


TP53 Mutation-Mediated Immune Evasion in Cancer: Mechanisms and Therapeutic Implications


Abstract
Simple Summary
Abstract
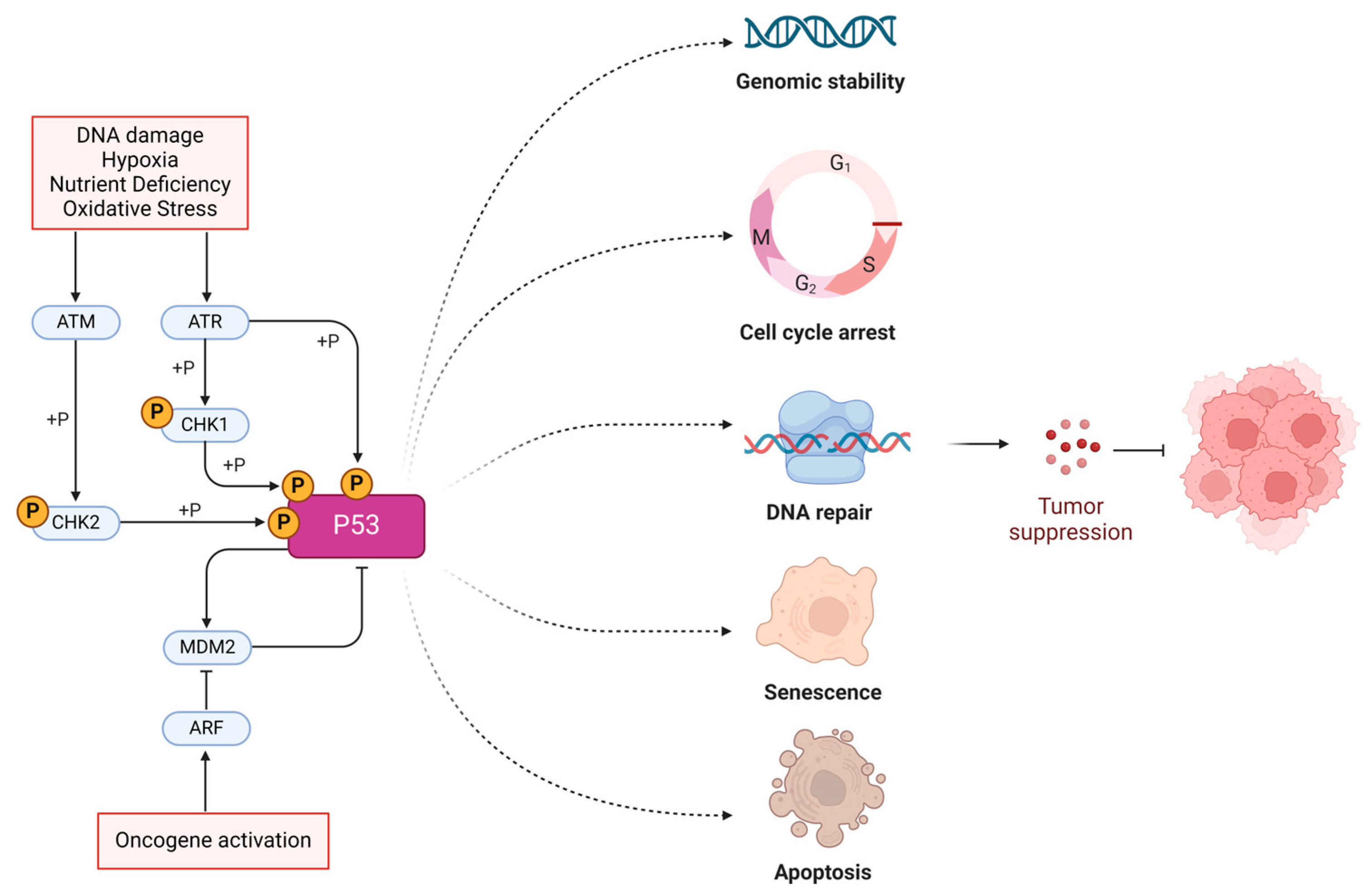
1. Introduction
2. TP53 Gene Structure, Mutation Spectrum, and Etiology
- Exon 1–2: Encodes the transactivation domain, which is responsible for activating the transcription of target genes.
- Exon 3–4: Contains sequences that contribute to the proline-rich region, which is important for the apoptotic function of p53.
- Exon 5–8: Encodes the DNA-binding domain, a crucial region that allows the p53 protein to bind to specific DNA sequences and regulate gene expression. This region is also the most frequently mutated in cancers.
- Exon 9: Encodes part of the oligomerization domain, which is essential for the tetramer formation of the p53 protein.
- Exon 10–11: Encodes the remaining part of the oligomerization domain and the C-terminal regulatory domain, which is involved in the regulation of p53’s activity.
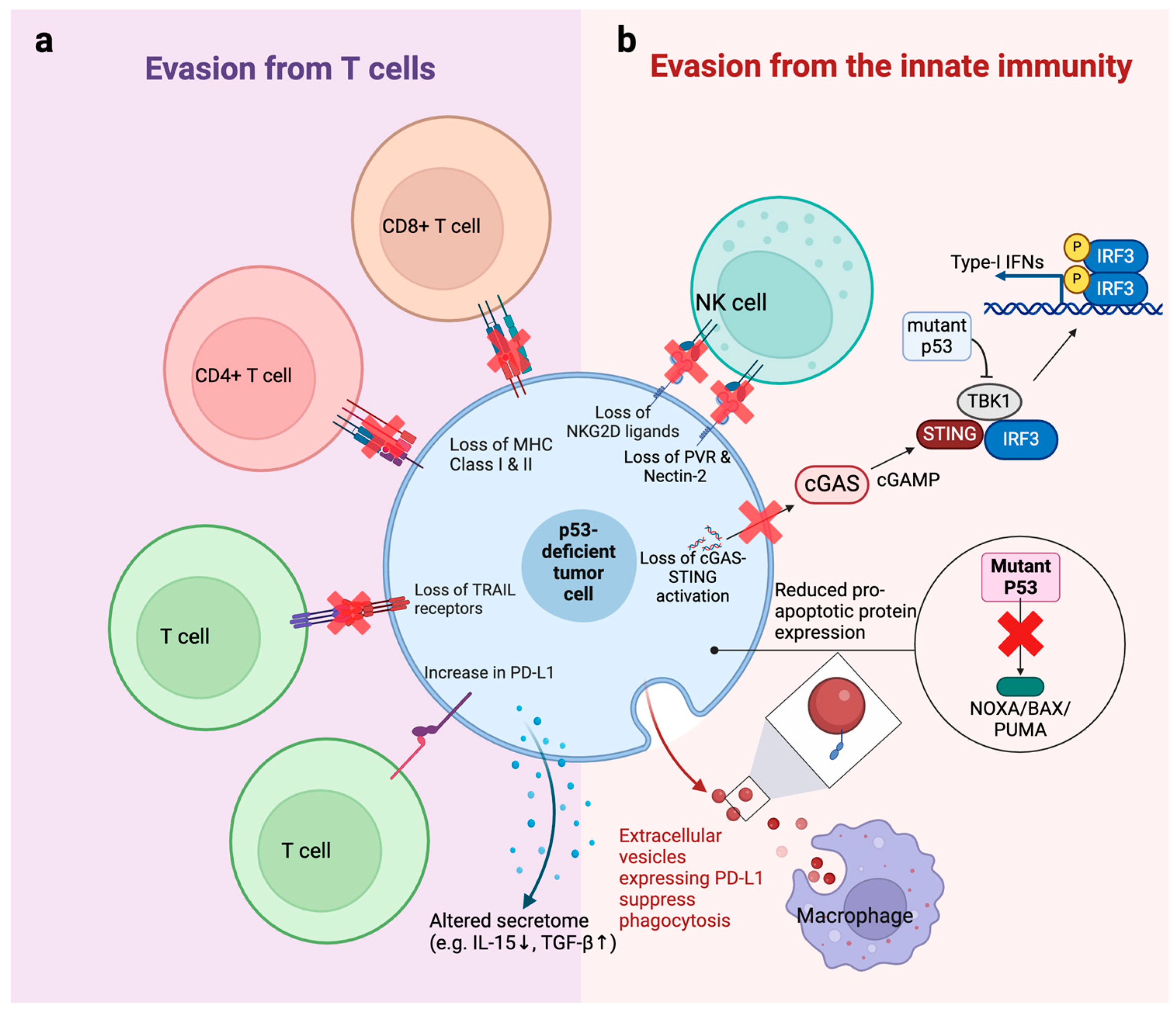
3. Mechanisms of Immune Evasion Caused by TP53 Mutations
3.1. Evasion from T Cells
3.2. Evasion from the Innate Immune System
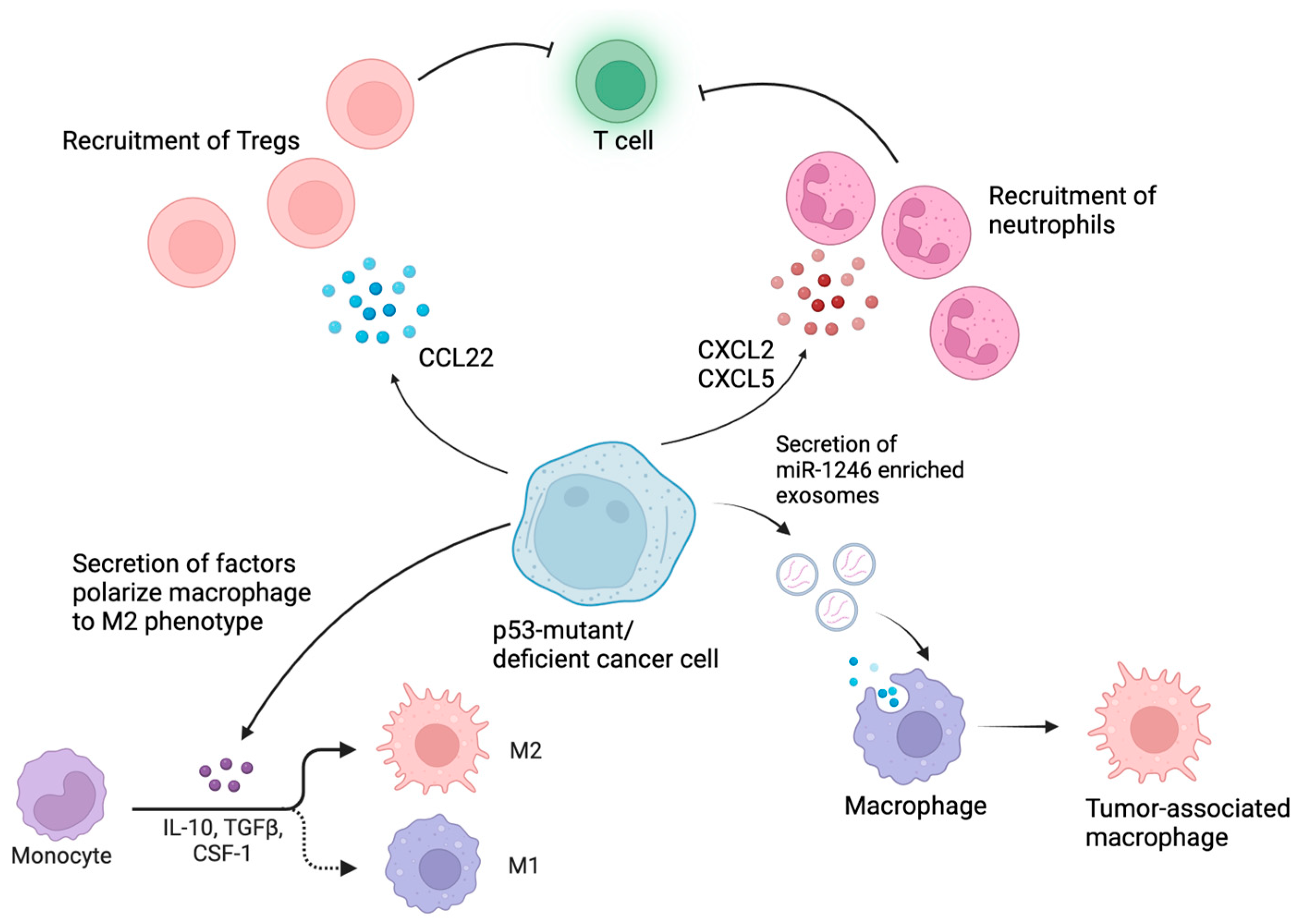
4. p53-Mutant Tumor Cells Create an Immunosuppressive TME to Promote Immune Evasion
4.1. p53 Mutations Reprogram Myeloid Cells in the TME into Immunosuppressive Phenotypes
4.2. p53-Mutant Cancers Recruit Tregs to Cause Immunosuppression
5. Clinical Evidence of Immune Evasion in p53-Mutant Cancers
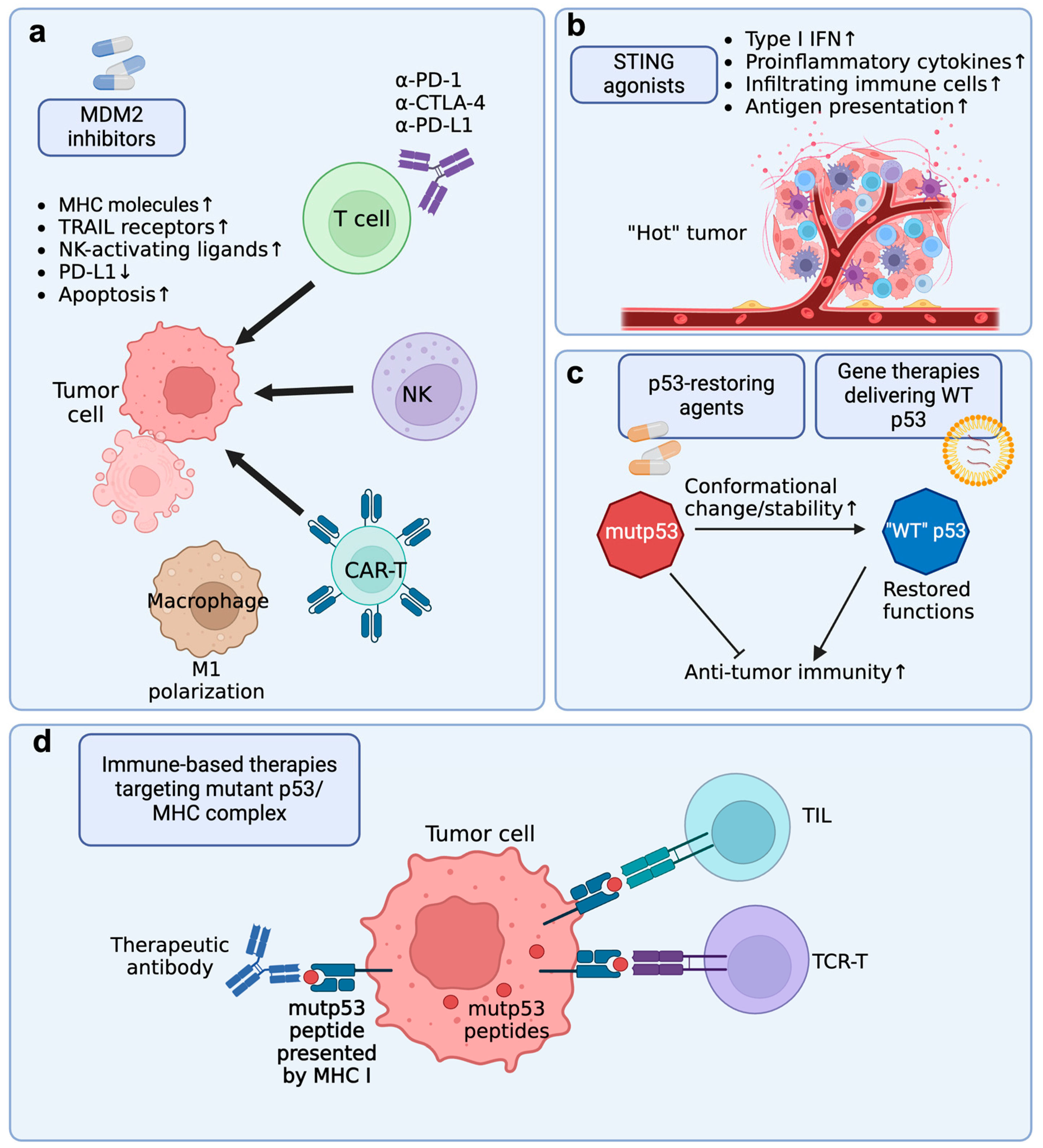
6. Strategies Enhancing the Efficacy of Immune Response by Targeting p53
6.1. MDM2 Inhibitor + Immune Checkpoint Blockade (ICB)
6.2. MDM2 Inhibitor + Immune Cell Adoptive Transfer Therapy
6.3. MDM2 Inhibitors + HSCT
6.4. STING Agonists + Immunotherapies
6.5. Targeting Immune Evasion of p53-Mutant Cancers by p53-Restoring Agents
6.6. Restoring Function of Mutant p53 with Gene Therapies
6.7. Targeting Mutant p53 by Immune-Based Therapies
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Correction Statement
References
- Ge, T.; Gu, X.; Jia, R.; Ge, S.; Chai, P.; Zhuang, A.; Fan, X. Crosstalk between metabolic reprogramming and epigenetics in cancer: Updates on mechanisms and therapeutic opportunities. Cancer Commun. 2022, 42, 1049–1082. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, S.; Liu, B.; Pant, V.; Celii, F.; Chau, G.; Elizondo-Fraire, A.C.; Yang, P.; You, M.J.; El-Naggar, A.K.; et al. Somatic Trp53 mutations differentially drive breast cancer and evolution of metastases. Nat. Commun. 2018, 9, 3953. [Google Scholar] [CrossRef]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; BièChe, I.; et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Piecuch, A.; Sady, M.; Gajewski, Z.; Flis, S. Gain of Function (GOF) Mutant p53 in Cancer-Current Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 13287. [Google Scholar] [CrossRef]
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2021, 8, 607670. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Geske, F.J.; Nelson, A.C.; Lieberman, R.; Strange, R.; Sun, T.; E Gerschenson, L. DNA repair is activated in early stages of p53-induced apoptosis. Cell Death Differ. 2000, 7, 393–401. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Yang, L.V.; Abrams, S.L.; Steelman, L.S.; Follo, M.Y.; Cocco, L.; Ratti, S.; Martelli, A.M.; Augello, G.; Cervello, M. Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells 2022, 11, 2155. [Google Scholar] [CrossRef]
- Zhu, K.-L.; Su, F.; Yang, J.-R.; Xiao, R.-W.; Wu, R.-Y.; Cao, M.-Y.; Ling, X.-L.; Zhang, T. TP53 to mediate immune escape in tumor microenvironment: An overview of the research progress. Mol. Biol. Rep. 2024, 51, 205. [Google Scholar] [CrossRef]
- Zhou, X.; Santos, G.S.; Zhan, Y.; Oliveira, M.M.S.; Rezaei, S.; Singh, M.; Peuget, S.; Westerberg, L.S.; Johnsen, J.I.; Selivanova, G. Mutant p53 gain of function mediates cancer immune escape that is counteracted by APR-246. Br. J. Cancer 2022, 127, 2060–2071. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef]
- Saha, M.N.; Qiu, L.; Chang, H. Targeting p53 by small molecules in hematological malignancies. J. Hematol. Oncol. 2013, 6, 23. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Lepre, M.G.; Omar, S.I.; Grasso, G.; Morbiducci, U.; Deriu, M.A.; Tuszynski, J.A. Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA. Molecules 2017, 22, 1358. [Google Scholar] [CrossRef] [PubMed]
- Pintus, S.; Ivanisenko, N.; Demenkov, P.; Ivanisenko, T.; Ramachandran, S.; Kolchanov, N.; Ivanisenko, V. The substitutions G245C and G245D in the Zn(2+)-binding pocket of the p53 protein result in differences of conformational flexibility of the DNA-binding domain. J. Biomol. Struct. Dyn. 2013, 31, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Balasundaram, A.; Doss, C.G.P. Unraveling the Structural Changes in the DNA-Binding Region of Tumor Protein p53 (TP53) upon Hotspot Mutation p53 Arg248 by Comparative Computational Approach. Int. J. Mol. Sci. 2022, 23, 15499. [Google Scholar] [CrossRef] [PubMed]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function p53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, P.; Zeng, S.X.; Lu, H. It takes a team: A gain-of-function story of p53-R249S. J. Mol. Cell Biol. 2019, 11, 277–283. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, M.; Fang, Y.; Li, J.; Chen, Y.; Jiao, S. TP53 R273C Mutation Is Associated with Poor Prognosis in LGG Patients. Front. Genet. 2022, 13, 720651. [Google Scholar] [CrossRef]
- Garg, A.; Hazra, J.P.; Sannigrahi, M.K.; Rakshit, S.; Sinha, S. Variable Mutations at the p53-R273 Oncogenic Hotspot Position Leads to Altered Properties. Biophys. J. 2019, 118, 720–728. [Google Scholar] [CrossRef]
- Calhoun, S.; Daggett, V. Structural effects of the L145Q, V157F, and R282W cancer-associated mutations in the p53 DNA-binding core domain. Biochemistry 2011, 50, 5345–5353. [Google Scholar] [CrossRef]
- Zhang, M.; Zhuang, G.; Sun, X.; Shen, Y.; Wang, W.; Li, Q.; Di, W. TP53 mutation-mediated genomic instability induces the evolution of chemoresistance and recurrence in epithelial ovarian cancer. Diagn. Pathol. 2017, 12, 16. [Google Scholar] [CrossRef]
- Kucab, J.E.; Phillips, D.H.; Arlt, V.M. Linking environmental carcinogen exposure to TP53 mutations in human tumours using the human TP53 knock-in (Hupki) mouse model. FEBS J. 2010, 277, 2567–2583. [Google Scholar]
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li-Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7, a026187. [Google Scholar] [CrossRef]
- Travé, G.; Zanier, K. HPV-mediated inactivation of tumor suppressor p53. Cell Cycle 2016, 15, 2231–2232. [Google Scholar] [CrossRef]
- Zong, Z.; Xie, F.; Wang, S.; Wu, X.; Zhang, Z.; Yang, B.; Zhou, F. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell 2024, 187, 2375–2392.e33. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, E.; Vorholt, D.; Blakemore, S.J.; Sackey, B.; Nolte, J.L.; Barbarino, V.; Schmitz, J.H.; Nickel, N.; Bachurski, D.; Lobastova, L.; et al. Extracellular vesicles and PDL1 suppress macrophages inducing therapy resistance in TP53-deficient B-cell malignancies. Blood 2022, 139, 3617–3629. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 2023, 83, 266–280.e6. [Google Scholar] [CrossRef]
- Efe, G.; Dunbar, K.J.; Sugiura, K.; Cunningham, K.; Carcamo, S.; Karaiskos, S.; Tang, Q.; Cruz-Acuña, R.; Resnick-Silverman, L.; Peura, J.; et al. p53 Gain-of-Function Mutation Induces Metastasis via BRD4-Dependent CSF-1 Expression. Cancer Discov. 2023, 13, 2632–2651. [Google Scholar] [CrossRef]
- Siolas, D.; Vucic, E.; Kurz, E.; Hajdu, C.; Bar-Sagi, D. Gain-of-function p53(R172H) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep. 2021, 36, 109578. [Google Scholar] [CrossRef] [PubMed]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e5. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Patel, R.S.; Cook, D.R.; Choi, M.Y.; Patil, A.; Liang, A.C.; Li, M.Z.; Haigis, K.M.; Elledge, S.J. The adaptive immune system is a major driver of selection for tumor suppressor gene inactivation. Science 2021, 373, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Garrido, F. The Challenges of HLA Class I Loss in Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2022, 28, 5021–5029. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Zhu, K.; Wang, J.; Zhu, J.; Jiang, J.; Shou, J.; Chen, X. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 1999, 18, 7740–7747. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Schmidt, D.; Lowinus, T.; Ryoo, J.; Dopfer, E.P.; Nunez, N.G.; Costa-Pereira, S.; Toffalori, C.; Punta, M.; Fetsch, V.; et al. Targeting MDM2 enhances antileukemia immunity after allogeneic transplantation via MHC-II and TRAIL-R1/2 upregulation. Blood 2022, 140, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Schmaltz, C.; Alpdogan, O.; Kappel, B.J.; Muriglan, S.J.; Rotolo, J.A.; Ongchin, J.; Willis, L.M.; Greenberg, A.S.; Eng, J.M.; Crawford, J.M.; et al. T cells require TRAIL for optimal graft-versus-tumor activity. Nat. Med. 2002, 8, 1433–1437. [Google Scholar] [CrossRef]
- Costa, C.; Indovina, P.; Mattioli, E.; Forte, I.M.; Iannuzzi, C.A.; Luzzi, L.; Bellan, C.; De Summa, S.; Bucci, E.; Di Marzo, D.; et al. P53-regulated miR-320a targets PDL1 and is downregulated in malignant mesothelioma. Cell Death Dis. 2020, 11, 748. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, M.; Giesler, S.; Richtsfeld, S.; Costa-Pereira, S.; Rindlisbacher, L.; Wertheimer, T.; Braun, L.M.; Andrieux, G.; Duquesne, S.; Pfeifer, D.; et al. MDM2 Inhibition Enhances Immune Checkpoint Inhibitor Efficacy by Increasing IL15 and MHC Class II Production. Mol. Cancer Res. 2023, 21, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Pawlowsky, L.; Marcinek, A.; Brauchle, B.; Muth, A.; Kazerani, M.; Petrera, A.; Kischel, R.; Emhardt, A.J.; Rothenberg-Thurley, M.; et al. The Battle within: AML´s p53 Strategies to Evade T-Cell Attack. Blood 2023, 142 (Suppl. S1), 1411. [Google Scholar] [CrossRef]
- Liao, N.-S.; Bix, M.; Zijlstra, M.; Jaenisch, R.; Raulet, D. MHC Class I Deficiency: Susceptibility to Natural Killer (NK) Cells and Impaired NK Activity. Science 1991, 253, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2022, 23, 90–105. [Google Scholar] [CrossRef]
- Veneziani, I.; Infante, P.; Ferretti, E.; Melaiu, O.; Battistelli, C.; Lucarini, V.; Compagnone, M.; Nicoletti, C.; Castellano, A.; Petrini, S.; et al. Nutlin-3a Enhances Natural Killer Cell-Mediated Killing of Neuroblastoma by Restoring p53-Dependent Expression of Ligands for NKG2D and DNAM-1 Receptors. Cancer Immunol. Res. 2021, 9, 170–183. [Google Scholar] [CrossRef]
- Pan, R.; Ryan, J.; Pan, D.; Wucherpfennig, K.W.; Letai, A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell 2022, 185, 1521–1538.e18. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Weiskopf, K.; Weissman, I.L. Macrophages are critical effectors of antibody therapies for cancer. mAbs 2015, 7, 303–310. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef] [PubMed]
- Onkar, S.S.; Carleton, N.M.; Lucas, P.C.; Bruno, T.C.; Lee, A.V.; Vignali, D.A.; Oesterreich, S. The Great Immune Escape: Understanding the Divergent Immune Response in Breast Cancer Subtypes. Cancer Discov. 2022, 13, 23–40. [Google Scholar] [CrossRef]
- Christofides, A.; Strauss, L.; Yeo, A.; Cao, C.; Charest, A.; Boussiotis, V.A. The complex role of tumor-infiltrating macrophages. Nat. Immunol. 2022, 23, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ng, D.S.W.; Mah, W.-C.; Almeida, F.F.; A Rahmat, S.; Rao, V.K.; Leow, S.C.; Laudisi, F.; Peh, M.T.; Goh, A.M.; et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. 2014, 22, 1081–1093. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef]
- Abolhalaj, M.; Sincic, V.; Lilljebjörn, H.; Sandén, C.; Aab, A.; Hägerbrand, K.; Ellmark, P.; Borrebaeck, C.A.K.; Fioretos, T.; Lundberg, K. Transcriptional profiling demonstrates altered characteristics of CD8(+) cytotoxic T-cells and regulatory T-cells in TP53-mutated acute myeloid leukemia. Cancer Med. 2022, 11, 3023–3032. [Google Scholar] [CrossRef]
- Shouval, R.; Tomas, A.A.; Fein, J.A.; Flynn, J.R.; Markovits, E.; Mayer, S.; Afuye, A.O.; Alperovich, A.; Anagnostou, T.; Besser, M.J.; et al. Impact of TP53 Genomic Alterations in Large B-Cell Lymphoma Treated With CD19-Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2022, 40, 369–381. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, L.; Chen, P.; Gao, X.; Wang, S.; Li, F.; Dou, L.; Gao, C.; Li, Y.; Liu, D. Haematologic malignancies with unfavourable gene mutations benefit from donor lymphocyte infusion with/without decitabine for prophylaxis of relapse after allogeneic HSCT: A pilot study. Cancer Med. 2021, 10, 3165–3176. [Google Scholar] [CrossRef]
- Middeke, J.M.; Herold, S.; Rücker-Braun, E.; Berdel, W.E.; Stelljes, M.; Kaufmann, M.; Schäfer-Eckart, K.; Baldus, C.D.; Stuhlmann, R.; Ho, A.D.; et al. TP53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2016, 172, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Nakamura, Y.; Kitao, H.; Shimokawa, M.; Kotani, D.; Bando, H.; Nishina, T.; Yamada, T.; Yuki, S.; Narita, Y.; et al. Mutational spectrum of TP53 gene correlates with nivolumab treatment efficacy in advanced gastric cancer (TP53MUT study). Br. J. Cancer 2023, 129, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shan, Q.; Guo, J.; Han, X.; Zhao, C.; Li, H.; Wang, Z. PDL1 high expression without TP53, KEAP1 and EPHA5 mutations could better predict survival for patients with NSCLC receiving atezolizumab. Lung Cancer 2021, 151, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, J.W.; Nho, N.T.; Kim, C.; Chen, Z.; Li, S.; Hill, C.; Ramalingam, S.S.; Sica, G.; Owonikoko, T.K. Impact of TP53 mutations on efficacy of PD-1 targeted immunotherapy in non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36 (Suppl. S15), e21090. [Google Scholar] [CrossRef]
- Zhu, M.; Kim, J.; Deng, Q.; Ricciuti, B.; Alessi, J.V.; Eglenen-Polat, B.; Bender, M.E.; Huang, H.-C.; Kowash, R.R.; Cuevas, I.; et al. Loss of p53 and mutational heterogeneity drives immune resistance in an autochthonous mouse lung cancer model with high tumor mutational burden. Cancer Cell 2023, 41, 1731–1748.e8. [Google Scholar] [CrossRef]
- Kim, J.; Jung, J.; Kim, K.; Lee, J.; Im, Y. TP53 mutations predict poor response to immunotherapy in patients with metastatic solid tumors. Cancer Med. 2023, 12, 12438–12451. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2022, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Forde, P.M.; Emens, L.A.; Yarchoan, M.; Smith, K.N.; Pardoll, D.M. Neoadjuvant immune checkpoint blockade: A window of opportunity to advance cancer immunotherapy. Cancer Cell 2023, 41, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer 2019, 7, 327. [Google Scholar] [CrossRef]
- McKean, M.; Tolcher, A.W.; Reeves, J.A.; Chmielowski, B.; Shaheen, M.F.; Beck, J.T.; Orloff, M.M.; Somaiah, N.; Van Tine, B.A.; Drabick, J.J.; et al. Newly updated activity results of alrizomadlin (APG-115), a novel MDM2/p53 inhibitor, plus pembrolizumab: Phase 2 study in adults and children with various solid tumors. J. Clin. Oncol. 2022, 40, 9517. [Google Scholar] [CrossRef]
- Focaccetti, C.; Benvenuto, M.; Pighi, C.; Vitelli, A.; Napolitano, F.; Cotugno, N.; Fruci, D.; Palma, P.; Rossi, P.; Bei, R.; et al. DNAM-1-chimeric receptor-engineered NK cells, combined with Nutlin-3a, more effectively fight neuroblastoma cells in vitro: A proof-of-concept study. Front. Immunol. 2022, 13, 886319. [Google Scholar] [CrossRef] [PubMed]
- Roshandel, E.; Tavakoli, F.; Parkhideh, S.; Akhlaghi, S.S.; Ardakani, M.T.; Soleimani, M. Post-hematopoietic stem cell transplantation relapse: Role of checkpoint inhibitors. Health Sci. Rep. 2022, 5, e536. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Yuan, Y.; La Marca, J.E.; Young, S.; Chang, C.; Whelan, L.; Ross, A.M.; Fischer, K.C.; Pomilio, G.; Morris, R.; et al. Putting the STING back into BH3-mimetic drugs for TP53-mutant blood cancers. Cancer Cell 2024, 42, 850–868. [Google Scholar] [CrossRef]
- Park, H.; Shapiro, G.; Gao, X.; Mahipal, A.; Starr, J.; Furqan, M.; Singh, P.; Ahrorov, A.; Gandhi, L.; Ghosh, A.; et al. Phase Ib study of eprenetapopt (APR-246) in combination with pembrolizumab in patients with advanced or metastatic solid tumors. ESMO Open 2022, 7, 100573. [Google Scholar] [CrossRef]
- Xiao, S.; Shi, F.; Song, H.; Cui, J.; Zheng, D.; Zhang, H.; Tan, K.; Wu, J.; Chen, X.; Wu, J.; et al. Characterization of the generic mutant p53-rescue compounds in a broad range of assays. Cancer Cell 2024, 42, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-S.; Harford, J.B.; Moghe, M.; Rait, A.; Chang, E.H. Combination with SGT-53 overcomes tumor resistance to a checkpoint inhibitor. OncoImmunology 2018, 7, e1484982. [Google Scholar] [CrossRef]
- Leung, C.P.; Barve, M.A.; Wu, M.-S.; Pirollo, K.F.; Strauss, J.F.; Liao, W.-C.; Yang, S.-H.; Nunan, R.A.; Adams, J.; Harford, J.B.; et al. A phase II trial combining tumor-targeting TP53 gene therapy with gemcitabine/nab-paclitaxel as a second-line treatment for metastatic pancreatic cancer. J. Clin. Oncol. 2021, 39 (Suppl. S15), 413. [Google Scholar] [CrossRef]
- Kong, N.; Tao, W.; Ling, X.; Wang, J.; Xiao, Y.; Shi, S.; Ji, X.; Shajii, A.; Gan, S.T.; Kim, N.Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, eaaw1565. [Google Scholar] [CrossRef]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.-C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Vale, N.R.; Zacharakis, N.; Krishna, S.; Yu, Z.; Gasmi, B.; Gartner, J.J.; Sindiri, S.; Malekzadeh, P.; Deniger, D.C.; et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor-Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res. 2022, 10, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, 1009. [Google Scholar] [CrossRef]
- Cui, Y.; Guo, G. Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef]
- Jaiswal, S. Clonal hematopoiesis and nonhematologic disorders. Blood 2020, 136, 1606–1614. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Type | Location | Amino Acid Change | Phenotypic Effect | LOF/GOF * | Examples of Associated Cancers | Citations |
|---|---|---|---|---|---|---|
| Missense | Exon 5 | R175H | Impaired DNA-binding ability, induced genetic instability | LOF | Breast, Lung, Ovarian | [21] |
| Missense | Exon 6 | Y220C | Altered protein conformation and DNA-binding ability | LOF | Breast, Lung, Head and Neck | [20,22] |
| Missense | Exon 7 | G245S | Altered DNA-binding domain | LOF | Ovarian, Breast, Lung | [23] |
| Missense | Exon 7 | G245D | Disrupted DNA-binding domain | LOF | Colorectal, Breast, Ovarian | [24] |
| Missense | Exon 8 | R248L | Reduced DNA-binding capacity | LOF | Various Cancers | [25] |
| Missense | Exon 8 | R248Q | Reduced DNA-binding capacity | LOF | Ovarian, Esophageal, Colorectal | [9] |
| Missense | Exon 8 | R248W | Reduced DNA-binding capacity | LOF and GOF | Breast, Colorectal, Pancreatic | [26] |
| Missense | Exon 8 | R249S | Reduced DNA-binding capacity | LOF and GOF | Liver, Ovarian, Lung | [27] |
| Missense | Exon 8 | R273C | Disrupted DNA-binding domain | LOF | Bladder, Lung, Colorectal | [28] |
| Missense | Exon 8 | R273L | Alters DNA-binding domain | LOF and GOF | Various Cancers | [29] |
| Missense | Exon 10 | R282W | Disruption of tetramerization and DNA-binding | LOF | Sarcoma, Colon, Brain | [30] |
| Mutation Type | Immunoregulatory Effects | Affected Immune Cells | Tumor Type | Cell Line or Primary Tumor | References |
|---|---|---|---|---|---|
| Deletion | Reduced ERAP1 and MHC 1 | T cells | Colon cancer | HCT116 | [36] |
| Upregulation of PD-L1 | T cells | Colon cancer, lung cancer | HCT116, H1299 | [37] | |
| Increased release of extracellular vesicles carrying PD-L1 | Macrophages | B cell malignancies | Patient primary tumors, Eμ-TCL1 | [38] | |
| Reduced cGAS/STING activation | Various immune cells | Lung cancer, colon cancer | A549, H1299, CT26 | [39] | |
| R172H (mouse) | Increased M2 polarization of macrophages caused by upregulation of CSF1 in mutant tumor cells | Macrophages | Esophageal squamous cell carcinoma | Chemically induced primary tumor in mice | [40] |
| Increased release of CXCL2, which causes neutrophil infiltration | Neutrophils | Pancreatic ductal adenocarcinoma | Primary murine tumor | [41] | |
| R175H | Reduced ERAP1 and MHC 1 | T cells | Colon cancer | HCT116 | [36] |
| Reduced ligands for NKG2D | NK cells | Lung cancer | H1299 | [42] | |
| G245D | Reduced ERAP1 and MHC 1 | T cells | Colon cancer | HCT116 | [36] |
| R248W | Reprogramming of macrophages into M2 phenotype | TME | Colon cancer | HCT116 | [43] |
| R249S (mouse) | Reduced cGAS/STING activation | Various immune cells | Lung cancer | 4T1 | [44] |
| R273C | Reduced ERAP1 and MHC 1 | T cells | Colon cancer | HCT116 | [36] |
| R280T | Reduced ERAP1 and MHC 1 | T cells | Colon cancer | HCT116 | [36] |
| Cancer Type | Immunotherapy | Findings | References |
|---|---|---|---|
| DLBCL | Anti-CD19 CAR-T | p53 alterations are associated with lower OR rate, shorter OS, and shorter PFS | [71] |
| AML, MDS | Hematopoietic stem cell transplantation (HSCT) | p53 mutations associated with lower 3-year RFS | [72,73,74] |
| Solid tumors (gastric cancer, colorectal cancer, breast cancer, NSCLC, etc.) | Immune checkpoint blockade (ICB) | p53 mutations correlate with poor efficacy of ICB | [75,76,77,78,79] |
| AML | Flotetuzumab (CD123 × CD3 bispecific) | Higher objective response rates in TP53-mutated cases | [80] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Tan, J.Y.M.; Chitkara, N.; Bhatt, S. TP53 Mutation-Mediated Immune Evasion in Cancer: Mechanisms and Therapeutic Implications. Cancers 2024, 16, 3069. https://doi.org/10.3390/cancers16173069
Wang C, Tan JYM, Chitkara N, Bhatt S. TP53 Mutation-Mediated Immune Evasion in Cancer: Mechanisms and Therapeutic Implications. Cancers. 2024; 16(17):3069. https://doi.org/10.3390/cancers16173069
Chicago/Turabian StyleWang, Chuqi, Jordan Yong Ming Tan, Nishtha Chitkara, and Shruti Bhatt. 2024. "TP53 Mutation-Mediated Immune Evasion in Cancer: Mechanisms and Therapeutic Implications" Cancers 16, no. 17: 3069. https://doi.org/10.3390/cancers16173069
APA StyleWang, C., Tan, J. Y. M., Chitkara, N., & Bhatt, S. (2024). TP53 Mutation-Mediated Immune Evasion in Cancer: Mechanisms and Therapeutic Implications. Cancers, 16(17), 3069. https://doi.org/10.3390/cancers16173069