 ± 0.51 * | 9.8 ± 0.64 *** | TD | 13.74 ± 0.52 * | 11.91 ± 0.3 *** | 10.298 ± 0.51 ** |
| Catastrophe frequency (µm−1 ± SEM) | 0.12 ± 0.016 | 0.29 ± 0.016 *** | 0.21 ± 0.01 ** | 0.89 ± 0.06 *** | TD | 0.20 ± 0.02 * | 0.28 ± 0.03 * | 0.89 ± 0.13 ** |
| Catastrophe frequency (min−1 ± SEM) | 1.56 ± 0.21 | 2.38 ± 0.24 | 1.65 ± 0.21 | 2.52 ± 0.3 * | TD | 1.53 ± 0.11 | 1.2 ± 0.13 | 1.85 ± 0.14 |
| Antibody Name | Pathway(s) | HeLa | MDA-MB-231 | ||
| ESE-15-One | ESE-16 | ESE-15-One | ESE-16 | ||
| MAPK/Erk signaling, cytoskeleton | |||||
| P-Ezrin (T567)/radixin (T564)/moesin (T558) | PI3K pathway, MAPK/Erk signaling, cytoskeleton | ↑↑↑ over 24 h | ↑↑↑ over 24 h | ↑↑ over 24 h | ↑↑ over 24 h |
| P-Shc (Y239/240) | Tyrosine kinase & MAPK/Erk signaling | ↓ 6–24 h | ↓ 6–24 h | - | - |
| P-p38 MAPK (T180/Y182) | MAPK/Erk signaling | ↑ 1–2 h | ↑ 1–2 h | - | ↑ 1–2 h |
| Angiogenesis, matrix metalloproteases, extra-cellular matrix | |||||
| TIMP2 | Angiogenesis, matrix metalloproteases, extra-cellular matrix | ↑↑ over 24 h | ↑↑ over 24 h | ↑↑ over 24 h | ↑↑ over 24 h |
| P-VEGF R2 (Y1214) | Angiogenesis | ↑↑ over 24 h | ↑↑ over 24 h | ↑↑ over 24 h | ↑↑ over24 h |
| Tyrosine kinase signaling, adhesion | |||||
| P-Fyn (Y528)/Src (Y530) | Tyrosine kinase signaling, SRC family | ↓ 6–24 h | ↓ over 12–24 h | - | - |
| P-FAK (Y861) | Tyrosine kinase signaling, Adhesion | ↑ 1–2 h | ↑ 1–24 h | ↑ 1–2 h | ↑ 1–24 h |
| Proteins apparently not affected by compound exposure | |||||
| E-cadherin, ROCK-I/ROK beta, P-Met, TYMP | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercier, A.E.; Joubert, A.M.; Prudent, R.; Viallet, J.; Desroches-Castan, A.; De Koning, L.; Mabeta, P.; Helena, J.; Pepper, M.S.; Lafanechère, L. Sulfamoylated Estradiol Analogs Targeting the Actin and Microtubule Cytoskeletons Demonstrate Anti-Cancer Properties In Vitro and In Ovo. Cancers 2024, 16, 2941. https://doi.org/10.3390/cancers16172941
Mercier AE, Joubert AM, Prudent R, Viallet J, Desroches-Castan A, De Koning L, Mabeta P, Helena J, Pepper MS, Lafanechère L. Sulfamoylated Estradiol Analogs Targeting the Actin and Microtubule Cytoskeletons Demonstrate Anti-Cancer Properties In Vitro and In Ovo. Cancers. 2024; 16(17):2941. https://doi.org/10.3390/cancers16172941
Chicago/Turabian StyleMercier, Anne Elisabeth, Anna Margaretha Joubert, Renaud Prudent, Jean Viallet, Agnes Desroches-Castan, Leanne De Koning, Peace Mabeta, Jolene Helena, Michael Sean Pepper, and Laurence Lafanechère. 2024. "Sulfamoylated Estradiol Analogs Targeting the Actin and Microtubule Cytoskeletons Demonstrate Anti-Cancer Properties In Vitro and In Ovo" Cancers 16, no. 17: 2941. https://doi.org/10.3390/cancers16172941
APA StyleMercier, A. E., Joubert, A. M., Prudent, R., Viallet, J., Desroches-Castan, A., De Koning, L., Mabeta, P., Helena, J., Pepper, M. S., & Lafanechère, L. (2024). Sulfamoylated Estradiol Analogs Targeting the Actin and Microtubule Cytoskeletons Demonstrate Anti-Cancer Properties In Vitro and In Ovo. Cancers, 16(17), 2941. https://doi.org/10.3390/cancers16172941