
The Protective Effects of Mcl-1 on Mitochondrial Damage and Oxidative Stress in Imiquimod-Induced Cancer Cell Death

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Cellular, Mitochondrial ROS and Lipid ROS Assays
2.4. Cell Viability and DNA Content Assays
2.5. Immunoblotting
2.6. Mitochondrial Oxygen Consumption Assay
2.7. Confocal Imaging for Colocalization of Mcl-1 and Mitochondria
2.8. Confocal Imaging for Assessment of Mitochondrial Morphology
2.9. Confocal Imaging for the Colocalization of Mitochondria and Lysosomes
2.10. Statistical Analysis
3. Results
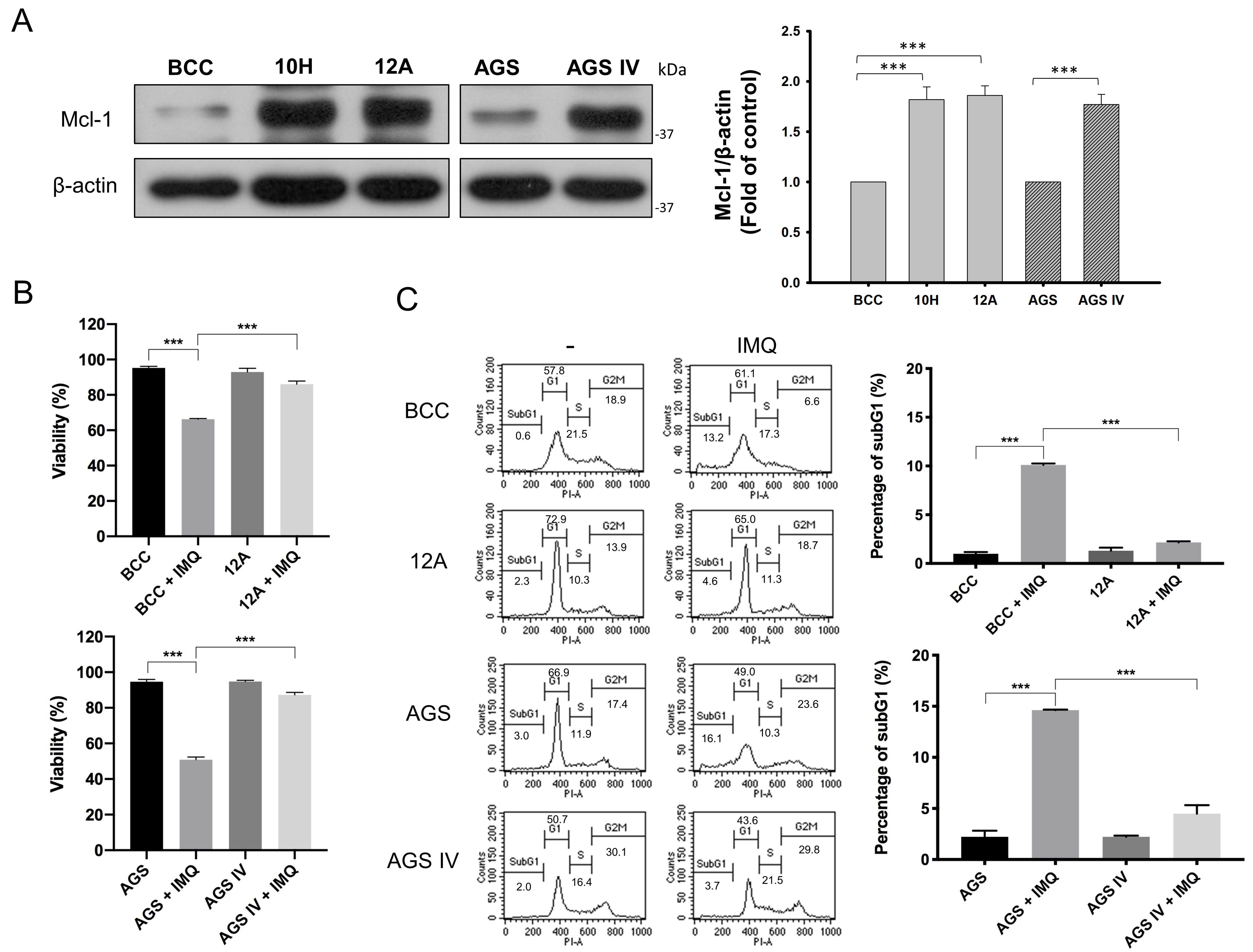
3.1. Mcl-1 Overexpression Diminished IMQ-Induced Cancer Cell Death
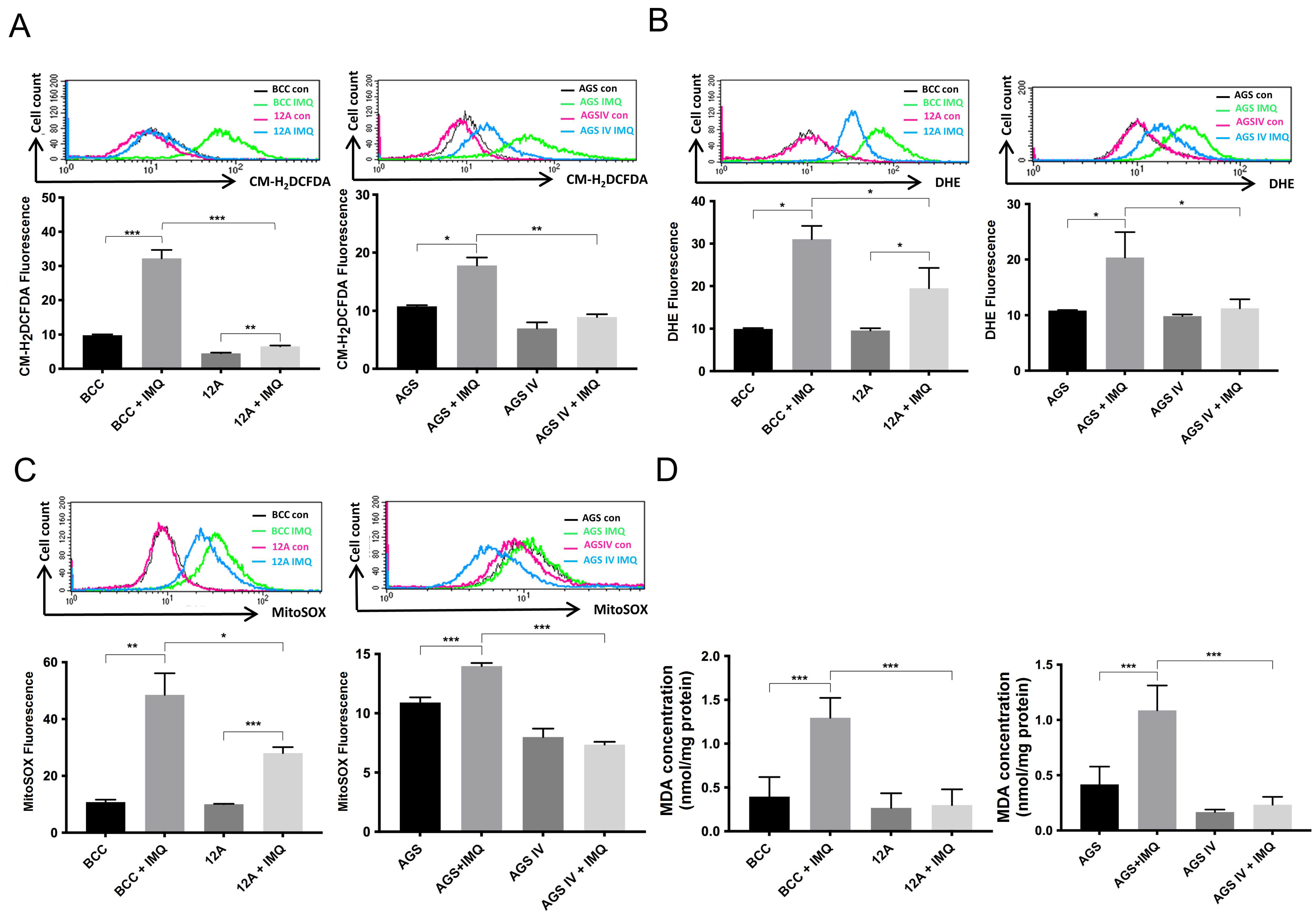
3.2. Overexpressed Mcl-1 Moderated IMQ-Induced Oxidative Stress Levels in Cancer Cells
3.3. Mcl-1 Overexpression Increased the Mitochondrial Oxygen Consumption Rate in Cancer Cells
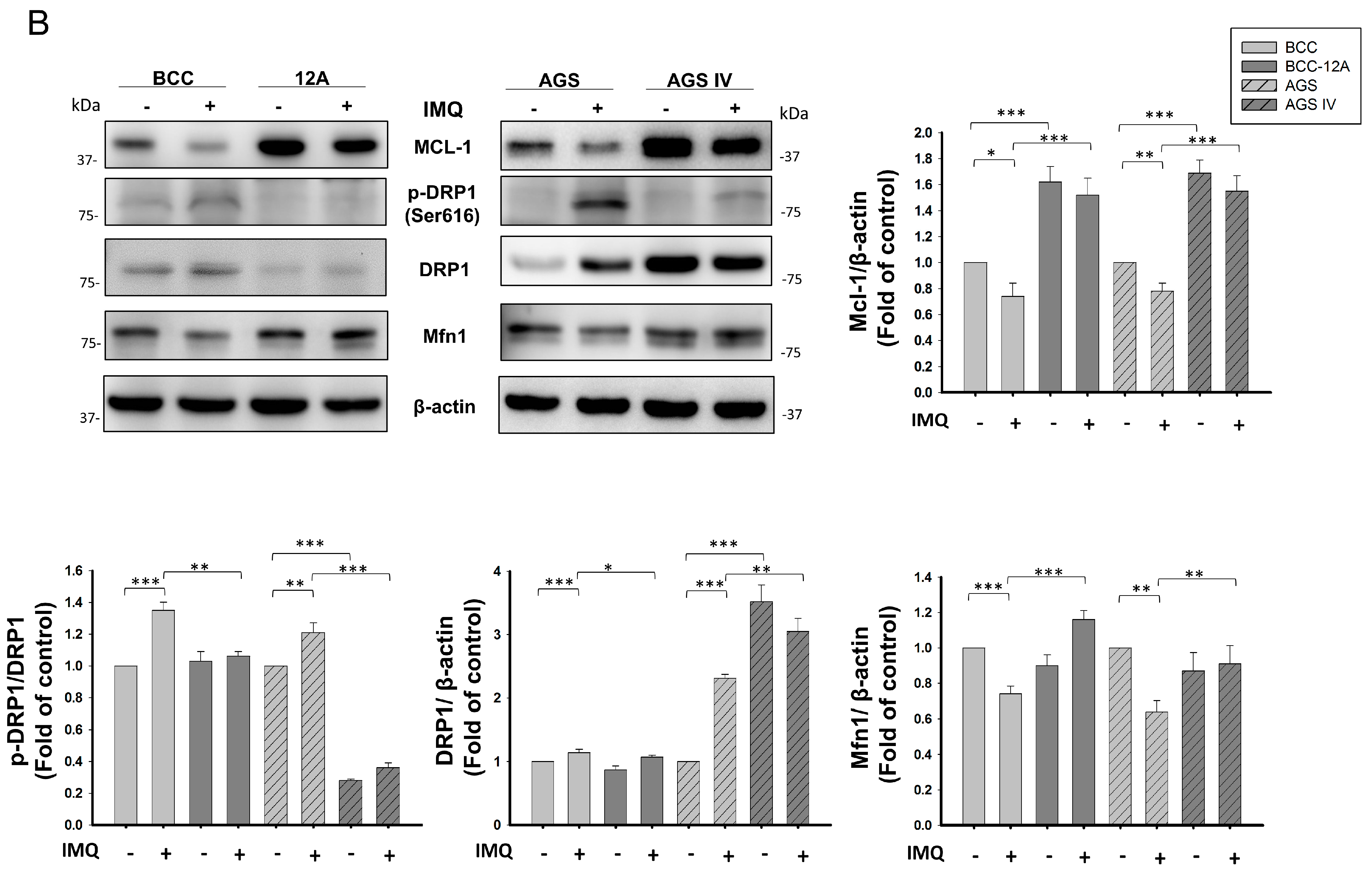
3.4. Mcl-1 Overexpression Stabilized Mitochondrial Dynamics in Cancer Cells after IMQ Treatment
3.5. Mcl-1 Overexpression Inhibited IMQ-Induced Mitophagy in Cancer Cells
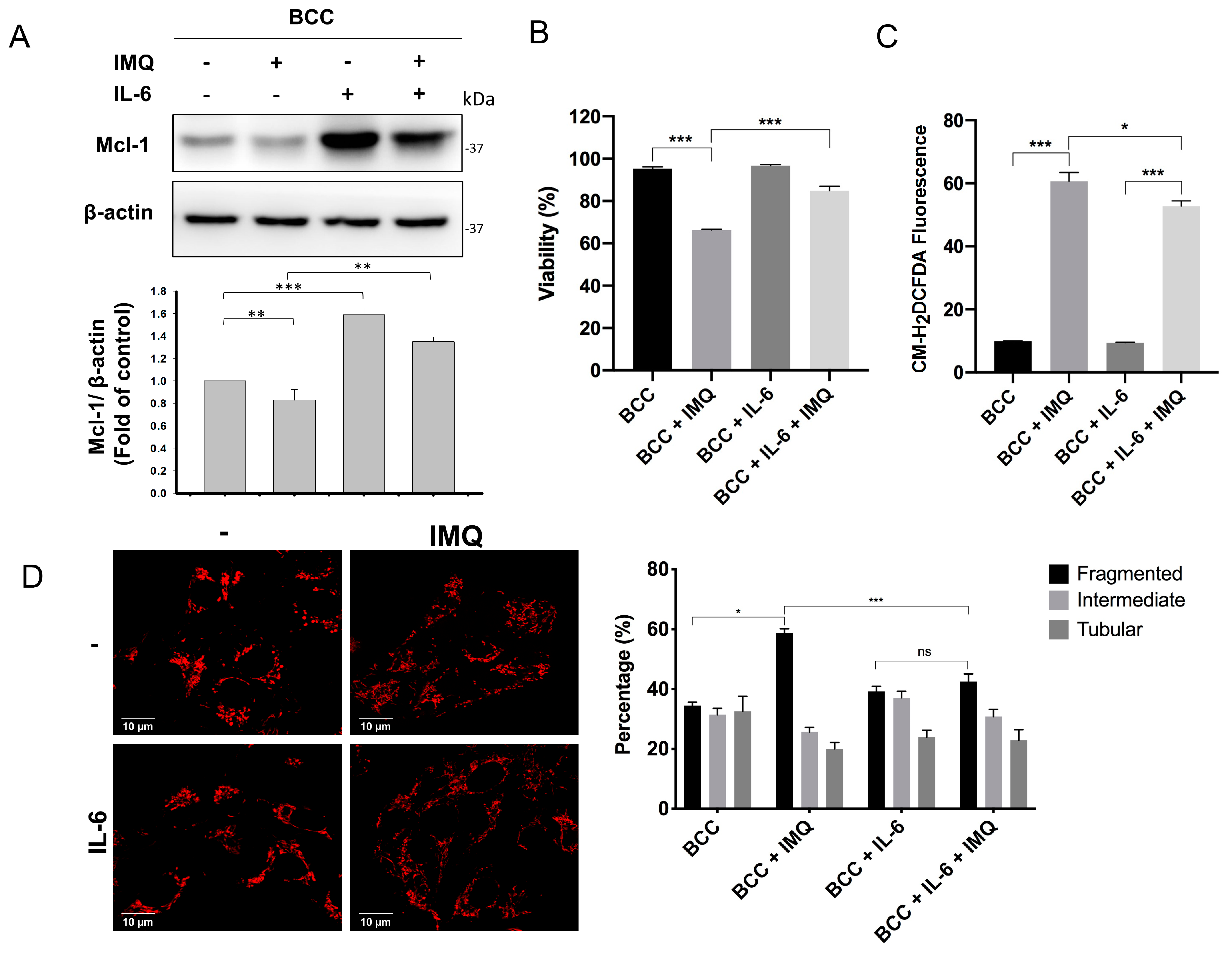
3.6. Exogenous IL-6 Treatment Increased Mcl-1 Levels and Reduced Cell Death and ROS Generation in IMQ-Treated BCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.W. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia 2002, 16, 444–454. [Google Scholar] [CrossRef]
- Michels, J.; Johnson, P.W.; Packham, G. Mcl-1. Int. J. Biochem. Cell Biol. 2005, 37, 267–271. [Google Scholar] [CrossRef]
- Shieh, J.J.; Liu, K.T.; Huang, S.W.; Chen, Y.J.; Hssssieh, T.Y. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J. Investig. Dermatol. 2009, 129, 2497–2506. [Google Scholar] [CrossRef]
- Morciano, G.; Giorgi, C.; Balestra, D.; Marchi, S.; Perrone, D.; Pinotti, M.; Pinton, P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell 2016, 27, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Fofaria, N.M.; Frederick, D.T.; Sullivan, R.J.; Flaherty, K.T.; Srivastava, S.K. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget 2015, 6, 40535–40556. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Das, S.G.; Li, Y.; Xing, C. Overexpression of Mcl-1 confers multidrug resistance, whereas topoisomerase IIbeta downregulation introduces mitoxantrone-specific drug resistance in acute myeloid leukemia. Mol. Pharmacol. 2013, 84, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Chang, C.C.; Lin, C.C.; Tsai, J.J.; Chen, Y.J.; Wu, C.Y.; Liu, K.T.; Shieh, J.J. Mcl-1 determines the imiquimod-induced apoptosis but not imiquimod-induced autophagy in skin cancer cells. J. Dermatol. Sci. 2012, 65, 170–178. [Google Scholar] [CrossRef]
- Huang, S.W.; Kao, J.K.; Wu, C.Y.; Wang, S.T.; Lee, H.C.; Liang, S.M.; Chen, Y.J.; Shieh, J.J. Targeting aerobic glycolysis and HIF-1alpha expression enhance imiquimod-induced apoptosis in cancer cells. Oncotarget 2014, 5, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Modugno, M.; Banfi, P.; Gasparri, F.; Borzilleri, R.; Carter, P.; Cornelius, L.; Gottardis, M.; Lee, V.; Mapelli, C.; Naglich, J.G.; et al. Mcl-1 antagonism is a potential therapeutic strategy in a subset of solid cancers. Exp. Cell Res. 2015, 332, 267–277. [Google Scholar] [CrossRef]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Yu, C.F.; Bieber, T.; Allam, J.P. Toll-like receptor 7 agonists and skin. Drug News Perspect. 2008, 21, 158–165. [Google Scholar]
- Walter, A.; Schafer, M.; Cecconi, V.; Matter, C.; Urosevic-Maiwald, M.; Belloni, B.; Schonewolf, N.; Dummer, R.; Bloch, W.; Werner, S.; et al. Aldara activates TLR7-independent immune defence. Nat. Commun. 2013, 4, 1560. [Google Scholar] [CrossRef] [PubMed]
- Schon, M.P.; Schon, M. Imiquimod: Mode of action. Br. J. Dermatol. 2007, 157 (Suppl. S2), 8–13. [Google Scholar] [CrossRef]
- Gross, C.J.; Mishra, R.; Schneider, K.S.; Medard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.A.; Fromm, S.; et al. K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef]
- Chang, S.H.; Huang, S.W.; Wang, S.T.; Chung, K.C.; Hsieh, C.W.; Kao, J.K.; Chen, Y.J.; Wu, C.Y.; Shieh, J.J. Imiquimod-induced autophagy is regulated by ER stress-mediated PKR activation in cancer cells. J. Dermatol. Sci. 2017, 87, 138–148. [Google Scholar] [CrossRef]
- Huang, S.W.; Chang, S.H.; Mu, S.W.; Jiang, H.Y.; Wang, S.T.; Kao, J.K.; Huang, J.L.; Wu, C.Y.; Chen, Y.J.; Shieh, J.J. Imiquimod activates p53-dependent apoptosis in a human basal cell carcinoma cell line. J. Dermatol. Sci. 2016, 81, 182–191. [Google Scholar] [CrossRef]
- Huang, S.W.; Liu, K.T.; Chang, C.C.; Chen, Y.J.; Wu, C.Y.; Tsai, J.J.; Lu, W.C.; Wang, Y.T.; Liu, C.M.; Shieh, J.J. Imiquimod simultaneously induces autophagy and apoptosis in human basal cell carcinoma cells. Br. J. Dermatol. 2010, 163, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.T.; Huang, S.W.; Kao, J.K.; Liang, S.M.; Chen, Y.J.; Chen, Y.Y.; Wu, C.Y.; Shieh, J.J. Imiquimod-induced AMPK activation causes translation attenuation and apoptosis but not autophagy. J. Dermatol. Sci. 2015, 78, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Chuang, K.C.; Chang, C.R.; Chang, S.H.; Huang, S.W.; Chuang, S.M.; Li, Z.Y.; Wang, S.T.; Kao, J.K.; Chen, Y.J.; Shieh, J.J. Imiquimod-induced ROS production disrupts the balance of mitochondrial dynamics and increases mitophagy in skin cancer cells. J. Dermatol. Sci. 2020, 98, 152–162. [Google Scholar] [CrossRef]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [CrossRef]
- Yen, H.T.; Chiang, L.C.; Wen, K.H.; Tsai, C.C.; Yu, C.L.; Yu, H.S. The expression of cytokines by an established basal cell carcinoma cell line (BCC-1/KMC) compared with cultured normal keratinocytes. Arch. Dermatol. Res. 1996, 288, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Scorrano, L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Taher, M.Y.; Davies, D.M.; Maher, J. The role of the interleukin (IL)-6/IL-6 receptor axis in cancer. Biochem. Soc. Trans. 2018, 46, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Jee, S.H.; Chiu, H.C.; Tsai, T.F.; Tsai, W.L.; Liao, Y.H.; Chu, C.Y.; Kuo, M.L. The phosphotidyl inositol 3-kinase/Akt signal pathway is involved in interleukin-6-mediated Mcl-1 upregulation and anti-apoptosis activity in basal cell carcinoma cells. J. Investig. Dermatol. 2002, 119, 1121–1127. [Google Scholar] [CrossRef]
- Jee, S.H.; Shen, S.C.; Chiu, H.C.; Tsai, W.L.; Kuo, M.L. Overexpression of interleukin-6 in human basal cell carcinoma cell lines increases anti-apoptotic activity and tumorigenic potency. Oncogene 2001, 20, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci. CMLS 2009, 66, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Wu, C.Y.; Chuang, K.C.; Huang, S.W.; Li, Z.Y.; Wang, S.T.; Lai, Z.L.; Chang, C.C.; Chen, Y.J.; Wong, T.W.; et al. Imiquimod Accelerated Antitumor Response by Targeting Lysosome Adaptation in Skin Cancer Cells. J. Investig. Dermatol. 2021, 141, 2219–2228.e8. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Pfannenstiel, L.; Demelash, A.; Phoon, Y.P.; Mayell, C.; Cabrera, C.; Liu, C.; Zhao, J.; Dermawan, J.; Patil, D.; et al. MCL1 nuclear translocation induces chemoresistance in colorectal carcinoma. Cell Death Dis. 2022, 13, 63. [Google Scholar] [CrossRef]
- Bosc, C.; Selak, M.A.; Sarry, J.E. Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment. Cell Metab. 2017, 26, 705–707. [Google Scholar] [CrossRef]
- Liu, A.S.; Yu, H.Y.; Yang, Y.L.; Xue, F.Y.; Chen, X.; Zhang, Y.; Zhou, Z.Y.; Zhang, B.; Li, L.; Sun, C.Z.; et al. A Chemotherapy-Driven Increase in Mcl-1 Mediates the Effect of miR-375 on Cisplatin Resistance in Osteosarcoma Cells. OncoTargets Ther. 2019, 12, 11667–11677. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.Y.; Lee, B.S.; Chang, J.W.; Park, J.K.; Han, J.H.; Kim, Y.S.; Shin, Y.S.; Byeon, H.K.; Kim, C.H. Downregulation of Nrf2 by the combination of TRAIL and Valproic acid induces apoptotic cell death of TRAIL-resistant papillary thyroid cancer cells via suppression of Bcl-xL. Cancer Lett. 2016, 372, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ruefli-Brasse, A.; Reed, J.C. Therapeutics targeting Bcl-2 in hematological malignancies. Biochem. J. 2017, 474, 3643–3657. [Google Scholar] [CrossRef] [PubMed]
- Senichkin, V.V.; Streletskaia, A.Y.; Zhivotovsky, B.; Kopeina, G.S. Molecular Comprehension of Mcl-1: From Gene Structure to Cancer Therapy. Trends Cell Biol. 2019, 29, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Widden, H.; Placzek, W.J. The multiple mechanisms of MCL1 in the regulation of cell fate. Commun. Biol. 2021, 4, 1029. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Le Gouill, S.; Podar, K.; Harousseau, J.L.; Anderson, K.C. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle 2004, 3, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Hwang, C.S.; Yin, J.H.; Chen, S.D.; Yang, D.I. Oncostatin M-dependent Mcl-1 induction mediated by JAK1/2-STAT1/3 and CREB contributes to bioenergetic improvements and protective effects against mitochondrial dysfunction in cortical neurons. Biochim. Biophys. Acta 2015, 1853, 2306–2325. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, S.-H.; Chuang, K.-C.; Li, Z.-Y.; Chang, M.-C.; Liu, K.-T.; Hsu, C.-S.; Huang, S.-W.; Chung, M.-C.; Wang, S.-C.; Chen, Y.-J.; et al. The Protective Effects of Mcl-1 on Mitochondrial Damage and Oxidative Stress in Imiquimod-Induced Cancer Cell Death. Cancers 2024, 16, 3060. https://doi.org/10.3390/cancers16173060
Chang S-H, Chuang K-C, Li Z-Y, Chang M-C, Liu K-T, Hsu C-S, Huang S-W, Chung M-C, Wang S-C, Chen Y-J, et al. The Protective Effects of Mcl-1 on Mitochondrial Damage and Oxidative Stress in Imiquimod-Induced Cancer Cell Death. Cancers. 2024; 16(17):3060. https://doi.org/10.3390/cancers16173060
Chicago/Turabian StyleChang, Shu-Hao, Kai-Cheng Chuang, Zheng-Yi Li, Mao-Chia Chang, Kuang-Ting Liu, Chien-Sheng Hsu, Shi-Wei Huang, Mu-Chi Chung, Shih-Chung Wang, Yi-Ju Chen, and et al. 2024. "The Protective Effects of Mcl-1 on Mitochondrial Damage and Oxidative Stress in Imiquimod-Induced Cancer Cell Death" Cancers 16, no. 17: 3060. https://doi.org/10.3390/cancers16173060