
Resolution of Optimal Mitochondrial and Nuclear DNA Enrichment in Target-Panel Sequencing and Physiological Mitochondrial DNA Copy Number Estimation in Liver Cancer and Non-Liver Cancer Subjects
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Method
2.1. Sample Collection, DNA Extraction, Library Construction
2.2. Next-Generation Sequencing and Data Preprocessing
2.3. Data Processing and Sequencing Quality Estimation
3. Results
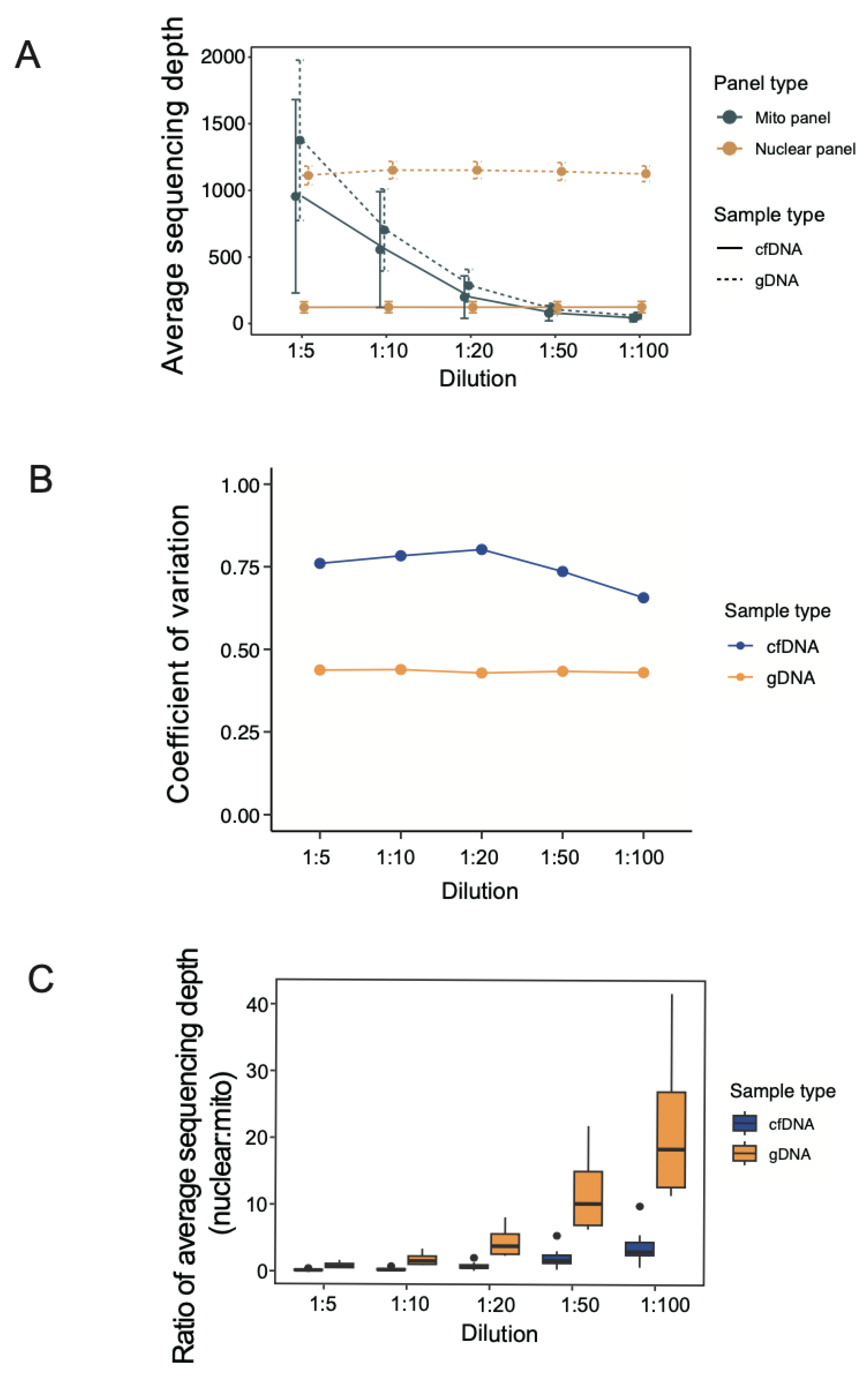
3.1. Sequencing Read Count under Different Dilutions of mtDNA Probes
3.2. Average Sequencing Depth under Different Dilutions of mtDNA Probes
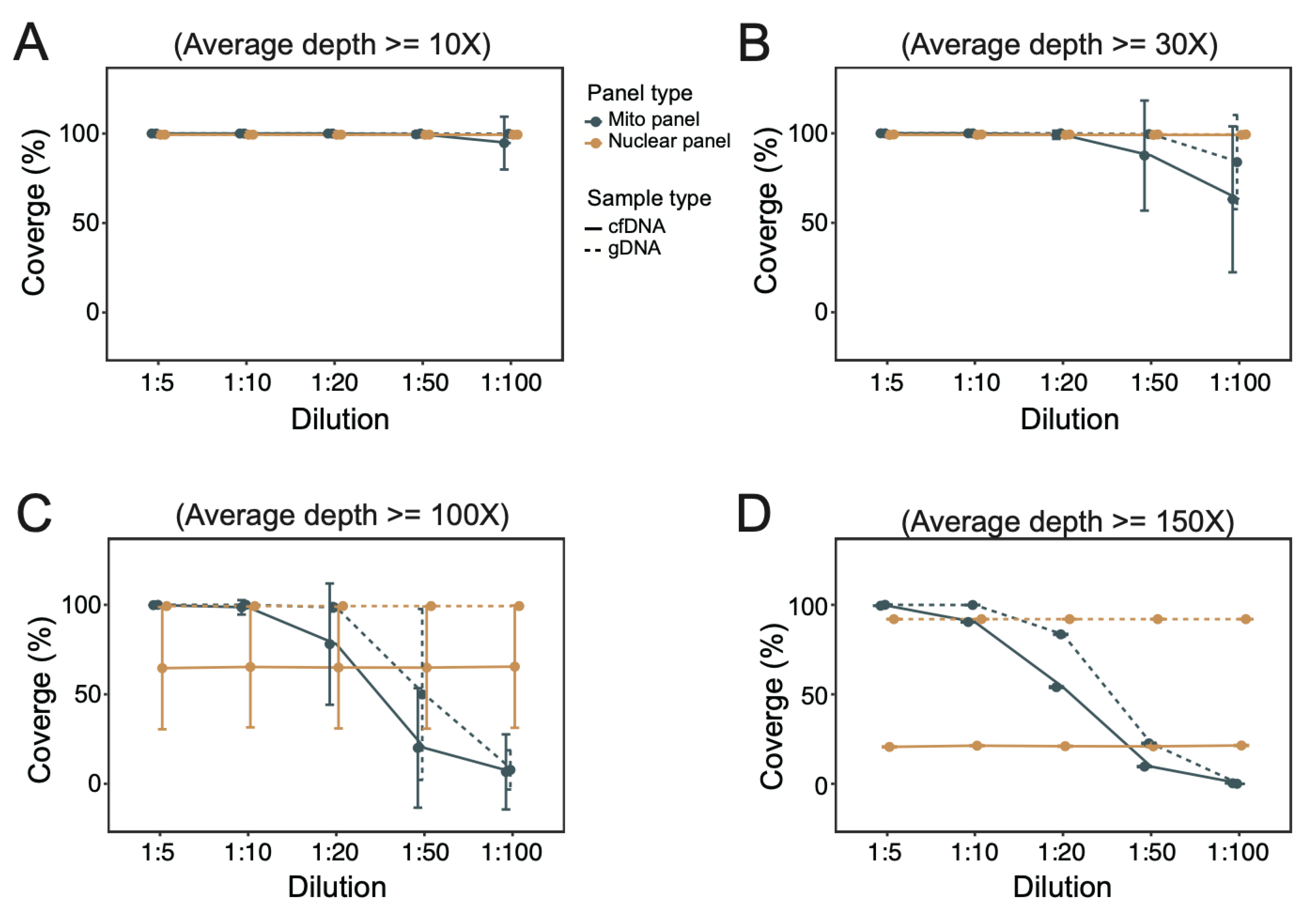
3.3. Coverage of Targeted Regions under Different Dilutions of mtDNA Probes
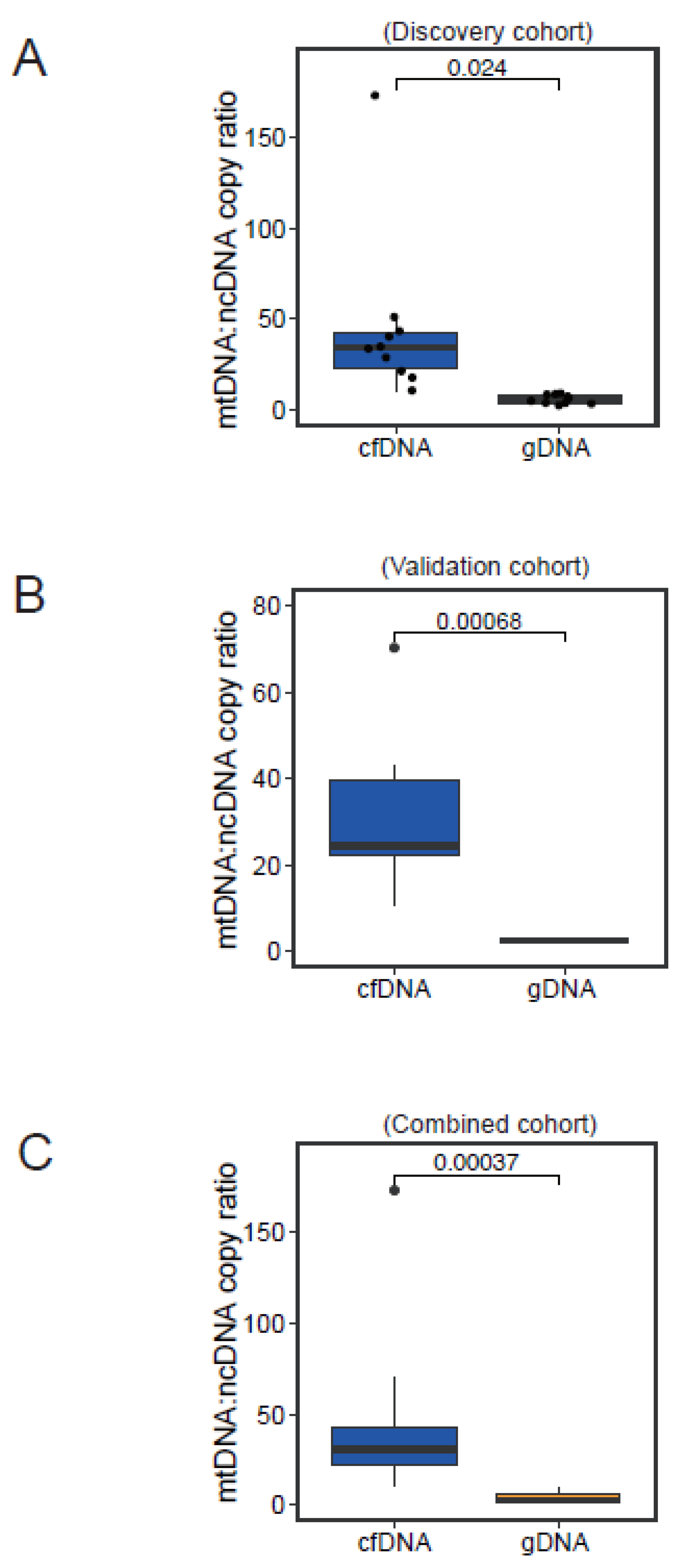
3.4. Normal Physiological Range of Variation in mtDNA:ncDNA Copy Ratio
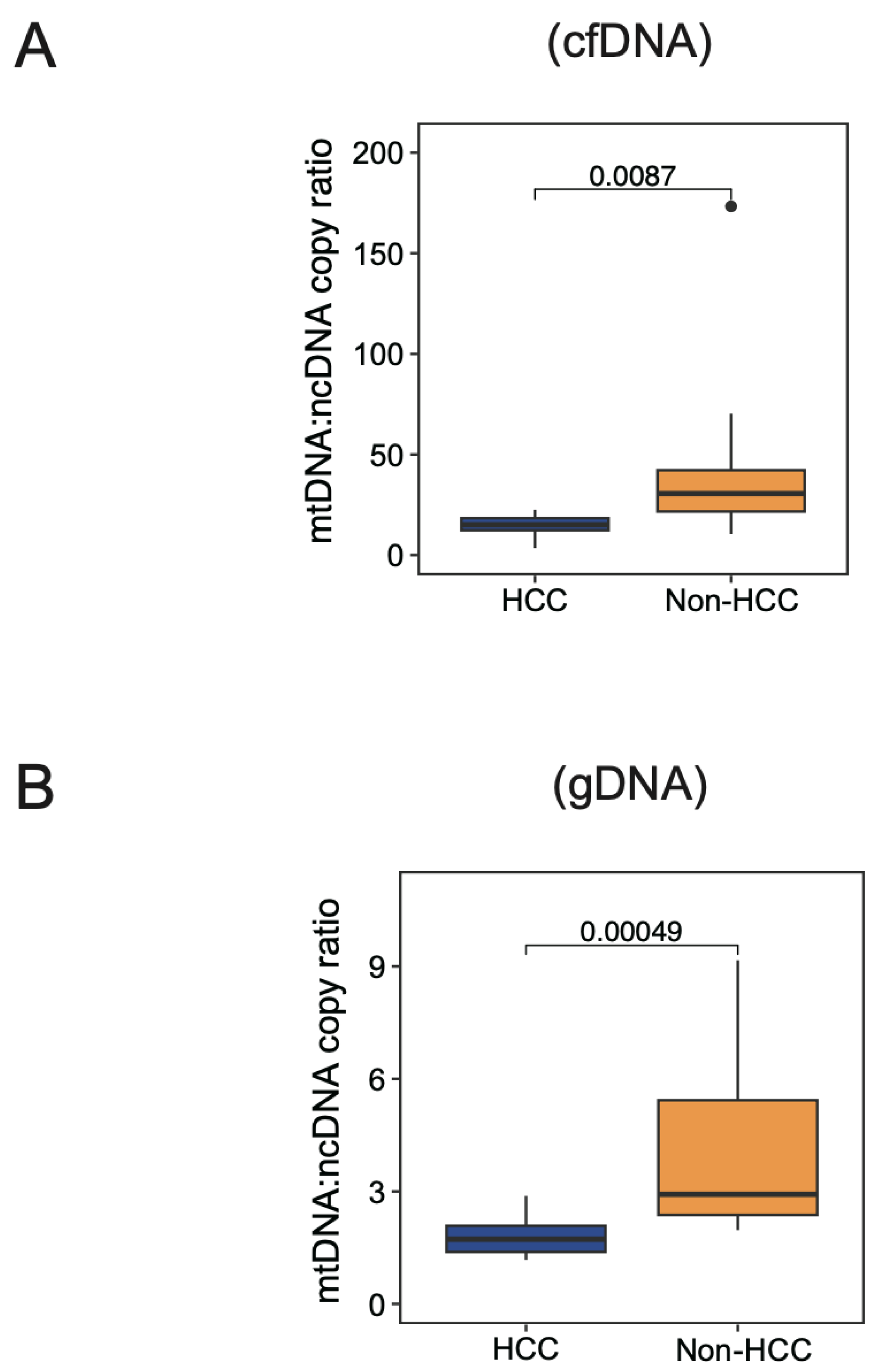
3.5. Physiological Range of Variation in mtDNA:ncDNA Copy Ratio in HCC Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monzel, A.S.; Enriquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Loyola-Machado, A.C.; Guerra, M.T.; Nathanson, M.H. Mitochondrial calcium signaling in cholangiocarcinoma. Hepatoma Res. 2023, 9, 25. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Zhang, D.; Wang, Y.; Qin, H.; Yang, W.; Cui, R.; Sheng, J. Mitochondrial Quality Control in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 713721. [Google Scholar] [CrossRef]
- Lozano-Rosas, M.G.; Chávez, E.; Aparicio-Cadena, A.R.; Velasco-Loyden, G.; Sánchez, V.C.d. Mitoepigenetics and hepatocellular carcinoma. Hepatoma Res. 2018, 4, 19. [Google Scholar] [CrossRef]
- Umetsu, S.; Inui, A.; Kobayashi, S.; Shimura, M.; Uehara, T.; Uchida, H.; Irie, R.; Sogo, T.; Komatsu, H.; Yoshioka, T.; et al. First cases of MPV17 related mitochondrial DNA depletion syndrome with compound heterozygous mutations in p.R50Q/p.R50W: A case report. Hepatoma Res. 2020, 6, 1. [Google Scholar] [CrossRef]
- Yin, C.; Li, D.Y.; Guo, X.; Cao, H.Y.; Chen, Y.B.; Zhou, F.; Ge, N.J.; Liu, Y.; Guo, S.S.; Zhao, Z.; et al. NGS-based profiling reveals a critical contributing role of somatic D-loop mtDNA mutations in HBV-related hepatocarcinogenesis. Ann. Oncol. 2019, 30, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Tsui, Y.M.; Ho, D.W.; Ng, I.O. Liquid Biopsy Using Cell-Free or Circulating Tumor DNA in the Management of Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Zhou, Q.; Dao, D.Y.; Lo, Y.M.D. Circulating biomarkers in the diagnosis and management of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, B.; Tastekin, D.; Guzelbulut, F.; Akgoz, M.; Pence, S. Quantification of cell-free circulating mitochondrial DNA copy number variation in hepatocellular carcinoma. Rev. Assoc. Med. Bras. 2022, 68, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, Y.; Li, S.; Liu, H.; Xu, F.; Lai, X.; Zhang, Q.; Xu, J.; Wan, S. Circulating Cell-Free mtDNA Content as a Non-invasive Prognostic Biomarker in HCC Patients Receiving TACE and Traditional Chinese Medicine. Front. Genet. 2021, 12, 719451. [Google Scholar] [CrossRef] [PubMed]
- Giosa, D.; Lombardo, D.; Musolino, C.; Chines, V.; Raffa, G.; Casuscelli di Tocco, F.; D’Aliberti, D.; Caminiti, G.; Saitta, C.; Alibrandi, A.; et al. Mitochondrial DNA is a target of HBV integration. Commun. Biol. 2023, 6, 684. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Jenkins, E.C.; Lechuga-Vieco, A.V.; Nie, K.; Fiel, M.I.; Rialdi, A.; Guccione, E.; Enriquez, J.A.; Sia, D.; Lujambio, A.; et al. The portrait of liver cancer is shaped by mitochondrial genetics. Cell Rep. 2022, 38, 110254. [Google Scholar] [CrossRef]
- Rausser, S.; Trumpff, C.; McGill, M.A.; Junker, A.; Wang, W.; Ho, S.H.; Mitchell, A.; Karan, K.R.; Monk, C.; Segerstrom, S.C.; et al. Mitochondrial phenotypes in purified human immune cell subtypes and cell mixtures. Elife 2021, 10, e70899. [Google Scholar] [CrossRef]
- Picard, M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion 2021, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Zabala, C.; Mansilla, S.; De Brun, L.; Martinez, J.; Garau, M.; Rivas, G.; Acosta, C.; Lens, D.; Cerisola, A.; et al. Blood cell respiration rates and mtDNA copy number: A promising tool for the diagnosis of mitochondrial disease. Mitochondrion 2021, 61, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Kanai, M.; Durham, T.J.; Tsuo, K.; McCoy, J.G.; Kotrys, A.V.; Zhou, W.; Chinnery, P.F.; Karczewski, K.J.; Calvo, S.E.; et al. Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature 2023, 620, 839–848. [Google Scholar] [CrossRef]
- Rosa, H.S.; Ajaz, S.; Gnudi, L.; Malik, A.N. A case for measuring both cellular and cell-free mitochondrial DNA as a disease biomarker in human blood. FASEB J. 2020, 34, 12278–12288. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, K.; Guo, S.; Wang, Y.; Ji, X.; Yuan, Q.; Su, L.; Guo, X.; Gu, X.; Xing, J. NGS-based accurate and efficient detection of circulating cell-free mitochondrial DNA in cancer patients. Mol. Ther. Nucleic Acids 2021, 23, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.W.H.; Chan, L.K.; Chiu, Y.T.; Xu, I.M.J.; Poon, R.T.P.; Cheung, T.T.; Tang, C.N.; Tang, V.W.L.; Lo, I.L.O.; Lam, P.W.Y.; et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017, 66, 1496–1506. [Google Scholar] [CrossRef]
- Kim, M.; Mahmood, M.; Reznik, E.; Gammage, P.A. Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer 2022, 8, 1046–1059. [Google Scholar] [CrossRef]
- Ji, X.; Guo, W.; Gu, X.; Guo, S.; Zhou, K.; Su, L.; Yuan, Q.; Liu, Y.; Guo, X.; Huang, Q.; et al. Mutational profiling of mtDNA control region reveals tumor-specific evolutionary selection involved in mitochondrial dysfunction. EBioMedicine 2022, 80, 104058. [Google Scholar] [CrossRef]
- Miyata, Y.; Mukae, Y.; Harada, J.; Matsuda, T.; Mitsunari, K.; Matsuo, T.; Ohba, K.; Sakai, H. Pathological and Pharmacological Roles of Mitochondrial Reactive Oxygen Species in Malignant Neoplasms: Therapies Involving Chemical Compounds, Natural Products, and Photosensitizers. Molecules 2020, 25, 5252. [Google Scholar] [CrossRef]
- Bai, R.K.; Chang, J.; Yeh, K.T.; Lou, M.A.; Lu, J.F.; Tan, D.J.; Liu, H.; Wong, L.J. Mitochondrial DNA content varies with pathological characteristics of breast cancer. J. Oncol. 2011, 2011, 496189. [Google Scholar] [CrossRef]
- Ding, J.; Sidore, C.; Butler, T.J.; Wing, M.K.; Qian, Y.; Meirelles, O.; Busonero, F.; Tsoi, L.C.; Maschio, A.; Angius, A.; et al. Assessing Mitochondrial DNA Variation and Copy Number in Lymphocytes of ~2,000 Sardinians Using Tailored Sequencing Analysis Tools. PLoS Genet. 2015, 11, e1005306. [Google Scholar]
- Mengel-From, J.; Thinggaard, M.; Dalgard, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, M.; Hubner, A.; Li, M.; Madea, B.; Stoneking, M. Age-Related and Heteroplasmy-Related Variation in Human mtDNA Copy Number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef] [PubMed]
- Andreu, A.L.; Martinez, R.; Marti, R.; Garcia-Arumi, E. Quantification of mitochondrial DNA copy number: Pre-analytical factors. Mitochondrion 2009, 9, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Longchamps, R.J.; Castellani, C.A.; Yang, S.Y.; Newcomb, C.E.; Sumpter, J.A.; Lane, J.; Grove, M.L.; Guallar, E.; Pankratz, N.; Taylor, K.D.; et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS ONE 2020, 15, e0228166. [Google Scholar] [CrossRef]
- Czarny, P.; Wigner, P.; Strycharz, J.; Swiderska, E.; Synowiec, E.; Szatkowska, M.; Sliwinska, A.; Talarowska, M.; Szemraj, J.; Su, K.P.; et al. Mitochondrial DNA copy number, damage, repair and degradation in depressive disorder. World J. Biol. Psychiatry 2020, 21, 91–101. [Google Scholar] [CrossRef]
- Al-Kafaji, G.; Bakheit, H.F.; Alharbi, M.A.; Farahat, A.A.; Jailani, M.; Ebrahin, B.H.; Bakhiet, M. Mitochondrial DNA Copy Number in Peripheral Blood as a Potential Non-invasive Biomarker for Multiple Sclerosis. Neuromol. Med. 2020, 22, 304–313. [Google Scholar] [CrossRef]
- Zole, E.; Ranka, R. Mitochondrial DNA copy number and telomere length in peripheral blood mononuclear cells in comparison with whole blood in three different age groups. Arch. Gerontol. Geriatr. 2019, 83, 131–137. [Google Scholar] [CrossRef]
- Ortega-Vazquez, A.; Sanchez-Badajos, S.; Ramirez-Garcia, M.A.; Alvarez-Luquin, D.; Lopez-Lopez, M.; Adalid-Peralta, L.V.; Monroy-Jaramillo, N. Longitudinal Changes in Mitochondrial DNA Copy Number and Telomere Length in Patients with Parkinson’s Disease. Genes 2023, 14, 1913. [Google Scholar] [CrossRef]
- Mizuno, G.; Yamada, H.; Tsuboi, Y.; Munetsuna, E.; Yamazaki, M.; Ando, Y.; Kageyama, I.; Nouchi, Y.; Teshigawara, A.; Hattori, Y.; et al. Low mitochondrial DNA copy number in peripheral blood mononuclear cells is associated with future mortality risk: A long-term follow-up study from Japan. J. Nutr. Health Aging 2024, 28, 100013. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, X.-Y.; Tsui, Y.-M.; Tam, I.K.-K.; Li, P.-M.; Cheung, G.C.-H.; Lee, J.M.-F.; Ng, I.O.-L.; Ho, D.W.-H. Resolution of Optimal Mitochondrial and Nuclear DNA Enrichment in Target-Panel Sequencing and Physiological Mitochondrial DNA Copy Number Estimation in Liver Cancer and Non-Liver Cancer Subjects. Cancers 2024, 16, 3012. https://doi.org/10.3390/cancers16173012
Lyu X-Y, Tsui Y-M, Tam IK-K, Li P-M, Cheung GC-H, Lee JM-F, Ng IO-L, Ho DW-H. Resolution of Optimal Mitochondrial and Nuclear DNA Enrichment in Target-Panel Sequencing and Physiological Mitochondrial DNA Copy Number Estimation in Liver Cancer and Non-Liver Cancer Subjects. Cancers. 2024; 16(17):3012. https://doi.org/10.3390/cancers16173012
Chicago/Turabian StyleLyu, Xue-Ying, Yu-Man Tsui, Ivan Ka-Kit Tam, Po-Man Li, Gary Cheuk-Hang Cheung, Joyce Man-Fong Lee, Irene Oi-Lin Ng, and Daniel Wai-Hung Ho. 2024. "Resolution of Optimal Mitochondrial and Nuclear DNA Enrichment in Target-Panel Sequencing and Physiological Mitochondrial DNA Copy Number Estimation in Liver Cancer and Non-Liver Cancer Subjects" Cancers 16, no. 17: 3012. https://doi.org/10.3390/cancers16173012