
Chondroitin Sulfate Proteoglycan 4 (CSPG4) as an Emerging Target for Immunotherapy to Treat Melanoma

 , , , , ,
, , , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
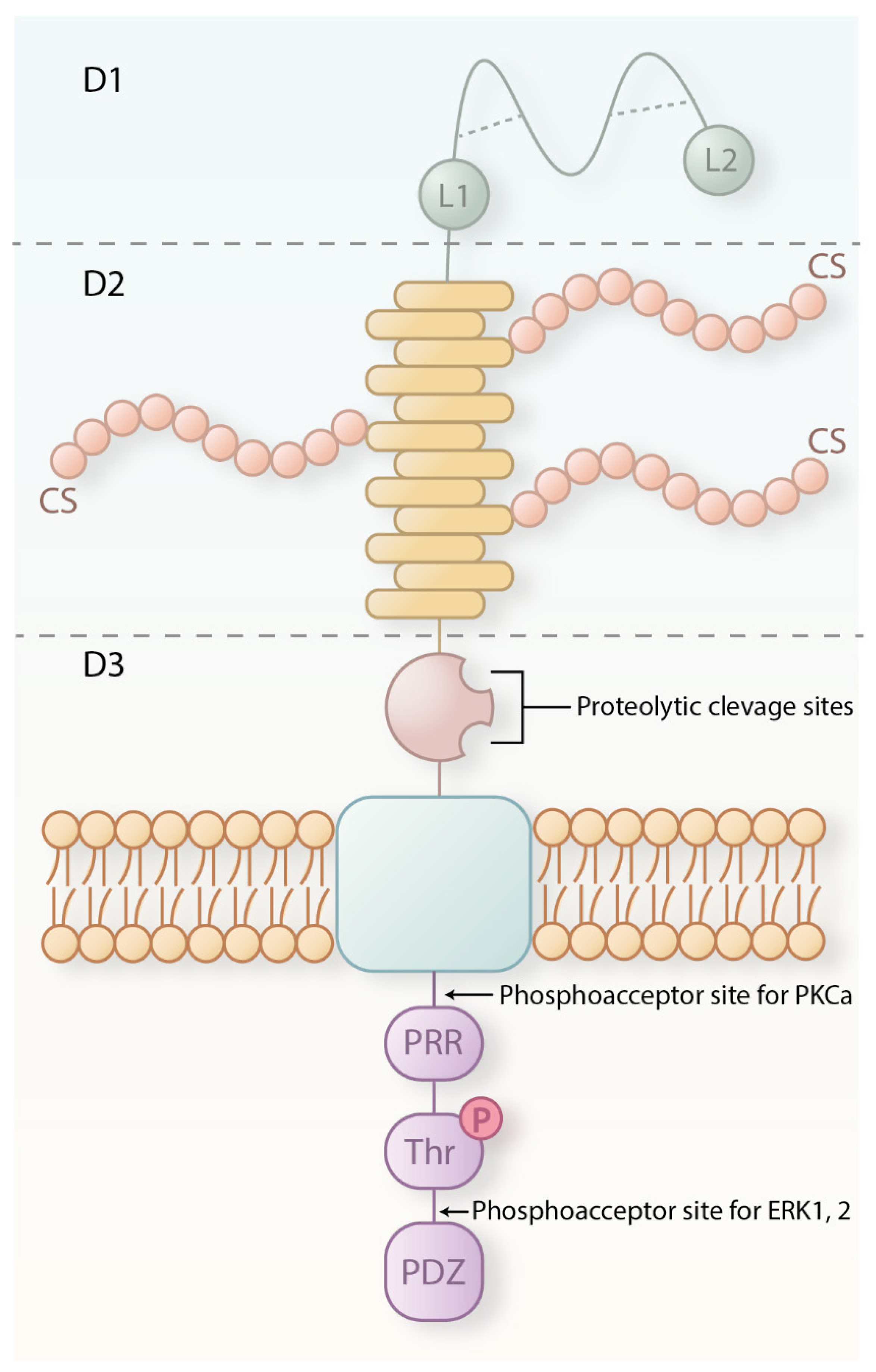
2. CSPG4 Structure, Expression and Functions in Health
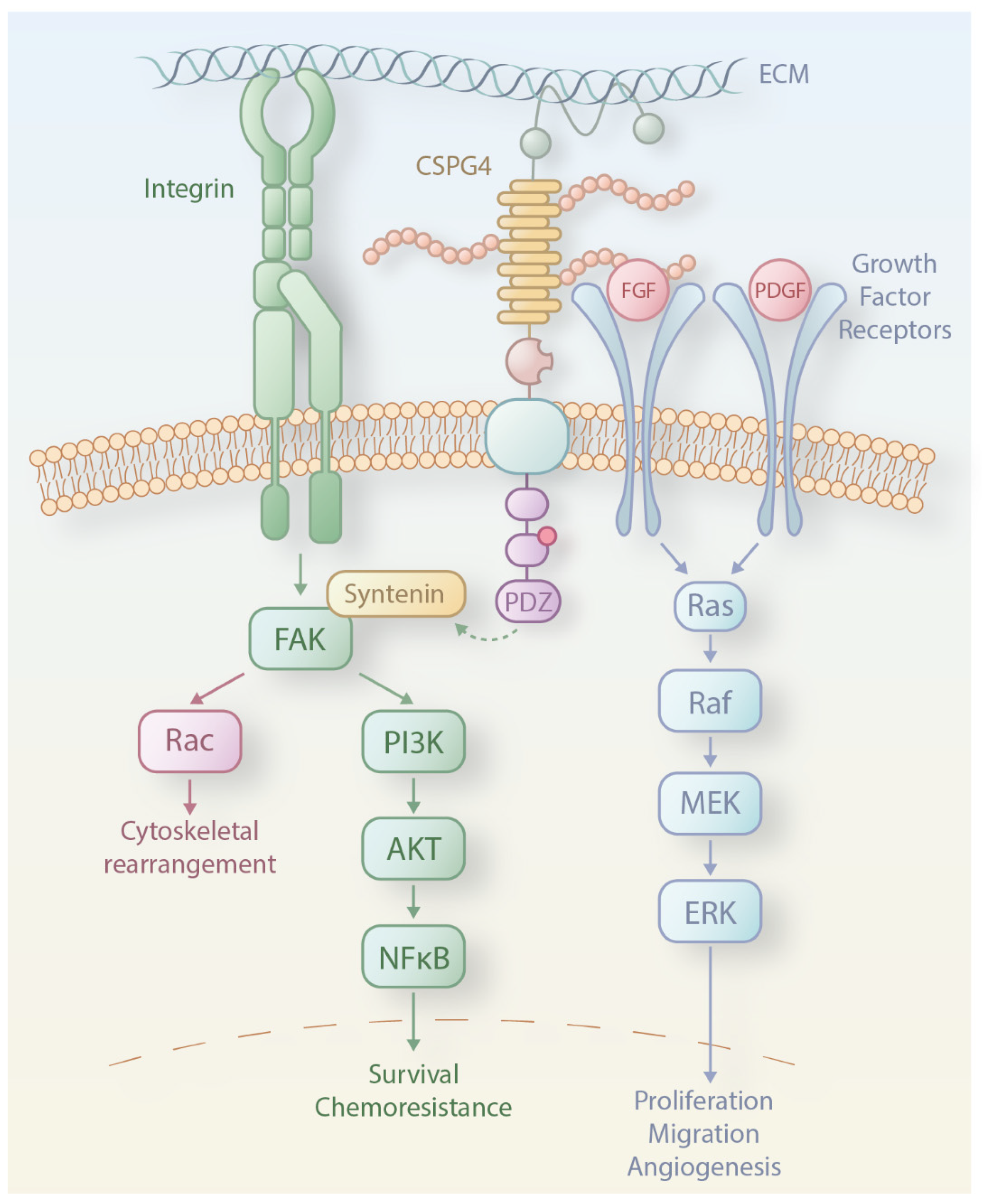
3. CSPG4 Expression and Functions in Melanoma
4. CSPG4 and the Immune Response
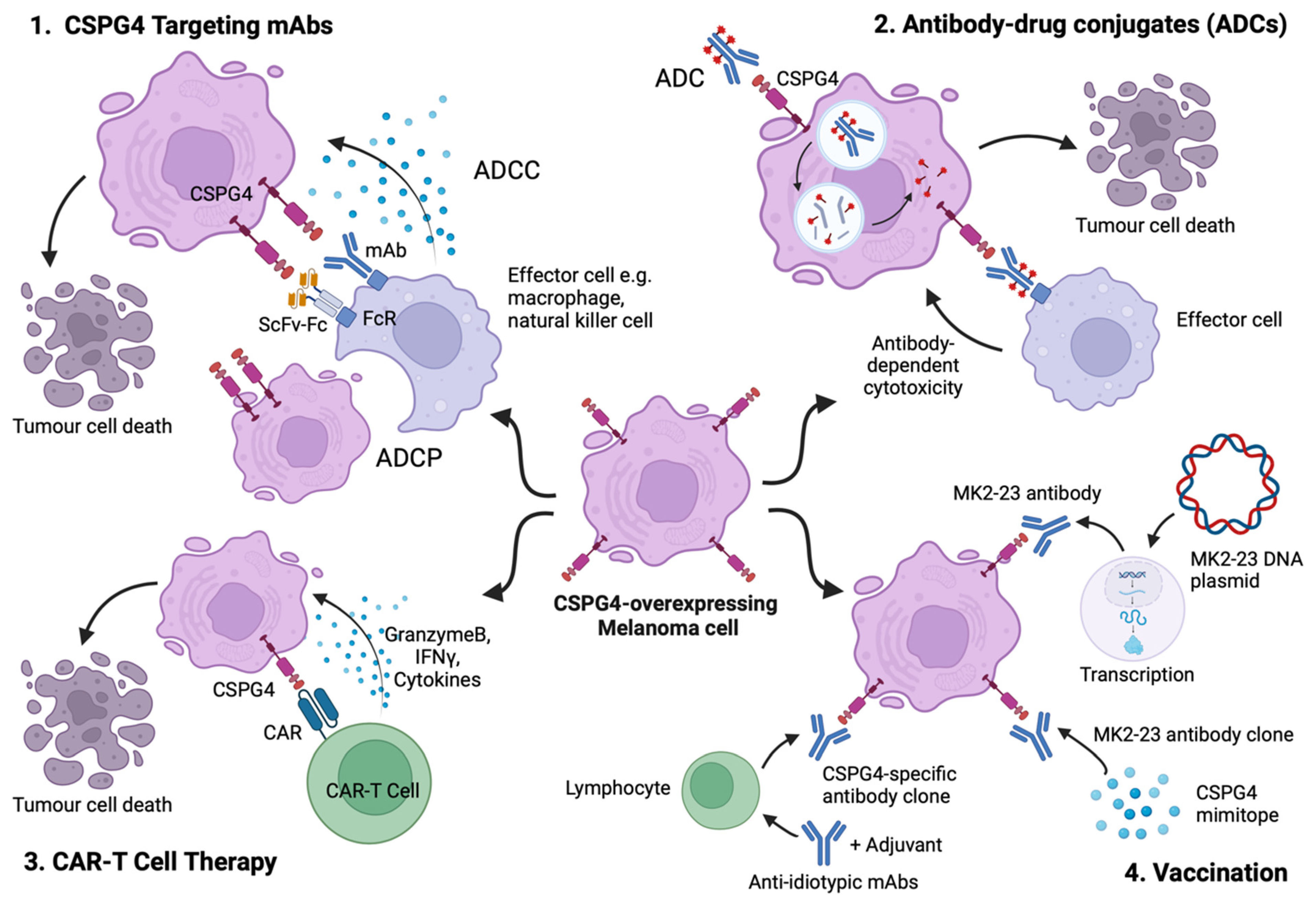
5. CSPG4 as a Target for Cancer Immunotherapy
5.1. Targeting CSPG4 Using Monoclonal Antibodies
5.2. Antibody–Drug Conjugates (ADCs) Directed at CSPG4-Expressing Melanoma
5.3. CSPG4-Specific Chimeric Antigen Receptors (CARs)
5.4. Anti-Idiotypic and Mimotope Vaccines Inducing Humoral Responses against CSPG4
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Melanoma Skin Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/melanoma-skin-cancer#heading-Three (accessed on 22 June 2024).
- Non-Melanoma Skin Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/non-melanoma-skin-cancer (accessed on 22 June 2024).
- Melanoma Survival. Available online: https://www.cancerresearchuk.org/about-cancer/melanoma/survival (accessed on 22 June 2024).
- Wolchok, J.D. How Is Immunotherapy for Melanoma Changing the Outlook for Patients? Available online: https://www.cancerresearch.org/cancer-types/melanoma (accessed on 12 November 2023).
- Rashid, S.; Shaughnessy, M.; Tsao, H. Melanoma classification and management in the era of molecular medicine. Dermatol. Clin. 2023, 41, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Savoia, P.; Zavattaro, E.; Cremona, O. Clinical Implications of Acquired BRAF Inhibitors Resistance in Melanoma. Int. J. Mol. Sci. 2020, 21, 9730. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Lee, N.; Zakka, L.R.; Mihm, M.C.; Schatton, T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology 2016, 48, 177–187. [Google Scholar] [CrossRef]
- Pitcovski, J.; Shahar, E.; Aizenshtein, E.; Gorodetsky, R. Melanoma antigens and related immunological markers. Crit. Rev. Oncol. Hematol. 2017, 115, 36–49. [Google Scholar] [CrossRef]
- Kreidieh, F.Y.; Tawbi, H.A. The introduction of LAG-3 checkpoint blockade in melanoma: Immunotherapy landscape beyond PD-1 and CTLA-4 inhibition. Ther. Adv. Med. Oncol. 2023, 15, 17588359231186027. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chavez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suarez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.S.; Imai, K.; Natali, P.G.; Ferrone, S. Distribution and molecular characterization of a cell-surface and a cytoplasmic antigen detectable in human melanoma cells with monoclonal antibodies. Int. J. Cancer 1981, 28, 293–300. [Google Scholar] [CrossRef] [PubMed]
- CSPG4 Tissue Expression. Available online: https://www.proteinatlas.org/ENSG00000173546 (accessed on 22 June 2024).
- Ghali, L.; Wong, S.T.; Tidman, N.; Quinn, A.; Philpott, M.P.; Leigh, I.M. Epidermal and hair follicle progenitor cells express melanoma-associated chondroitin sulfate proteoglycan core protein. J. Investig. Dermatol. 2004, 122, 433–442. [Google Scholar] [CrossRef]
- Schlingemann, R.O.; Rietveld, F.J.; de Waal, R.M.; Ferrone, S.; Ruiter, D.J. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am. J. Pathol. 1990, 136, 1393–1405. [Google Scholar] [PubMed]
- Maciag, P.C.; Seavey, M.M.; Pan, Z.K.; Ferrone, S.; Paterson, Y. Cancer immunotherapy targeting the high molecular weight melanoma-associated antigen protein results in a broad antitumor response and reduction of pericytes in the tumor vasculature. Cancer Res. 2008, 68, 8066–8075. [Google Scholar] [CrossRef] [PubMed]
- Stallcup, W.B.; Huang, F.J. A role for the NG2 proteoglycan in glioma progression. Cell Adhes. Migr. 2008, 2, 192–201. [Google Scholar] [CrossRef]
- Nicolosi, P.A.; Dallatomasina, A.; Perris, R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015, 5, 530–544. [Google Scholar] [CrossRef]
- Price, M.A.; Colvin Wanshura, L.E.; Yang, J.; Carlson, J.; Xiang, B.; Li, G.; Ferrone, S.; Dudek, A.Z.; Turley, E.A.; McCarthy, J.B. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011, 24, 1148–1157. [Google Scholar] [CrossRef]
- Nishiyama, A.; Dahlin, K.J.; Prince, J.T.; Johnstone, S.R.; Stallcup, W.B. The primary structure of NG2, a novel membrane-spanning proteoglycan. J. Cell Biol. 1991, 114, 359–371. [Google Scholar] [CrossRef]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef]
- Harrer, D.C.; Dörrie, J.; Schaft, N. CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities-Merits and Challenges. Int. J. Mol. Sci. 2019, 20, 5942. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, J.; Makagiansar, I.T.; Stallcup, W.B. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol. Biol. Cell 2004, 15, 3580–3590. [Google Scholar] [CrossRef] [PubMed]
- Makagiansar, I.T.; Williams, S.; Mustelin, T.; Stallcup, W.B. Differential phosphorylation of NG2 proteoglycan by ERK and PKCalpha helps balance cell proliferation and migration. J. Cell Biol. 2007, 178, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef] [PubMed]
- Barritt, D.S.; Pearn, M.T.; Zisch, A.H.; Lee, S.S.; Javier, R.T.; Pasquale, E.B.; Stallcup, W.B. The multi-PDZ domain protein MUPP1 is a cytoplasmic ligand for the membrane-spanning proteoglycan NG2. J. Cell Biochem. 2000, 79, 213–224. [Google Scholar] [CrossRef]
- Yang, J.; Price, M.A.; Neudauer, C.L.; Wilson, C.; Ferrone, S.; Xia, H.; Iida, J.; Simpson, M.A.; McCarthy, J.B. Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK activation by distinct mechanisms. J. Cell Biol. 2004, 165, 881–891. [Google Scholar] [CrossRef]
- Nishiyama, A.; Lin, X.H.; Giese, N.; Heldin, C.H.; Stallcup, W.B. Interaction between NG2 proteoglycan and PDGF a-receptor on 02A progenitor cells is required for optimal response to PDGF. J. Neurosci. Res. 1996, 43, 315–330. [Google Scholar] [CrossRef]
- Kucharova, K.; Stallcup, W.B. The NG2 proteoglycan promotes oligodendrocyte progenitor proliferation and developmental myelination. Neuroscience 2010, 166, 185–194. [Google Scholar] [CrossRef]
- Tang, F.; Lord, M.S.; Stallcup, W.B.; Whitelock, J.M. Cell surface chondroitin sulphate proteoglycan 4 (CSPG4) binds to the basement membrane heparan sulphate proteoglycan, perlecan, and is involved in cell adhesion. J. Biochem. 2018, 163, 399–412. [Google Scholar] [CrossRef]
- Van Sinderen, M.; Cuman, C.; Winship, A.; Menkhorst, E.; Dimitriadis, E. The chrondroitin sulfate proteoglycan (CSPG4) regulates human trophoblast function. Placenta 2013, 34, 907–912. [Google Scholar] [CrossRef]
- Sakry, D.; Neitz, A.; Singh, J.; Frischknecht, R.; Marongiu, D.; Biname, F.; Perera, S.S.; Endres, K.; Lutz, B.; Radyushkin, K.; et al. Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. 2014, 12, e1001993. [Google Scholar] [CrossRef] [PubMed]
- Legg, J.; Jensen, U.B.; Broad, S.; Leigh, I.; Watt, F.M. Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003, 130, 6049–6063. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol. Biol. Lett. 2017, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Rolih, V.; Barutello, G.; Iussich, S.; De Maria, R.; Quaglino, E.; Buracco, P.; Cavallo, F.; Riccardo, F. CSPG4: A prototype oncoantigen for translational immunotherapy studies. J. Transl. Med. 2017, 15, 151. [Google Scholar] [CrossRef]
- Grossauer, A.; Uranowska, K.; Kitzwögerer, M.; Mostegel, M.; Breiteneder, H.; Hafner, C. Immunohistochemical detection of the chondroitin sulfate proteoglycan 4 protein in primary and metastatic melanoma. Oncol. Lett. 2023, 26, 382. [Google Scholar] [CrossRef]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef]
- Chauhan, J.; Grandits, M.; Palhares, L.; Mele, S.; Nakamura, M.; Lopez-Abente, J.; Crescioli, S.; Laddach, R.; Romero-Clavijo, P.; Cheung, A.; et al. Anti-cancer pro-inflammatory effects of an IgE antibody targeting the melanoma-associated antigen chondroitin sulfate proteoglycan 4. Nat. Commun. 2023, 14, 2192. [Google Scholar] [CrossRef]
- Vergilis, I.J.; Szarek, M.; Ferrone, S.; Reynolds, S.R. Presence and prognostic significance of melanoma-associated antigens CYT-MAA and HMW-MAA in serum of patients with melanoma. J. Investig. Dermatol. 2005, 125, 526–531. [Google Scholar] [CrossRef]
- Hsu, S.-H.C.; Nadesan, P.; Puviindran, V.; Stallcup, W.B.; Kirsch, D.G.; Alman, B.A. Effects of chondroitin sulfate proteoglycan 4 (NG2/CSPG4) on soft-tissue sarcoma growth depend on tumor developmental stage. J. Biol. Chem. 2018, 293, 2466–2475. [Google Scholar] [CrossRef]
- Wang, J.; Svendsen, A.; Kmiecik, J.; Immervoll, H.; Skaftnesmo, K.O.; Planagumà, J.; Reed, R.K.; Bjerkvig, R.; Miletic, H.; Enger, P.Ø.; et al. Targeting the NG2/CSPG4 Proteoglycan Retards Tumour Growth and Angiogenesis in Preclinical Models of GBM and Melanoma. PLoS ONE 2011, 6, e23062. [Google Scholar] [CrossRef]
- Wang, X.; Osada, T.; Wang, Y.; Yu, L.; Sakakura, K.; Katayama, A.; McCarthy, J.B.; Brufsky, A.; Chivukula, M.; Khoury, T.; et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J. Natl. Cancer Inst. 2010, 102, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-cadherin cell surface regulation in cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Price, M.A.; Li, G.Y.; Bar-Eli, M.; Salgia, R.; Jagedeeswaran, R.; Carlson, J.H.; Ferrone, S.; Turley, E.A.; McCarthy, J.B. Melanoma proteoglycan modifies gene expression to stimulate tumor cell motility, growth, and epithelial-to-mesenchymal transition. Cancer Res. 2009, 69, 7538–7547. [Google Scholar] [CrossRef]
- Cooney, C.A.; Jousheghany, F.; Yao-Borengasser, A.; Phanavanh, B.; Gomes, T.; Kieber-Emmons, A.M.; Siegel, E.R.; Suva, L.J.; Ferrone, S.; Kieber-Emmons, T.; et al. Chondroitin sulfates play a major role in breast cancer metastasis: A role for CSPG4 and CHST11 gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. 2011, 13, R58. [Google Scholar] [CrossRef]
- Iida, J.; Pei, D.; Kang, T.; Simpson, M.A.; Herlyn, M.; Furcht, L.T.; McCarthy, J.B. Melanoma chondroitin sulfate proteoglycan regulates matrix metalloproteinase-dependent human melanoma invasion into type I collagen. J. Biol. Chem. 2001, 276, 18786–18794. [Google Scholar] [CrossRef]
- Erfurt, C.; Sun, Z.; Haendle, I.; Schuler-Thurner, B.; Heirman, C.; Thielemans, K.; van der Bruggen, P.; Schuler, G.; Schultz, E.S. Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J. Immunol. 2007, 178, 7703–7709. [Google Scholar] [CrossRef]
- Yu, L.; Favoino, E.; Wang, Y.; Ma, Y.; Deng, X.; Wang, X. The CSPG4-specific monoclonal antibody enhances and prolongs the effects of the BRAF inhibitor in melanoma cells. Immunol. Res. 2011, 50, 294–302. [Google Scholar] [CrossRef]
- Chekenya, M.; Krakstad, C.; Svendsen, A.; Netland, I.A.; Staalesen, V.; Tysnes, B.B.; Selheim, F.; Wang, J.; Sakariassen, P.; Sandal, T.; et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene 2008, 27, 5182–5194. [Google Scholar] [CrossRef]
- Volz, N.B.; Stintzing, S.; Zhang, W.; Yang, D.; Ning, Y.; Wakatsuki, T.; El-Khoueiry, R.E.; Li, J.E.; Kardosh, A.; Loupakis, F.; et al. Genes involved in pericyte-driven tumor maturation predict treatment benefit of first-line FOLFIRI plus bevacizumab in patients with metastatic colorectal cancer. Pharmacogenomics J. 2015, 15, 69–76. [Google Scholar] [CrossRef]
- Gao, Q.; Lu, J.; Huo, Y.; Baby, N.; Ling, E.A.; Dheen, S.T. NG2, a member of chondroitin sulfate proteoglycans family mediates the inflammatory response of activated microglia. Neuroscience 2010, 165, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Akiyama, H.; Harikai, N.; Toyoda, H.; Toida, T.; Maitani, T.; Imanari, T. Effect of chondroitin sulfate on murine splenocytes sensitized with ovalbumin. Immunol. Lett. 2002, 84, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Roehrl, M.H. Glycosaminoglycans are a potential cause of rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2002, 99, 14362–14367. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, J.; Tykocinski, M.L. Differential effects of chondroitin sulfates A and B on monocyte and B-cell activation: Evidence for B-cell activation via a CD44-dependent pathway. Blood 1998, 92, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, E.; Yoshihara, R.; Tai, A.; Yamamoto, I.; Gohda, E. PKC- and PI3K-dependent but ERK-independent proliferation of murine splenic B cells stimulated by chondroitin sulfate B. Immunol. Lett. 2005, 99, 80–84. [Google Scholar] [CrossRef]
- Yang, R.; Yan, Z.; Chen, F.; Hansson, G.K.; Kiessling, R. Hyaluronic acid and chondroitin sulphate A rapidly promote differentiation of immature DC with upregulation of costimulatory and antigen-presenting molecules, and enhancement of NF-kappaB and protein kinase activity. Scand. J. Immunol. 2002, 55, 2–13. [Google Scholar] [CrossRef]
- Rolls, A.; Cahalon, L.; Bakalash, S.; Avidan, H.; Lider, O.; Schwartz, M. A sulfated disaccharide derived from chondroitin sulfate proteoglycan protects against inflammation-associated neurodegeneration. FASEB J. 2006, 20, 547–549. [Google Scholar] [CrossRef]
- Palhares, L.; Brito, A.S.; de Lima, M.A.; Nader, H.B.; London, J.A.; Barsukov, I.L.; Andrade, G.P.V.; Yates, E.A.; Chavante, S.F. A further unique chondroitin sulfate from the shrimp Litopenaeus vannamei with antithrombin activity that modulates acute inflammation. Carbohydr. Polym. 2019, 222, 115031. [Google Scholar] [CrossRef]
- Palhares, L.; Barbosa, J.S.; Scortecci, K.C.; Rocha, H.A.O.; Brito, A.S.; Chavante, S.F. In vitro antitumor and anti-angiogenic activities of a shrimp chondroitin sulfate. Int. J. Biol. Macromol. 2020, 162, 1153–1165. [Google Scholar] [CrossRef]
- Imai, K.; Molinaro, G.A.; Ferrone, S. Monoclonal antibodies to human melanoma-associated antigens. Transplant. Proc. 1980, 12, 380–383. [Google Scholar]
- Hafner, C.; Breiteneder, H.; Ferrone, S.; Thallinger, C.; Wagner, S.; Schmidt, W.M.; Jasinska, J.; Kundi, M.; Wolff, K.; Zielinski, C.C.; et al. Suppression of human melanoma tumor growth in SCID mice by a human high molecular weight-melanoma associated antigen (HMW-MAA) specific monoclonal antibody. Int. J. Cancer 2005, 114, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Uranowska, K.; Samadaei, M.; Kalic, T.; Pinter, M.; Breiteneder, H.; Hafner, C. A chondroitin sulfate proteoglycan 4-specific monoclonal antibody inhibits melanoma cell invasion in a spheroid model. Int. J. Oncol. 2021, 59, 70. [Google Scholar] [CrossRef] [PubMed]
- Obrist, R.; Schmidli, J.; Muller, R.; Gallati, H.; Obrecht, J.P. Acute and subacute toxicity of chemotactic conjugates between monoclonal antibody and fMet-Leu-Phe in humans: A phase I clinical trial. Cancer Immunol. Immunother. 1991, 32, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef]
- Karagiannis, P.; Gilbert, A.E.; Josephs, D.H.; Ali, N.; Dodev, T.; Saul, L.; Correa, I.; Roberts, L.; Beddowes, E.; Koers, A.; et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J. Clin. Investig. 2013, 123, 1457–1474. [Google Scholar] [CrossRef]
- Williams, I.P.; Crescioli, S.; Sow, H.S.; Bax, H.J.; Hobbs, C.; Ilieva, K.M.; French, E.; Pellizzari, G.; Cox, V.; Josephs, D.H.; et al. In vivo safety profile of a CSPG4-directed IgE antibody in an immunocompetent rat model. mAbs 2020, 12, 1685349. [Google Scholar] [CrossRef]
- Kondrashov, A.; Sapkota, S.; Sharma, A.; Riano, I.; Kurzrock, R.; Adashek, J.J. Antibody-Drug Conjugates in Solid Tumor Oncology: An Effectiveness Payday with a Targeted Payload. Pharmaceutics 2023, 15, 2160. [Google Scholar] [CrossRef]
- Esapa, B.; Jiang, J.; Cheung, A.; Chenoweth, A.; Thurston, D.E.; Karagiannis, S.N. Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers. Cancers 2023, 15, 1845. [Google Scholar] [CrossRef]
- Yang, H.M.; Reisfeld, R.A. Doxorubicin conjugated with a monoclonal antibody directed to a human melanoma-associated proteoglycan suppresses the growth of established tumor xenografts in nude mice. Proc. Natl. Acad. Sci. USA 1988, 85, 1189–1193. [Google Scholar] [CrossRef]
- Ghose, T.; Ferrone, S.; Blair, A.H.; Kralovec, Y.; Temponi, M.; Singh, M.; Mammen, M. Regression of human melanoma xenografts in nude mice injected with methotrexate linked to monoclonal antibody 225.28 to human high molecular weight-melanoma associated antigen. Cancer Immunol. Immunother. 1991, 34, 90–96. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Rizvi, S.M.; Jager, M.J.; Conway, R.M.; Billson, F.A.; Allen, B.J.; Madigan, M.C. In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4365–4371. [Google Scholar] [CrossRef] [PubMed]
- Raja, C.; Graham, P.; Abbas Rizvi, S.M.; Song, E.; Goldsmith, H.; Thompson, J.; Bosserhoff, A.; Morgenstern, A.; Apostolidis, C.; Kearsley, J.; et al. Interim analysis of toxicity and response in phase 1 trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2007, 6, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.J.; Singla, A.A.; Rizvi, S.M.; Graham, P.; Bruchertseifer, F.; Apostolidis, C.; Morgenstern, A. Analysis of patient survival in a Phase I trial of systemic targeted alpha-therapy for metastatic melanoma. Immunotherapy 2011, 3, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Falvo, E.; Tremante, E.; Fraioli, R.; Leonetti, C.; Zamparelli, C.; Boffi, A.; Morea, V.; Ceci, P.; Giacomini, P. Antibody-drug conjugates: Targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale 2013, 5, 12278–12285. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Crescioli, S.; Mele, S.; Sachouli, E.; Cheung, A.; Chui, C.K.; Andriollo, P.; Jackson, P.J.M.; Lacy, K.E.; Spicer, J.F.; et al. A Novel Antibody-Drug Conjugate (ADC) Delivering a DNA Mono-Alkylating Payload to Chondroitin Sulfate Proteoglycan (CSPG4)-Expressing Melanoma. Cancers 2020, 12, 1029. [Google Scholar] [CrossRef]
- Eng, M.S.; Kaur, J.; Prasmickaite, L.; Engesaeter, B.O.; Weyergang, A.; Skarpen, E.; Berg, K.; Rosenblum, M.G.; Maelandsmo, G.M.; Hogset, A.; et al. Enhanced targeting of triple-negative breast carcinoma and malignant melanoma by photochemical internalization of CSPG4-targeting immunotoxins. Photochem. Photobiol. Sci. 2018, 17, 539–551. [Google Scholar] [CrossRef]
- Schwenkert, M.; Birkholz, K.; Schwemmlein, M.; Kellner, C.; Kugler, M.; Peipp, M.; Nettelbeck, D.M.; Schuler-Thurner, B.; Schaft, N.; Dorrie, J.; et al. A single chain immunotoxin, targeting the melanoma-associated chondroitin sulfate proteoglycan, is a potent inducer of apoptosis in cultured human melanoma cells. Melanoma Res. 2008, 18, 73–84. [Google Scholar] [CrossRef]
- de Bruyn, M.; Rybczynska, A.A.; Wei, Y.; Schwenkert, M.; Fey, G.H.; Dierckx, R.A.; van Waarde, A.; Helfrich, W.; Bremer, E. Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP)-targeted delivery of soluble TRAIL potently inhibits melanoma outgrowth in vitro and in vivo. Mol. Cancer 2010, 9, 301. [Google Scholar] [CrossRef]
- Burns, W.R.; Zhao, Y.; Frankel, T.L.; Hinrichs, C.S.; Zheng, Z.; Xu, H.; Feldman, S.A.; Ferrone, S.; Rosenberg, S.A.; Morgan, R.A. A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res. 2010, 70, 3027–3033. [Google Scholar] [CrossRef]
- Reinhold, U.; Liu, L.; Lüdtke-Handjery, H.C.; Heuser, C.; Hombach, A.; Wang, X.; Tilgen, W.; Ferrone, S.; Abken, H. Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J. Investig. Dermatol. 1999, 112, 744–750. [Google Scholar] [CrossRef]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del Vecchio, M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Wiesinger, M.; März, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Uslu, U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. Int. J. Mol. Sci. 2018, 19, 2365. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, M.; März, J.; Kummer, M.; Schuler, G.; Dörrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers 2019, 11, 1198. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Wang, Q.J.; Rosenberg, S.A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: Tumor escape by antigen loss and loss of MHC expression. J. Immunother. 2004, 27, 184–190. [Google Scholar] [CrossRef]
- Märkl, F.; Benmebarek, M.R.; Keyl, J.; Cadilha, B.L.; Geiger, M.; Karches, C.; Obeck, H.; Schwerdtfeger, M.; Michaelides, S.; Briukhovetska, D.; et al. Bispecific antibodies redirect synthetic agonistic receptor modified T cells against melanoma. J. Immunother. Cancer 2023, 11, e006436. [Google Scholar] [CrossRef]
- Kropp, K.N.; Fatho, M.; Huduti, E.; Faust, M.; Lübcke, S.; Lennerz, V.; Paschen, A.; Theobald, M.; Wölfel, T.; Wölfel, C. Targeting the melanoma-associated antigen CSPG4 with HLA-C*07:01-restricted T-cell receptors. Front. Immunol. 2023, 14, 1245559. [Google Scholar] [CrossRef]
- Feldman, S.A.; Assadipour, Y.; Kriley, I.; Goff, S.L.; Rosenberg, S.A. Adoptive Cell Therapy--Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin. Oncol. 2015, 42, 626–639. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Teppert, K.; Winter, N.; Herbel, V.; Brandes, C.; Lennartz, S.; Engert, F.; Kaiser, A.; Schaser, T.; Lock, D. Combining CSPG4-CAR and CD20-CCR for treatment of metastatic melanoma. Front. Immunol. 2023, 14, 1178060. [Google Scholar] [CrossRef]
- Pinc, A.; Somasundaram, R.; Wagner, C.; Hörmann, M.; Karanikas, G.; Jalili, A.; Bauer, W.; Brunner, P.; Grabmeier-Pfistershammer, K.; Gschaider, M.; et al. Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol. Ther. 2012, 20, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, A.; Chen, Z.J.; Kageshita, T.; Yang, H.; Yamada, M.; Baskind, P.; Goldberg, N.; Puccio, C.; Ahmed, T.; Arlin, Z. Active specific immunotherapy in patients with melanoma. A clinical trial with mouse antiidiotypic monoclonal antibodies elicited with syngeneic anti-high-molecular-weight-melanoma-associated antigen monoclonal antibodies. J. Clin. Investig. 1990, 86, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, A.; Chen, Z.J.; Yang, H.; Wong, G.Y.; Ferrone, S. Human high molecular weight melanoma-associated antigen (HMW-MAA) mimicry by mouse anti-idiotypic monoclonal antibody MK2-23: Induction of humoral anti-HMW-MAA immunity and prolongation of survival in patients with stage IV melanoma. Proc. Natl. Acad. Sci. USA 1992, 89, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ko, E.C.; Peng, L.; Gillies, S.D.; Ferrone, S. Human high molecular weight melanoma-associated antigen mimicry by mouse anti-idiotypic monoclonal antibody MK2-23: Enhancement of immunogenicity of anti-idiotypic monoclonal antibody MK2-23 by fusion with interleukin 2. Cancer Res. 2005, 65, 6976–6983. [Google Scholar] [CrossRef]
- Barucca, A.; Capitani, M.; Cesca, M.; Tomassoni, D.; Kazmi, U.; Concetti, F.; Vincenzetti, L.; Concetti, A.; Venanzi, F.M. Recombinant DNA technology for melanoma immunotherapy: Anti-Id DNA vaccines targeting high molecular weight melanoma-associated antigen. Mol. Biotechnol. 2014, 56, 1032–1039. [Google Scholar] [CrossRef]
- Wagner, S.; Hafner, C.; Allwardt, D.; Jasinska, J.; Ferrone, S.; Zielinski, C.C.; Scheiner, O.; Wiedermann, U.; Pehamberger, H.; Breiteneder, H. Vaccination with a human high molecular weight melanoma-associated antigen mimotope induces a humoral response inhibiting melanoma cell growth in vitro. J. Immunol. 2005, 174, 976–982. [Google Scholar] [CrossRef]
- Riemer, A.B.; Hantusch, B.; Sponer, B.; Kraml, G.; Hafner, C.; Zielinski, C.C.; Scheiner, O.; Pehamberger, H.; Jensen-Jarolim, E. High-molecular-weight melanoma-associated antigen mimotope immunizations induce antibodies recognizing melanoma cells. Cancer Immunol. Immunother. 2005, 54, 677–684. [Google Scholar] [CrossRef]
- Luo, W.; Ko, E.; Hsu, J.C.; Wang, X.; Ferrone, S. Targeting melanoma cells with human high molecular weight-melanoma associated antigen-specific antibodies elicited by a peptide mimotope: Functional effects. J. Immunol. 2006, 176, 6046–6054. [Google Scholar] [CrossRef]
- Alfonso, S.; Valdes-Zayas, A.; Santiesteban, E.R.; Flores, Y.I.; Areces, F.; Hernandez, M.; Viada, C.E.; Mendoza, I.C.; Guerra, P.P.; Garcia, E.; et al. A randomized, multicenter, placebo-controlled clinical trial of racotumomab-alum vaccine as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin. Cancer Res. 2014, 20, 3660–3671. [Google Scholar] [CrossRef]
- MacLeod, M.K.; McKee, A.S.; David, A.; Wang, J.; Mason, R.; Kappler, J.W.; Marrack, P. Vaccine adjuvants aluminum and monophosphoryl lipid A provide distinct signals to generate protective cytotoxic memory CD8 T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7914–7919. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Du, G.; He, C.; Jiang, M.; Mou, X.; Xue, J.; Sun, X. Tumor cell membrane enveloped aluminum phosphate nanoparticles for enhanced cancer vaccination. J. Control. Release 2020, 326, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Mauldin, I.S.; Wages, N.A.; Stowman, A.M.; Wang, E.; Olson, W.C.; Deacon, D.H.; Smith, K.T.; Galeassi, N.; Teague, J.E.; Smolkin, M.E.; et al. Topical treatment of melanoma metastases with imiquimod, plus administration of a cancer vaccine, promotes immune signatures in the metastases. Cancer Immunol. Immunother. 2016, 65, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Saito, Y.; Iida-Norita, R.; Afroj, T.; Refaat, A.; Hazama, D.; Komori, S.; Ohata, S.; Takai, T.; Oduori, O.S.; Kotani, T.; et al. Preclinical evaluation of the efficacy of an antibody to human SIRPalpha for cancer immunotherapy in humanized mouse models. Front. Immunol. 2023, 14, 1294814. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef]
- Yang, J.; Price, M.A.; Wanshura, L.E.C.; He, J.; Yi, M.; Welch, D.R.; Li, G.; Conner, S.; Sachs, J.; Turley, E.A.; et al. Chondroitin sulfate proteoglycan 4 enhanced melanoma motility and growth requires a cysteine in the core protein transmembrane domain. Melanoma Res. 2019, 29, 365–375. [Google Scholar] [CrossRef]
- Gargett, T.; Truong, N.T.H.; Gardam, B.; Yu, W.; Ebert, L.M.; Johnson, A.; Yeo, E.C.F.; Wittwer, N.L.; Tapia Rico, G.; Logan, J.; et al. Safety and biological outcomes following a phase 1 trial of GD2-specific CAR-T cells in patients with GD2-positive metastatic melanoma and other solid cancers. J. Immunother. Cancer 2024, 12, e008659. [Google Scholar] [CrossRef]
- Lin, F.Y.; Stuckert, A.; Tat, C.; White, M.; Ruggieri, L.; Zhang, H.; Mehta, B.; Lapteva, N.; Mei, Z.; Major, A.; et al. Phase I Trial of GD2.CART Cells Augmented with Constitutive Interleukin-7 Receptor for Treatment of High-Grade Pediatric CNS Tumors. J. Clin. Oncol. 2024, 42, 2769–2779. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Pellegatta, S.; Savoldo, B.; Di Ianni, N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10, eaao2731. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Yong, J.; Zauner, R.; Wally, V.; Whitelock, J.; Sajinovic, M.; Kopecki, Z.; Liang, K.; Scott, K.F.; Mellick, A.S. Chondroitin Sulfate Proteoglycan 4 as a Marker for Aggressive Squamous Cell Carcinoma. Cancers 2022, 14, 5564. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, A.R.K.; Yang, J.; Price, M.A.; Forster, C.L.; Riddle, M.J.; Ebens, C.L.; Albert, F.W.; Giubellino, A.; McCarthy, J.B.; Tolar, J. Chondroitin sulfate proteoglycan 4 (CSPG4) increases invasion of recessive dystrophic epidermolysis bullosa-associated cutaneous squamous cell carcinoma by modifying TGFbeta signaling. Br. J. Dermatol. 2024, ljae295. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Habib, S.; Alexandru, M.; Chauhan, J.; Evan, T.; Troka, J.M.; Rahimi, A.; Esapa, B.; Tull, T.J.; Ng, W.Z.; et al. Chondroitin Sulfate Proteoglycan 4 (CSPG4) as an Emerging Target for Immunotherapy to Treat Melanoma. Cancers 2024, 16, 3260. https://doi.org/10.3390/cancers16193260
Chen X, Habib S, Alexandru M, Chauhan J, Evan T, Troka JM, Rahimi A, Esapa B, Tull TJ, Ng WZ, et al. Chondroitin Sulfate Proteoglycan 4 (CSPG4) as an Emerging Target for Immunotherapy to Treat Melanoma. Cancers. 2024; 16(19):3260. https://doi.org/10.3390/cancers16193260
Chicago/Turabian StyleChen, Xinyi, Shabana Habib, Madalina Alexandru, Jitesh Chauhan, Theodore Evan, Joanna M. Troka, Avigail Rahimi, Benjamina Esapa, Thomas J. Tull, Wen Zhe Ng, and et al. 2024. "Chondroitin Sulfate Proteoglycan 4 (CSPG4) as an Emerging Target for Immunotherapy to Treat Melanoma" Cancers 16, no. 19: 3260. https://doi.org/10.3390/cancers16193260
APA StyleChen, X., Habib, S., Alexandru, M., Chauhan, J., Evan, T., Troka, J. M., Rahimi, A., Esapa, B., Tull, T. J., Ng, W. Z., Fitzpatrick, A., Wu, Y., Geh, J. L. C., Lloyd-Hughes, H., Palhares, L. C. G. F., Adams, R., Bax, H. J., Whittaker, S., Jacków-Malinowska, J., & Karagiannis, S. N. (2024). Chondroitin Sulfate Proteoglycan 4 (CSPG4) as an Emerging Target for Immunotherapy to Treat Melanoma. Cancers, 16(19), 3260. https://doi.org/10.3390/cancers16193260