
Cisplatin Resistance and Metabolism: Simplification of Complexity

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
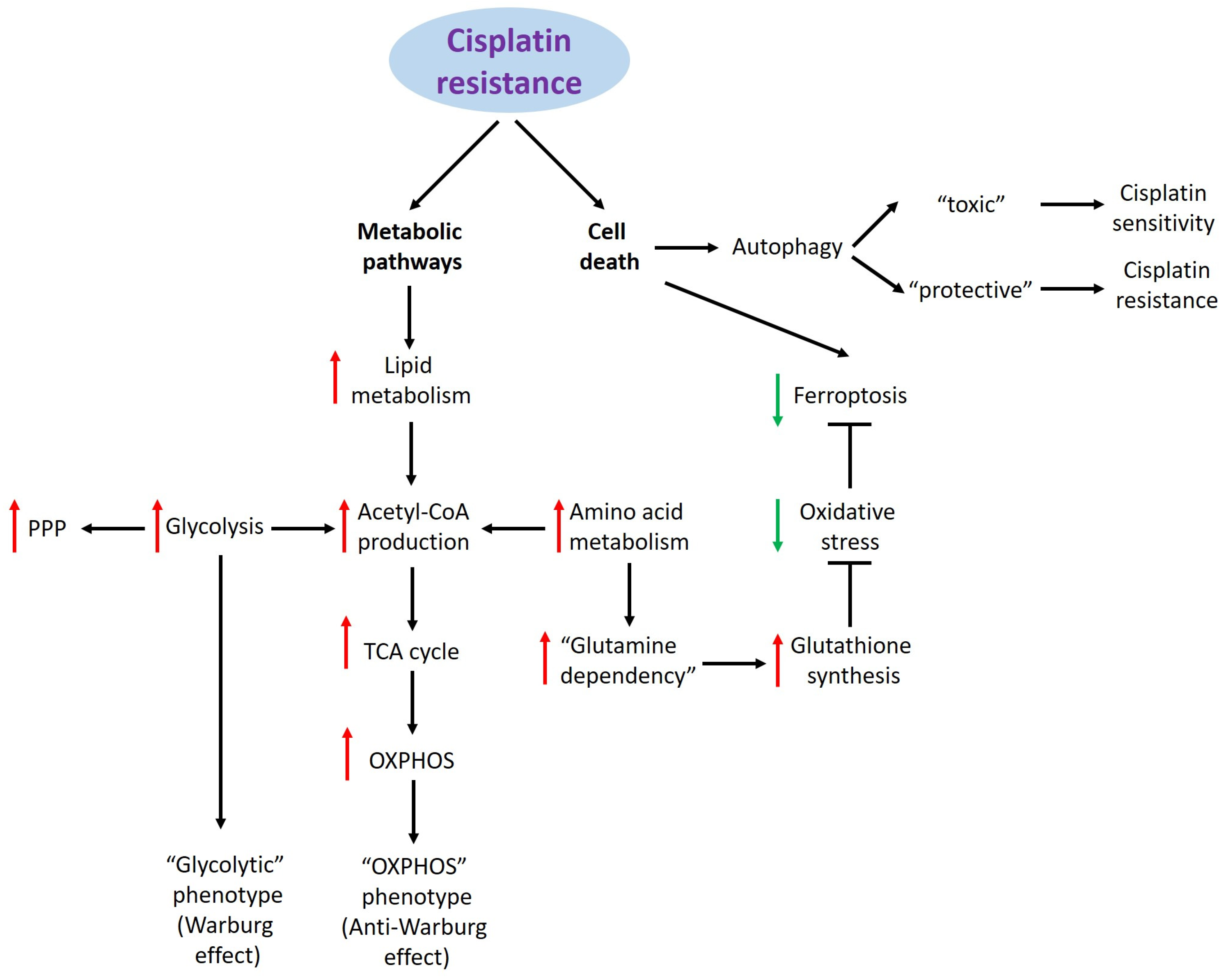
2. Cisplatin Resistance and Metabolic Pathways
2.1. Glucose Metabolism
2.2. Lipid Metabolism
2.3. Amino Acid Metabolism
3. Cisplatin Resistance and Cell Death
3.1. Ferroptosis
3.2. Autophagy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hussain, S.; Singh, A.; Nazir, S.U.; Tulsyan, S.; Khan, A.; Kumar, R.; Bashir, N.; Tanwar, P.; Mehrotra, R. Cancer Drug Resistance: A Fleet to Conquer. J. Cell Biochem. 2019, 120, 14213–14225. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Kopeina, G.S.; Imyanitov, E.N.; Zhivotovsky, B. Platinum Drugs and Taxanes: Can We Overcome Resistance? Cell Death Discov. 2021, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. Ther. 2019, 11, 97. [Google Scholar]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Scanlon, K.J.; Kashani-Sabet, M.; Tone, T.; Funato, T. Cisplatin Resistance in Human Cancers. Pharmacol. Ther. 1991, 52, 385–406. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Fu, R.; Zhao, B.; Chen, M.; Fu, X.; Zhang, Q.; Cui, Y.; Hu, X.; Zhou, W. Moving beyond Cisplatin Resistance: Mechanisms, Challenges, and Prospects for Overcoming Recurrence in Clinical Cancer Therapy. Med. Oncol. 2023, 41, 9. [Google Scholar] [CrossRef] [PubMed]
- Wangpaichitr, M.; Sullivan, E.J.; Theodoropoulos, G.; Wu, C.; You, M.; Feun, L.G.; Lampidis, T.J.; Kuo, M.T.; Savaraj, N. The Relationship of Thioredoxin-1 and Cisplatin Resistance: Its Impact on ROS and Oxidative Metabolism in Lung Cancer Cells. Mol. Cancer Ther. 2012, 11, 604–615. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, X.; Fu, J.; Xu, W.; Yuan, J. The Role of Tumour Metabolism in Cisplatin Resistance. Front. Mol. Biosci. 2021, 8, 691795. [Google Scholar] [CrossRef]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Links between Cancer Metabolism and Cisplatin Resistance. Int. Rev. Cell Mol. Biol. 2020, 354, 107–164. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Afonso, J.; Barbosa-Matos, C.; Silvestre, R.; Pereira-Vieira, J.; Gonçalves, S.M.; Mendes-Alves, C.; Parpot, P.; Pinto, J.; Carapito, Â.; Guedes de Pinho, P.; et al. Cisplatin-Resistant Urothelial Bladder Cancer Cells Undergo Metabolic Reprogramming beyond the Warburg Effect. Cancers 2024, 16, 1418. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Yang, F.; Zhou, C.; Chen, X.; Han, X.; Liu, X.; Ma, H.; Zheng, W. MCT1 Promotes the Cisplatin-Resistance by Antagonizing Fas in Epithelial Ovarian Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 2710–2718. [Google Scholar]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty Acid Synthase Overexpression: Target for Therapy and Reversal of Chemoresistance in Ovarian Cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Ralser, M.; Fan, J.; Rabinowitz, J.D. The Pentose Phosphate Pathway in Health and Disease. Nat. Metab. 2023, 5, 1275–1289. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Ancey, P.-B.; Contat, C.; Meylan, E. Glucose Transporters in Cancer—From Tumor Cells to the Tumor Microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Li, S.-J.; Yang, X.-N.; Qian, H.-Y. Antitumor Effects of WNT2B Silencing in GLUT1 Overexpressing Cisplatin Resistant Head and Neck Squamous Cell Carcinoma. Am. J. Cancer Res. 2015, 5, 300–308. [Google Scholar] [PubMed]
- Zhang, X.-Y.; Zhang, M.; Cong, Q.; Zhang, M.-X.; Zhang, M.-Y.; Lu, Y.-Y.; Xu, C.-J. Hexokinase 2 Confers Resistance to Cisplatin in Ovarian Cancer Cells by Enhancing Cisplatin-Induced Autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J.; et al. Enolase 1 Stimulates Glycolysis to Promote Chemoresistance in Gastric Cancer. Oncotarget 2017, 8, 47691–47708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cong, Q.; Zhang, X.-Y.; Zhang, M.-X.; Lu, Y.-Y.; Xu, C.-J. Pyruvate Dehydrogenase Kinase 1 Contributes to Cisplatin Resistance of Ovarian Cancer through EGFR Activation. J. Cell Physiol. 2019, 234, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.-L.; Park, J.Y.; Kim, E.H.; Jang, H.J.; Kwon, M. Activation of Mitochondrial Oxidation by PDK2 Inhibition Reverses Cisplatin Resistance in Head and Neck Cancer. Cancer Lett. 2016, 371, 20–29. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.-F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Fan, T.W.M.; Lane, A.N.; Higashi, R.M.; Farag, M.A.; Gao, H.; Bousamra, M.; Miller, D.M. Altered Regulation of Metabolic Pathways in Human Lung Cancer Discerned by (13)C Stable Isotope-Resolved Metabolomics (SIRM). Mol. Cancer 2009, 8, 41. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Bedi, M.; Ray, M.; Ghosh, A. Active Mitochondrial Respiration in Cancer: A Target for the Drug. Mol. Cell Biochem. 2022, 477, 345–361. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Theodoropoulos, G.; Nguyen, D.J.M.; Wu, C.; Spector, S.A.; Feun, L.G.; Savaraj, N. Cisplatin Resistance and Redox-Metabolic Vulnerability: A Second Alteration. Int. J. Mol. Sci. 2021, 22, 7379. [Google Scholar] [CrossRef]
- Jiang, Z.; He, J.; Zhang, B.; Wang, L.; Long, C.; Zhao, B.; Yang, Y.; Du, L.; Luo, W.; Hu, J.; et al. A Potential “Anti-Warburg Effect” in Circulating Tumor Cell-Mediated Metastatic Progression? Aging Dis. 2024, 16, 1–14. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive Oxygen Species in Cancer: Current Findings and Future Directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Shirato, A.; Kikugawa, T.; Miura, N.; Tanji, N.; Takemori, N.; Higashiyama, S.; Yokoyama, M. Cisplatin Resistance by Induction of Aldo-Keto Reductase Family 1 Member C2 in Human Bladder Cancer Cells. Oncol. Lett. 2014, 7, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-J.; Finkel, T.; Shen, D.-W.; Yin, J.-J.; Aszalos, A.; Gottesman, M.M. SIRT1 Contributes in Part to Cisplatin Resistance in Cancer Cells by Altering Mitochondrial Metabolism. Mol. Cancer Res. 2008, 6, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef]
- Buckley, A.M.; Bibby, B.A.; Dunne, M.R.; Kennedy, S.A.; Davern, M.B.; Kennedy, B.N.; Maher, S.G.; O’Sullivan, J. Characterisation of an Isogenic Model of Cisplatin Resistance in Oesophageal Adenocarcinoma Cells. Pharmaceuticals 2019, 12, 33. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and Metabolic Differences to Kill Cisplatin Resistant Lung Cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef]
- Gao, Y.; Dorn, P.; Liu, S.; Deng, H.; Hall, S.R.R.; Peng, R.-W.; Schmid, R.A.; Marti, T.M. Cisplatin-Resistant A549 Non-Small Cell Lung Cancer Cells Can Be Identified by Increased Mitochondrial Mass and Are Sensitive to Pemetrexed Treatment. Cancer Cell Int. 2019, 19, 317. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Yapryntseva, M.A.; Pervushin, N.V.; Tsvetcov, R.I.; Zhivotovsky, B.; Kopeina, G.S. Cancer Drug Resistance: Targeting Proliferation or Programmed Cell Death. Cells 2024, 13, 388. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid Metabolism Reprogramming and Its Potential Targets in Cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Uddin, S.; Jehan, Z.; Ahmed, M.; Alyan, A.; Al-Dayel, F.; Hussain, A.; Bavi, P.; Al-Kuraya, K.S. Overexpression of Fatty Acid Synthase in Middle Eastern Epithelial Ovarian Carcinoma Activates AKT and Its Inhibition Potentiates Cisplatin-Induced Apoptosis. Mol. Med. 2011, 17, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.-L. The Effect of FASN Inhibition on the Growth and Metabolism of a Cisplatin-Resistant Ovarian Carcinoma Model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Todor, I.N.; Lukyanova, N.Y.; Chekhun, V.F. The Lipid Content of Cisplatin- and Doxorubicin-Resistant MCF-7 Human Breast Cancer Cells. Exp. Oncol. 2012, 34, 97–100. [Google Scholar] [PubMed]
- Martinho, N.; Santos, T.C.B.; Florindo, H.F.; Silva, L.C. Cisplatin-Membrane Interactions and Their Influence on Platinum Complexes Activity and Toxicity. Front. Physiol. 2018, 9, 1898. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, X.-J.; Yang, P.; Zhao, M.; Lv, L.-X.; Zhang, G.-D.; Wang, Q.; Zhang, L. Alkylglyceronephosphate Synthase (AGPS) Alters Lipid Signaling Pathways and Supports Chemotherapy Resistance of Glioma and Hepatic Carcinoma Cell Lines. Asian Pac. J. Cancer Prev. 2014, 15, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Lee, S.; Zhu, W.-G.; Lee, O.-J.; Yun, S.J.; Kim, J.; Park, S. Glucose-Derived Acetate and ACSS2 as Key Players in Cisplatin Resistance in Bladder Cancer. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 413–421. [Google Scholar] [CrossRef]
- Obrist, F.; Michels, J.; Durand, S.; Chery, A.; Pol, J.; Levesque, S.; Joseph, A.; Astesana, V.; Pietrocola, F.; Wu, G.S.; et al. Metabolic Vulnerability of Cisplatin-Resistant Cancers. EMBO J. 2018, 37, e98597. [Google Scholar] [CrossRef]
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered Glutamine Metabolism in Platinum Resistant Ovarian Cancer. Oncotarget 2016, 7, 41637–41649. [Google Scholar] [CrossRef]
- Duan, G.; Shi, M.; Xie, L.; Xu, M.; Wang, Y.; Yan, H.; Zhuge, Y.; Zou, X. Increased Glutamine Consumption in Cisplatin-Resistant Cells Has a Negative Impact on Cell Growth. Sci. Rep. 2018, 8, 4067. [Google Scholar] [CrossRef]
- Witte, A.-B.; Anestål, K.; Jerremalm, E.; Ehrsson, H.; Arnér, E.S.J. Inhibition of Thioredoxin Reductase but Not of Glutathione Reductase by the Major Classes of Alkylating and Platinum-Containing Anticancer Compounds. Free Radic. Biol. Med. 2005, 39, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Rigas, B. The Thioredoxin System Mediates Redox-Induced Cell Death in Human Colon Cancer Cells: Implications for the Mechanism of Action of Anticancer Agents. Cancer Res. 2008, 68, 8269–8277. [Google Scholar] [CrossRef]
- Nguyen, D.J.M.; Theodoropoulos, G.; Li, Y.-Y.; Wu, C.; Sha, W.; Feun, L.G.; Lampidis, T.J.; Savaraj, N.; Wangpaichitr, M. Targeting the Kynurenine Pathway for the Treatment of Cisplatin-Resistant Lung Cancer. Mol. Cancer Res. 2020, 18, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ou, Y.; Zhou, Q.; Liang, Y.; Li, W.; Chen, Y.; Chen, W.; Wu, S.; Chen, Y.; Dai, X.; et al. Methionine Orchestrates the Metabolism Vulnerability in Cisplatin Resistant Bladder Cancer Microenvironment. Cell Death Dis. 2023, 14, 525. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberley, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic Silencing of Argininosuccinate Synthetase Confers Resistance to Platinum-Induced Cell Death but Collateral Sensitivity to Arginine Auxotrophy in Ovarian Cancer. Int. J. Cancer 2009, 125, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Kharkar, P.S.; Gandhi, N.S.; Kaur, E.; Dutt, S.; Nandave, M. Novel Analogs of Sulfasalazine as System Xc- Antiporter Inhibitors: Insights from the Molecular Modeling Studies. Drug Dev. Res. 2019, 80, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Merlos Rodrigo, M.A.; Jimenez Jimemez, A.M.; Haddad, Y.; Bodoor, K.; Adam, P.; Krizkova, S.; Heger, Z.; Adam, V. Metallothionein Isoforms as Double Agents—Their Roles in Carcinogenesis, Cancer Progression and Chemoresistance. Drug Resist. Updates 2020, 52, 100691. [Google Scholar] [CrossRef]
- Li, X.-F.; Hua, T.; Li, Y.; Tian, Y.-J.; Huo, Y.; Kang, S. The HSP70 Gene Predicts Prognosis and Response to Chemotherapy in Epithelial Ovarian Cancer. Ann. Transl. Med. 2021, 9, 806. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Sonego, M.; Pucci, B.; Addi, L.; Iannelli, F.; Capone, F.; Alfano, L.; Roca, M.S.; Milone, M.R.; Moccia, T.; et al. HSP90 Identified by a Proteomic Approach as Druggable Target to Reverse Platinum Resistance in Ovarian Cancer. Mol. Oncol. 2021, 15, 1005–1023. [Google Scholar] [CrossRef]
- Jordan, P.; Carmo-Fonseca, M. Molecular Mechanisms Involved in Cisplatin Cytotoxicity. Cell Mol. Life Sci. 2000, 57, 1229–1235. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 2022, 12, 858462. [Google Scholar] [CrossRef]
- Kapper, C.; Oppelt, P.; Arbeithuber, B.; Gyunesh, A.A.; Vilusic, I.; Stelzl, P.; Rezk-Füreder, M. Targeting Ferroptosis in Ovarian Cancer: Novel Strategies to Overcome Chemotherapy Resistance. Life Sci. 2024, 349, 122720. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhu, S.; Zhao, R.; Liu, W.; Jin, L.; Ren, X.; He, H. Targeting Ferroptosis as a Potential Strategy to Overcome the Resistance of Cisplatin in Oral Squamous Cell Carcinoma. Front. Pharmacol. 2024, 15, 1402514. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, K. The Induction of Ferroptosis by Impairing STAT3/Nrf2/GPx4 Signaling Enhances the Sensitivity of Osteosarcoma Cells to Cisplatin. Cell Biol. Int. 2019, 43, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Liu, Z.; Zhang, Z.; Zhang, Z. USP3 Promotes Cisplatin Resistance in Non-Small Cell Lung Cancer Cells by Suppressing ACOT7-Regulated Ferroptosis. Anticancer. Drugs 2024, 35, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.-L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of Ferroptotic Cell Death for Overcoming Cisplatin Resistance of Head and Neck Cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Lei, X.-Y.; He, K.-Y.; Guo, J.-R.; Liu, M.-J.; Li, J.-Q.; Li, Q.-T.; Jiang, Z.-H.; Zhang, L.; Wu, D.-H.; et al. HMGA1 Drives Chemoresistance in Esophageal Squamous Cell Carcinoma by Suppressing Ferroptosis. Cell Death Dis. 2024, 15, 158. [Google Scholar] [CrossRef]
- Niu, L.; Li, Y.; Huang, G.; Huang, W.; Fu, J.; Feng, L. FAM120A Deficiency Improves Resistance to Cisplatin in Gastric Cancer by Promoting Ferroptosis. Commun. Biol. 2024, 7, 399. [Google Scholar] [CrossRef]
- Deng, J.; Lin, X.; Qin, J.; Li, Q.; Zhang, Y.; Zhang, Q.; Ji, C.; Shen, S.; Li, Y.; Zhang, B.; et al. SPTBN2 Suppresses Ferroptosis in NSCLC Cells by Facilitating SLC7A11 Membrane Trafficking and Localization. Redox Biol. 2024, 70, 103039. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, X.; Chen, Y.; Tang, Q.; He, C.; Ding, X.; Hu, J.; Cai, Z.; Li, X.; Qiao, H.; et al. Targeting PAX8 Sensitizes Ovarian Cancer Cells to Ferroptosis by Inhibiting Glutathione Synthesis. Apoptosis 2024. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and Function. Genes. Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Zhu, L.; Zhu, Y.; Han, S.; Chen, M.; Song, P.; Dai, D.; Xu, W.; Jiang, T.; Feng, L.; Shin, V.Y.; et al. Impaired Autophagic Degradation of lncRNA ARHGAP5-AS1 Promotes Chemoresistance in Gastric Cancer. Cell Death Dis. 2019, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gewirtz, D.A. Is Autophagy Always a Barrier to Cisplatin Therapy? Biomolecules 2022, 12, 463. [Google Scholar] [CrossRef]
- Pervushin, N.V.; Kopeina, G.S.; Zhivotovsky, B. Bcl-B: An “Unknown” Protein of the Bcl-2 Family. Biol. Direct 2023, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Gogvadze, V.; Zhivotovsky, B. Mitophagy in Carcinogenesis and Cancer Treatment. Discov. Oncol. 2021, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, Y.; Liu, Z.; Wang, J.; Bai, J.; Du, R.; Long, M.; Shang, Z. Mitophagy-Mediated Tumor Dormancy Protects Cancer Cells from Chemotherapy. Biomedicines 2024, 12, 305. [Google Scholar] [CrossRef]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and Multidrug Resistance in Cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Patel, N.H.; Xu, J.; Saleh, T.; Wu, Y.; Lima, S.; Gewirtz, D.A. Influence of Nonprotective Autophagy and the Autophagic Switch on Sensitivity to Cisplatin in Non-Small Cell Lung Cancer Cells. Biochem. Pharmacol. 2020, 175, 113896. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The Four Faces of Autophagy: Implications for Cancer Therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- López-Portugués, C.; Montes-Bayón, M.; Díez, P. Biomarkers in Ovarian Cancer: Towards Personalized Medicine. Proteomes 2024, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Miserocchi, G.; Spadazzi, C.; Calpona, S.; De Rosa, F.; Usai, A.; De Vita, A.; Liverani, C.; Cocchi, C.; Vanni, S.; Calabrese, C.; et al. Precision Medicine in Head and Neck Cancers: Genomic and Preclinical Approaches. JPM 2022, 12, 854. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Biomarkers | Functions | Rate in (CR) Cisplatin-Resistant Cells | Tumor Models (References) |
|---|---|---|---|
| GLUTs | Glucose uptake | Controversial (GLUT1—Increased [21]/Decreased [14]; GLUT3—Increased [14]) | Head and neck squamous cell carcinoma (HNSCC) [21] Urothelial bladder cancer [14] |
| Hexokinase 2 (HK2) | Glucose-6-phosphate generation (1 step of glycolysis) | Increased | Ovarian cancer [22] |
| Enolase 1 (ENO1) | Phosphoenolpyruvate (9 step of glycolysis) | Increased | Gastric cancer [23] |
| Pyruvate dehydrogenase kinase 1 (PDK1) | Pyruvate dehydrogenase inhibition | Increased | Ovarian cancer [24] |
| Pyruvate dehydrogenase kinase 2 (PDK2) | Head and neck cancer [25] | ||
| Monocarboxylate transporters (MCTs) | Lactate acid efflux | Controversial (MCT1—Increased [15]; MCT4—Decreased [14]) | Ovarian cancer [15] Urothelial bladder cancer [14] |
| Biomarkers | Functions | Rate in Cisplatin-Resistant (CR) Cells | Tumor Models (References) |
|---|---|---|---|
| Alkylglyceronephosphate synthase (AGPS) | Phospholipid synthesis | Increased | Glioma [54] |
| Acyl-coenzyme A synthetase 2 (ACSS2) | Acetyl-CoA production | Increased | Bladder cancer [55] |
| Acetyl-CoA-carboxylase (ACC) | Fatty acid synthesis | Increased | Lung cancer [9] |
| Fatty acid synthase (FAS) | Fatty acid synthesis | Controversial (Increased [9]/Decreased [16]) | Lung cancer [9] Ovarian cancer [16] |
| Biomarkers | Functions | Rate in Cisplatin-Resistant (CR) Cells | Tumor Models (References) |
|---|---|---|---|
| Alanine-serine-cysteine transporter 2 (ASCT2/SLC1A5) | Glutamine transport | Increased | Ovarian cancer [57] |
| Glutaminase (GLS) | Glutamine hydrolysis to glutamate | Increased | Ovarian cancer [57] |
| Glutamate oxaloacetate transaminase 1 (GOT1) | Oxaloacetate generation | Increased | Various cancer models [58] |
| Glutamate dehydrogenase (GLUD1) | α-ketoglutarate generation | Decreased | Various cancer models [58] |
| Indoleamine 2,3-dioxygenase-1 (IDO1) | Tryptophan utilization | Increased | Lung cancer [61] |
| Argininosuccinate synthetase (ASS1) | Arginine synthesis | Decreased | Ovarian cancer [63] |
| Methionine adenosyl transferase IIa (MAT2A) | S-adenosylmethionine (SAM) generation | Increased | Bladder cancer [62] |
| Biomarkers | Signaling Pathway | Functions | Ferroptosis Regulation | Tumor Models (References) |
|---|---|---|---|---|
| Glutathione peroxidase 4 (GPX4) | STAT3/Nrf2 (transcriptional activation) | Lipid peroxidation protection | Inhibition | Osteosarcoma [75] Non-small cell lung cancer (NSCLC) [76] |
| Cystine/glutamate antiporter SLC7A11/xCT | Cystine transport | Inhibition | Head and neck cancer [77] | |
| HMGA1/ATF4 (transcriptional activation) | Esophageal squamous cell carcinoma (ESCC) [78] | |||
| FAM120A (translational activation) | Gastric cancer [79] | |||
| SPTBN2 (posttranslational activation) | NSCLC [80] | |||
| Glutamate–cysteine ligase (GCLC) | PAX8 (transcriptional activation) | Glutathione synthesis | Inhibition | Ovarian cancer [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pervushin, N.V.; Yapryntseva, M.A.; Panteleev, M.A.; Zhivotovsky, B.; Kopeina, G.S. Cisplatin Resistance and Metabolism: Simplification of Complexity. Cancers 2024, 16, 3082. https://doi.org/10.3390/cancers16173082
Pervushin NV, Yapryntseva MA, Panteleev MA, Zhivotovsky B, Kopeina GS. Cisplatin Resistance and Metabolism: Simplification of Complexity. Cancers. 2024; 16(17):3082. https://doi.org/10.3390/cancers16173082
Chicago/Turabian StylePervushin, Nikolay V., Maria A. Yapryntseva, Mikhail A. Panteleev, Boris Zhivotovsky, and Gelina S. Kopeina. 2024. "Cisplatin Resistance and Metabolism: Simplification of Complexity" Cancers 16, no. 17: 3082. https://doi.org/10.3390/cancers16173082