Mechanistic Insights on Microbiota-Mediated Development and Progression of Esophageal Cancer


Abstract
Simple Summary
Abstract
1. Introduction
1.1. Risk Factors
1.1.1. Common Non-Modifiable Risk Factors
1.1.2. Common Modifiable Risk Factors
1.1.3. GERD and BE
1.1.4. Highlighted Risk Factors in East Asia
1.1.5. Transition to Microbiota Focus
2. Epidemiology Findings
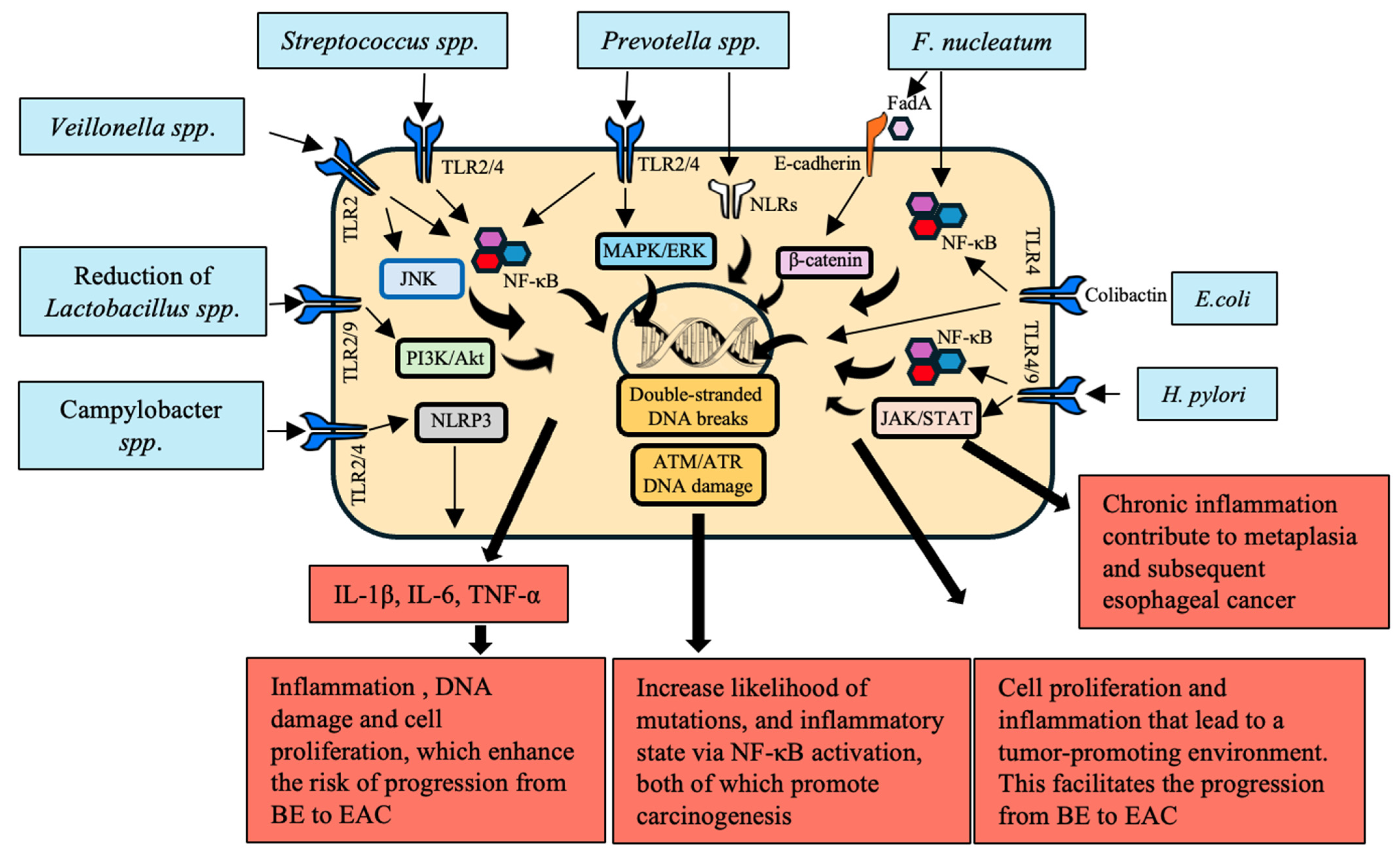
3. Chronic Inflammation
3.1. Immune Regulation by Microbiota
3.2. Activation of Inflammatory and Signaling Pathways
3.3. TME and Immune Reprogramming
4. Microbial Dysbiosis
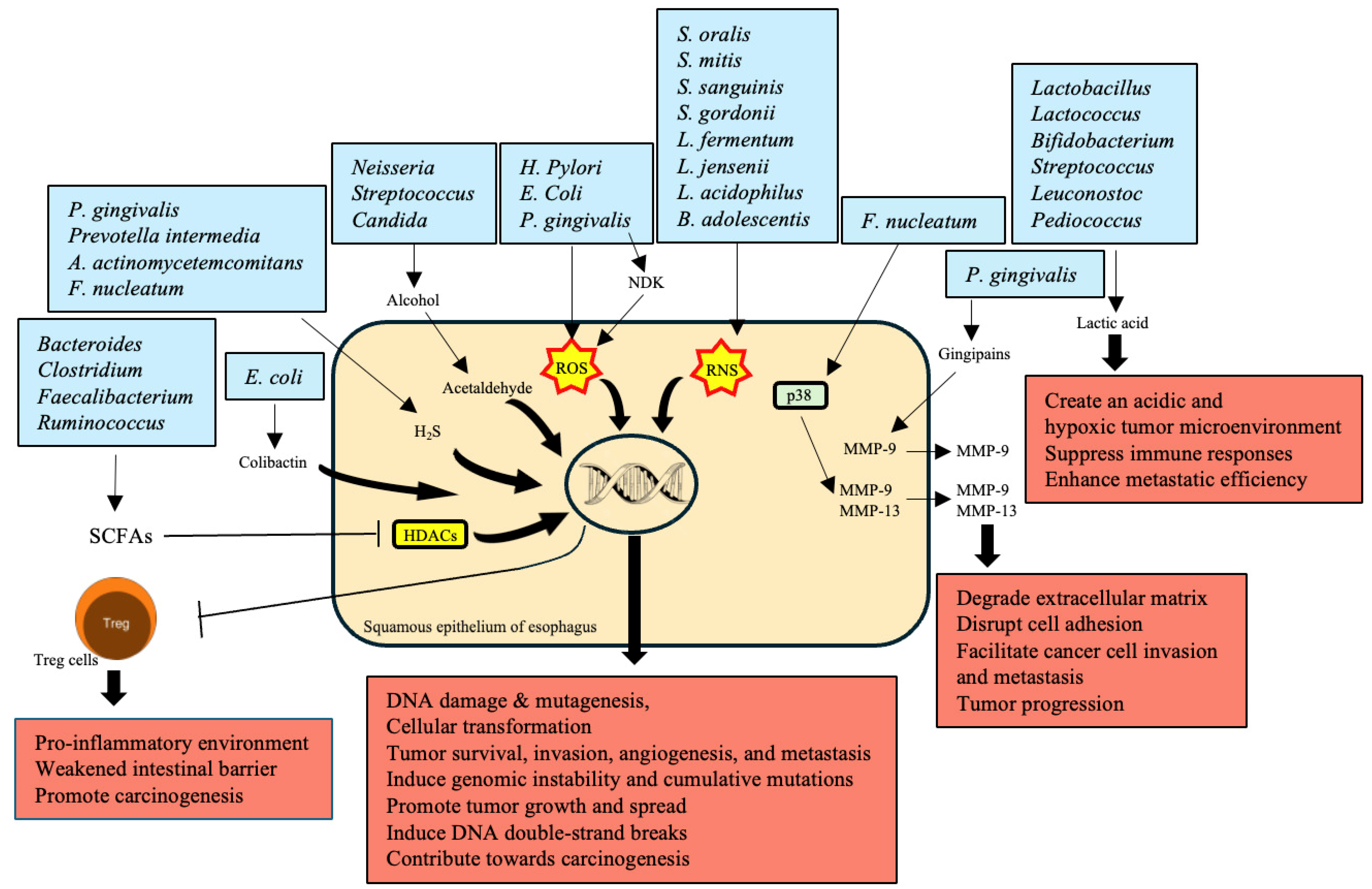
5. Production of Carcinogenic Metabolites
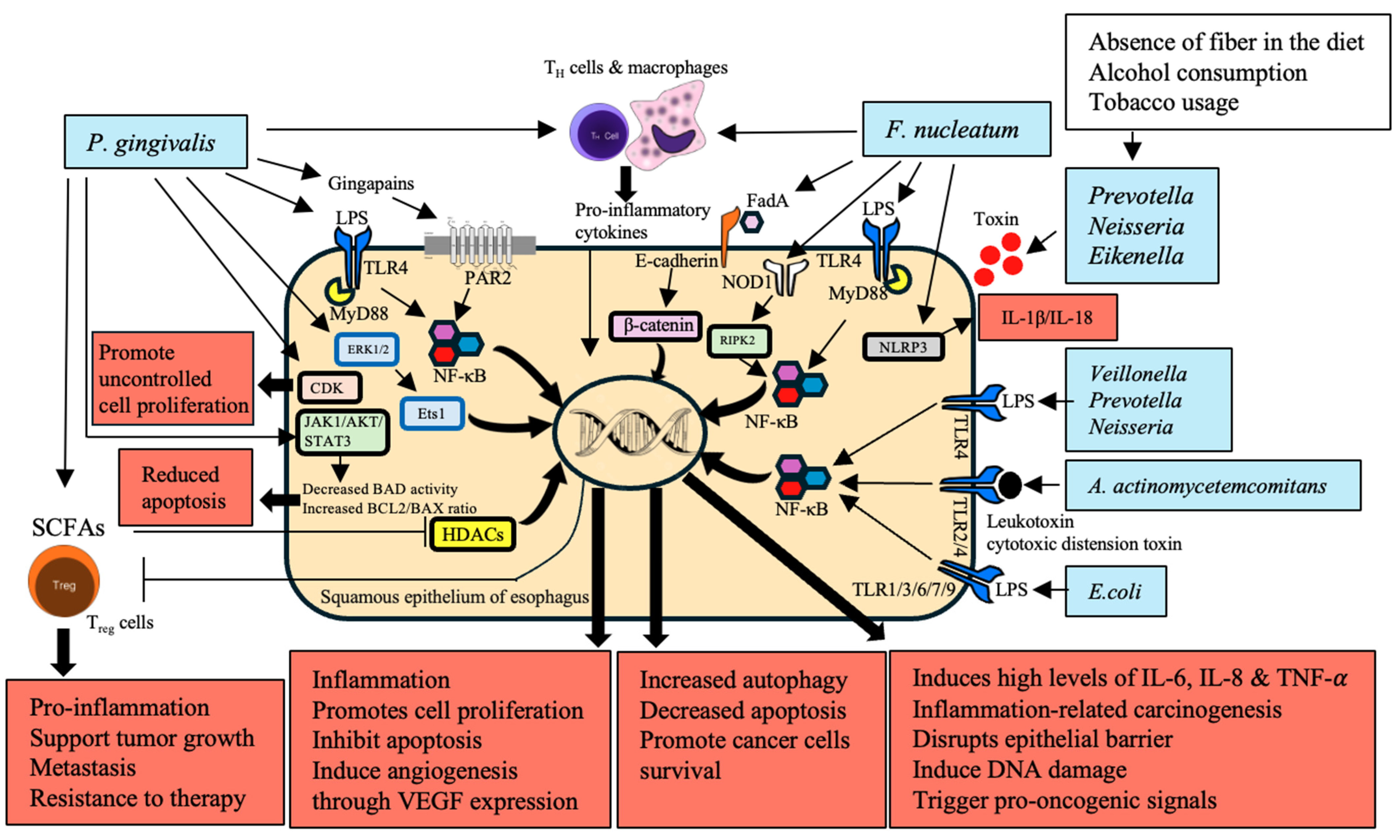
6. Direct Interaction with Epithelial Cells
7. Epigenetic Modifications
8. Interaction with GERD
- Campylobacter: It is overrepresented in GERD and BE patients, causing the induction of chronic inflammation and mucosa alteration that might contribute to EAC emergence [145].
- F. nucleatum: It binds to or invades epithelial cells, modulates the immune response, and promotes inflammation, which enhances the progression from BE to EAC through TLR activation [146].
- Prevotella: It is overrepresented in GERD, a precursor to BE and EAC, and it may facilitate chronic inflammation and mucosal damage [147].
- Rothia: It is increased in GERD and BE patients, causing chronic inflammation and mucosal damage that promotes progression to EAC [151].
- Capnocytophaga: It tends to be enriched in GERD and BE patients, mechanistically promoting chronic inflammation and esophageal mucosal changes, thereby creating conditions conducive to EAC [152].
9. Metabolic Changes and EC
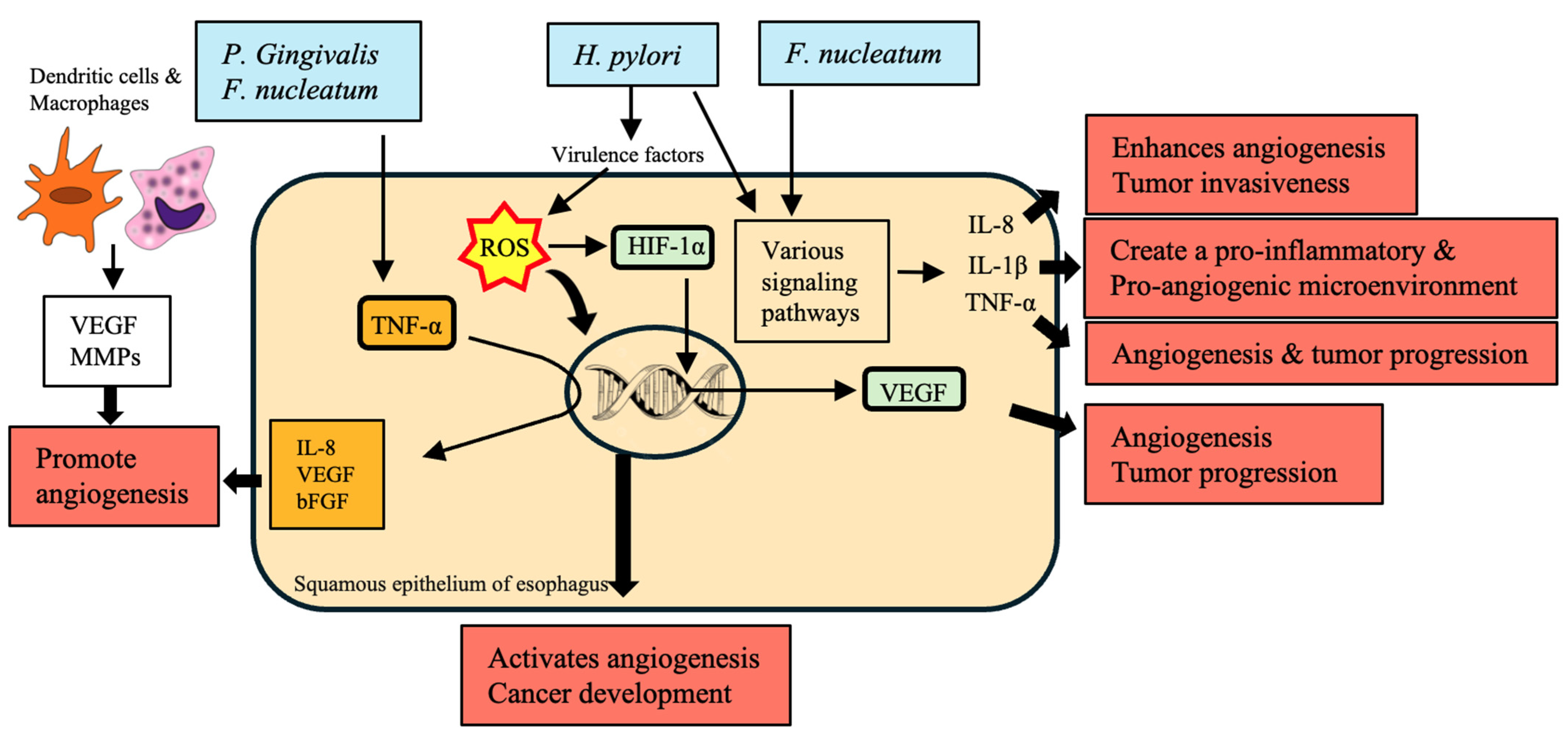
10. Angiogenesis
11. Future Directions
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Deboever, N.; Jones, C.M.; Yamashita, K.; Ajani, J.A.; Hofstetter, W.L. Advances in diagnosis and management of cancer of the esophagus. BMJ 2024, 385, e074962. [Google Scholar] [CrossRef]
- Coleman, H.G.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef]
- Lindkvist, B.; Johansen, D.; Stocks, T.; Concin, H.; Bjørge, T.; Almquist, M.; Häggström, C.; Engeland, A.; Hallmans, G.; Nagel, G.; et al. Metabolic risk factors for esophageal squamous cell carcinoma and adenocarcinoma: A prospective study of 580,000 subjects within the Me-Can project. BMC Cancer 2014, 14, 103. [Google Scholar] [CrossRef]
- Huang, J.; Lucero-Prisno, D.E., 3rd; Zhang, L.; Xu, W.; Wong, S.H.; Ng, S.C.; Wong, M.C.S. Updated epidemiology of gastrointestinal cancers in East Asia. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 271–287. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, Y.; Zhao, Y.; Huang, Y. The prognostic value of cyclooxygenase-2 expression in patients with esophageal cancer: Evidence from a meta-analysis. Onco Targets Ther. 2017, 10, 2893–2901. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res. 2017, 77, 6777–6787. [Google Scholar] [CrossRef]
- Baima, G.; Ribaldone, D.G.; Romano, F.; Aimetti, M.; Romandini, M. The Gum–Gut Axis: Periodontitis and the Risk of Gastrointestinal Cancers. Cancers 2023, 15, 4594. [Google Scholar] [CrossRef]
- Dong, E.; Duan, L.; Wu, B.U. Racial and Ethnic Minorities at Increased Risk for Gastric Cancer in a Regional US Population Study. Clin. Gastroenterol. Hepatol. 2017, 15, 511–517. [Google Scholar] [CrossRef]
- Nguyen, M.H.; Whittemore, A.S.; Garcia, R.T.; Tawfeek, S.A.; Ning, J.; Lam, S.; Wright, T.L.; Keeffe, E.B. Role of ethnicity in risk for hepatocellular carcinoma in patients with chronic hepatitis C and cirrhosis. Clin. Gastroenterol. Hepatol. 2004, 2, 820–824. [Google Scholar] [CrossRef]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, M.H.; Liptrot, S.; Paul, J.; Brown, I.L.; Morrison, D.; McColl, K.E. Oesophageal and gastric intestinal-type adenocarcinomas show the same male predominance due to a 17 year delayed development in females. Gut 2009, 58, 16–23. [Google Scholar] [CrossRef]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Gu, M.J.; Huang, Q.C.; Bao, C.Z.; Li, Y.J.; Li, X.Q.; Ye, D.; Ye, Z.H.; Chen, K.; Wang, J.B. Attributable causes of colorectal cancer in China. BMC Cancer 2018, 18, 38. [Google Scholar] [CrossRef]
- Li, Y.; Eshak, E.S.; Shirai, K.; Liu, K.; Dong, J.Y.; Iso, H.; Tamakoshi, A.; Group, J.S. Alcohol Consumption and Risk of Gastric Cancer: The Japan Collaborative Cohort Study. J. Epidemiol. 2021, 31, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, X.; Zhuang, M.; Yuan, Z.; Nie, S.; Lu, M.; Jin, L.; Ye, W. Smoking and alcohol drinking in relation to the risk of esophageal squamous cell carcinoma: A population-based case-control study in China. Sci. Rep. 2017, 7, 17249. [Google Scholar] [CrossRef] [PubMed]
- Im, P.K.; Millwood, I.Y.; Kartsonaki, C.; Chen, Y.; Guo, Y.; Du, H.; Bian, Z.; Lan, J.; Feng, S.; Yu, C.; et al. Alcohol drinking and risks of total and site-specific cancers in China: A 10-year prospective study of 0.5 million adults. Int. J. Cancer 2021, 149, 522–534. [Google Scholar] [CrossRef]
- Azeem, S.; Gillani, S.W.; Siddiqui, A.; Jandrajupalli, S.B.; Poh, V.; Syed Sulaiman, S.A. Diet and Colorectal Cancer Risk in Asia--a Systematic Review. Asian Pac. J. Cancer Prev. 2015, 16, 5389–5396. [Google Scholar] [CrossRef]
- Pham, N.M.; Mizoue, T.; Tanaka, K.; Tsuji, I.; Tamakoshi, A.; Matsuo, K.; Ito, H.; Wakai, K.; Nagata, C.; Sasazuki, S.; et al. Physical activity and colorectal cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn. J. Clin. Oncol. 2012, 42, 2–13. [Google Scholar] [CrossRef]
- Naing, C.; Lai, P.K.; Mak, J.W. Immediately modifiable risk factors attributable to colorectal cancer in Malaysia. BMC Public Health 2017, 17, 637. [Google Scholar] [CrossRef] [PubMed]
- Maret-Ouda, J.; Markar, S.R.; Lagergren, J. Gastroesophageal Reflux Disease: A Review. JAMA 2020, 324, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Corning, B.; Copland, A.P.; Frye, J.W. The Esophageal Microbiome in Health and Disease. Curr. Gastroenterol. Rep. 2018, 20, 39. [Google Scholar] [CrossRef]
- Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. [Google Scholar] [CrossRef]
- Sadafi, S.; Azizi, A.; Pasdar, Y.; Shakiba, E.; Darbandi, M. Risk factors for gastroesophageal reflux disease: A population-based study. BMC Gastroenterol. 2024, 24, 64. [Google Scholar] [CrossRef]
- Kim, S.E.; Lezama, M.M.; Schlottmann, F. Gastroesophageal Reflux Disease, Barrett’s Esophagus and Beyond. In Gastroesophageal Reflux Disease: From Pathophysiology to Treatment; Schlottmann, F., Herbella, F.A.M., Patti, M.G., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 147–158. [Google Scholar]
- Spechler, S.J. Barrett’s esophagus and esophageal adenocarcinoma: Pathogenesis, diagnosis, and therapy. Med. Clin. 2002, 86, 1423–1445. [Google Scholar] [CrossRef]
- Eslick, G.D.; Lim, L.L.; Byles, J.E.; Xia, H.H.; Talley, N.J. Association of Helicobacter pylori infection with gastric carcinoma: A meta-analysis. Am. J. Gastroenterol. 1999, 94, 2373–2379. [Google Scholar] [CrossRef]
- Hong, S.T.; Fang, Y. Clonorchis sinensis and clonorchiasis, an update. Parasitol. Int. 2012, 61, 17–24. [Google Scholar] [CrossRef]
- Qian, M.B.; Gan, X.Q.; Zhao, J.G.; Zheng, W.J.; Li, W.; Jiang, Z.H.; Zhu, T.J.; Zhou, X.N. Effectiveness of health education in improving knowledge, practice and belief related to clonorchiasis in children. Acta Trop. 2020, 207, 105436. [Google Scholar] [CrossRef]
- de Martel, C.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015, 62, 1190–1200. [Google Scholar] [CrossRef]
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tong, Y.; Yang, C.; Gan, Y.; Sun, H.; Bi, H.; Cao, S.; Yin, X.; Lu, Z. Consumption of hot beverages and foods and the risk of esophageal cancer: A meta-analysis of observational studies. BMC Cancer 2015, 15, 449. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.P.; Nie, G.J.; Chen, M.J.; Yaz, T.Y.; Guli, A.; Wuxur, A.; Huang, Q.Q.; Lin, Z.G.; Wu, J. Hot food and beverage consumption and the risk of esophageal squamous cell carcinoma: A case-control study in a northwest area in China. Medicine 2017, 96, e9325. [Google Scholar] [CrossRef]
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: Epidemiology and outcome. Clin. Epidemiol. 2014, 6, 99–109. [Google Scholar] [CrossRef]
- Li, X.; Zhu, S.; Zhang, T.; Chen, X. Association between oral microflora and gastrointestinal tumors (Review). Oncol. Rep. 2021, 46, 160. [Google Scholar] [CrossRef]
- Sharma, T.; Gupta, A.; Chauhan, R.; Bhat, A.A.; Nisar, S.; Hashem, S.; Akhtar, S.; Ahmad, A.; Haris, M.; Singh, M.; et al. Cross-talk between the microbiome and chronic inflammation in esophageal cancer: Potential driver of oncogenesis. Cancer Metastasis Rev. 2022, 41, 281–299. [Google Scholar] [CrossRef]
- Jiménez De Nunzio, S.; Portal-Núñez, S.; Arias Macías, C.M.; Bruna Del Cojo, M.; Adell-Pérez, C.; Latorre Molina, M.; Macías-González, M.; Adell-Pérez, A. Does a Dysbiotic Oral Microbiome Trigger the Risk of Chronic Inflammatory Disease? Curr. Treat. Options Allergy 2023, 10, 364–383. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef]
- Shao, D.; Vogtmann, E.; Liu, A.; Qin, J.; Chen, W.; Abnet, C.C.; Wei, W. Microbial characterization of esophageal squamous cell carcinoma and gastric cardia adenocarcinoma from a high-risk region of China. Cancer 2019, 125, 3993–4002. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, J.; Shen, Z.; Zhang, Z.; Wang, S. Characterization of Esophageal Microbiota in Patients With Esophagitis and Esophageal Squamous Cell Carcinoma. Front. Cell Infect. Microbiol. 2021, 11, 774330. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, S.; Ma, Z.; Liang, S.; Shan, T.; Zhang, M.; Zhu, X.; Zhang, P.; Liu, G.; Zhou, F.; et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect. Agents Cancer 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, C.H.; Jia, M.; Xing, X.; Gao, L.; Tsai, H.T.; Zhang, Z.; Liu, Z.; Zeng, B.; Yeung, S.J.; et al. Tumor-Associated Microbiota in Esophageal Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 641270. [Google Scholar] [CrossRef]
- Lin, Z.; Rao, W.; Xiang, Z.; Zeng, Q.; Liu, S.; Yu, K.; Zhou, J.; Wang, J.; Chen, W.; Chen, Y.; et al. Characteristics and interplay of esophageal microbiota in esophageal squamous cell carcinoma. BMC Cancer 2022, 22, 696. [Google Scholar] [CrossRef]
- Yamamura, K.; Baba, Y.; Nakagawa, S.; Mima, K.; Miyake, K.; Nakamura, K.; Sawayama, H.; Kinoshita, K.; Ishimoto, T.; Iwatsuki, M.; et al. Human Microbiome Fusobacterium Nucleatum in Esophageal Cancer Tissue Is Associated with Prognosis. Clin. Cancer Res. 2016, 22, 5574–5581. [Google Scholar] [CrossRef]
- Hu, M.; Bai, W.; Zhao, C.; Wang, J. Distribution of esophagus flora in esophageal squamous cell carcinoma and its correlation with clinicopathological characteristics. Transl. Cancer Res. 2020, 9, 3973–3985. [Google Scholar] [CrossRef]
- Li, M.; Shao, D.; Zhou, J.; Gu, J.; Qin, J.; Chen, W.; Wei, W. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin. J. Cancer Res. 2020, 32, 755–767. [Google Scholar] [CrossRef]
- Kovaleva, O.; Podlesnaya, P.; Rashidova, M.; Samoilova, D.; Petrenko, A.; Mochalnikova, V.; Kataev, V.; Khlopko, Y.; Plotnikov, A.; Gratchev, A. Prognostic Significance of the Microbiome and Stromal Cells Phenotype in Esophagus Squamous Cell Carcinoma. Biomedicines 2021, 9, 743. [Google Scholar] [CrossRef]
- Li, Z.; Shi, C.; Zheng, J.; Guo, Y.; Fan, T.; Zhao, H.; Jian, D.; Cheng, X.; Tang, H.; Ma, J. Fusobacterium nucleatum predicts a high risk of metastasis for esophageal squamous cell carcinoma. BMC Microbiol. 2021, 21, 301. [Google Scholar] [CrossRef]
- Zhang, B.; Xiao, Q.; Chen, H.; Zhou, T.; Yin, Y. Comparison of tumor-associated and nontumor-associated esophageal mucosa microbiota in patients with esophageal squamous cell carcinoma. Medicine 2022, 101, e30483. [Google Scholar] [CrossRef] [PubMed]
- Binder Gallimidi, A.; Fischman, S.; Revach, B.; Bulvik, R.; Maliutina, A.; Rubinstein, A.M.; Nussbaum, G.; Elkin, M. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget 2015, 6, 22613–22623. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Liu, J.; Guo, Y.; Li, C.; Wang, H.; Wang, H.; Zhao, H.; Pan, Y. Persistent Exposure to Porphyromonas gingivalis Promotes Proliferative and Invasion Capabilities, and Tumorigenic Properties of Human Immortalized Oral Epithelial Cells. Front. Cell Infect. Microbiol. 2017, 7, 57. [Google Scholar] [CrossRef]
- Inaba, H.; Sugita, H.; Kuboniwa, M.; Iwai, S.; Hamada, M.; Noda, T.; Morisaki, I.; Lamont, R.J.; Amano, A. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol. 2014, 16, 131–145. [Google Scholar] [CrossRef]
- Tortora, S.C.; Agurto, M.G.; Martello, L.A. The oral-gut-circulatory axis: From homeostasis to colon cancer. Front. Cell. Infect. Microbiol. 2023, 13, 1289452. [Google Scholar] [CrossRef]
- Okuyama, K.; Yanamoto, S. Oral Bacterial Contributions to Gingival Carcinogenesis and Progression. Cancer Prev. Res. 2023, 16, 199–209. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef]
- Rangan, P.; Mondino, A. Microbial short-chain fatty acids: A strategy to tune adoptive T cell therapy. J. Immunother. Cancer 2022, 10, e004147. [Google Scholar] [CrossRef]
- Nomoto, D.; Baba, Y.; Liu, Y.; Tsutsuki, H.; Okadome, K.; Harada, K.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Miyamoto, Y.; et al. Fusobacterium nucleatum promotes esophageal squamous cell carcinoma progression via the NOD1/RIPK2/NF-κB pathway. Cancer Lett. 2022, 530, 59–67. [Google Scholar] [CrossRef]
- Shi, T.; Wang, J.; Dong, J.; Hu, P.; Guo, Q. Periodontopathogens Porphyromonas gingivalis and Fusobacterium nucleatum and Their Roles in the Progression of Respiratory Diseases. Pathogens 2023, 12, 1110. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.H.; Kelly, L.A.; Kreft, R.; Barlek, M.; Omstead, A.N.; Matsui, D.; Boyd, N.H.; Gazarik, K.E.; Heit, M.I.; Nistico, L.; et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer 2016, 16, 52. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Maula, T.; Bao, K.; Lindholm, M.; Bostanci, N.; Oscarsson, J.; Ihalin, R.; Johansson, A. Virulence and Pathogenicity Properties of Aggregatibacter actinomycetemcomitans. Pathogens 2019, 8, 222. [Google Scholar] [CrossRef]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: Targeting IL-6 cytokine family. J. Immunother. Cancer 2023, 11, e007530. [Google Scholar] [CrossRef]
- Yáñez, L.; Soto, C.; Tapia, H.; Pacheco, M.; Tapia, J.; Osses, G.; Salinas, D.; Rojas-Celis, V.; Hoare, A.; Quest, A.F.G.; et al. Co-Culture of P. gingivalis and F. nucleatum Synergistically Elevates IL-6 Expression via TLR4 Signaling in Oral Keratinocytes. Int. J. Mol. Sci. 2024, 25, 3611. [Google Scholar] [CrossRef]
- Kamarajan, P.; Ateia, I.; Shin, J.M.; Fenno, J.C.; Le, C.; Zhan, L.; Chang, A.; Darveau, R.; Kapila, Y.L. Periodontal pathogens promote cancer aggressivity via TLR/MyD88 triggered activation of Integrin/FAK signaling that is therapeutically reversible by a probiotic bacteriocin. PLOS Pathog. 2020, 16, e1008881. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Yang, W.; Liu, S.; Mao, M.; Gong, Y.; Li, X.; Lei, T.; Liu, C.; Wu, S.; Hu, Q. T-cell infiltration and its regulatory mechanisms in cancers: Insights at single-cell resolution. J. Exp. Clin. Cancer Res. 2024, 43, 38. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Narikiyo, M.; Tanabe, C.; Yamada, Y.; Igaki, H.; Tachimori, Y.; Kato, H.; Muto, M.; Montesano, R.; Sakamoto, H.; Nakajima, Y.; et al. Frequent and preferential infection of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus in esophageal cancers. Cancer Sci. 2004, 95, 569–574. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2020, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, R.; Ma, L.; Liu, L.; Lai, X.; Yang, D.; Wei, J.; Ma, D.; Li, Z. Porphyromonas gingivalis promotes the motility of esophageal squamous cell carcinoma by activating NF-kappaB signaling pathway. Microbes Infect. 2019, 21, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.; Bartfeld, S.; Munke, R.; Lange, C.; Ogilvie, L.A.; Friedrich, A.; Meyer, T.F. Activation of NF-κB by Neisseria gonorrhoeae is associated with microcolony formation and type IV pilus retraction. Cell. Microbiol. 2011, 13, 1168–1182. [Google Scholar] [CrossRef]
- Mikucki, A.; McCluskey, N.R.; Kahler, C.M. The Host-Pathogen Interactions and Epicellular Lifestyle of Neisseria meningitidis. Front. Cell Infect. Microbiol. 2022, 12, 862935. [Google Scholar] [CrossRef]
- Nobel, Y.R.; Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Lightdale, C.J.; Toussaint, N.C.; Abrams, J.A. Increasing Dietary Fiber Intake Is Associated with a Distinct Esophageal Microbiome. Clin. Transl. Gastroenterol. 2018, 9, 199. [Google Scholar] [CrossRef]
- Hameed, A. Human Immunity Against Campylobacter Infection. Immune Netw. 2019, 19, e38. [Google Scholar] [CrossRef]
- Liu, N.; Ando, T.; Ishiguro, K.; Maeda, O.; Watanabe, O.; Funasaka, K.; Nakamura, M.; Miyahara, R.; Ohmiya, N.; Goto, H. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett’s esophagus. BMC Infect. Dis. 2013, 13, 130. [Google Scholar] [CrossRef]
- Zhou, J.; Shrestha, P.; Qiu, Z.; Harman, D.G.; Teoh, W.C.; Al-Sohaily, S.; Liem, H.; Turner, I.; Ho, V. Distinct Microbiota Dysbiosis in Patients with Non-Erosive Reflux Disease and Esophageal Adenocarcinoma. J. Clin. Med. 2020, 9, 2162. [Google Scholar] [CrossRef]
- Chen, X.; Winckler, B.; Lu, M.; Cheng, H.; Yuan, Z.; Yang, Y.; Jin, L.; Ye, W. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. PLoS ONE 2015, 10, e0143603. [Google Scholar] [CrossRef]
- Conteh, A.R.; Huang, R. Targeting the gut microbiota by Asian and Western dietary constituents: A new avenue for diabetes. Toxicol. Res. 2020, 9, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O.; Lecomte, V.; Maloney, C.A.; Morris, M.J. Cross-talk among metabolic parameters, esophageal microbiota, and host gene expression following chronic exposure to an obesogenic diet. Sci. Rep. 2017, 7, 45753. [Google Scholar] [CrossRef]
- Lee, W.H.; Chen, H.M.; Yang, S.F.; Liang, C.; Peng, C.Y.; Lin, F.M.; Tsai, L.L.; Wu, B.C.; Hsin, C.H.; Chuang, C.Y.; et al. Bacterial alterations in salivary microbiota and their association in oral cancer. Sci. Rep. 2017, 7, 16540. [Google Scholar] [CrossRef] [PubMed]
- Osias, G.L.; Bromer, M.Q.; Thomas, R.M.; Friedel, D.; Miller, L.S.; Suh, B.; Lorber, B.; Parkman, H.P.; Fisher, R.S. Esophageal bacteria and Barrett’s esophagus: A preliminary report. Dig. Dis. Sci. 2004, 49, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Amir, I.; Konikoff, F.M.; Oppenheim, M.; Gophna, U.; Half, E.E. Gastric microbiota is altered in oesophagitis and Barrett’s oesophagus and further modified by proton pump inhibitors. Environ. Microbiol. 2014, 16, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.H.; Zhu, Y.; Chen, W.F.; Fu, P.Y.; Xu, J.Q.; Wang, T.Y.; Yao, L.; Liu, Z.Q.; Li, X.Q.; Zhang, Z.C.; et al. The role of type II esophageal microbiota in achalasia: Activation of macrophages and degeneration of myenteric neurons. Microbiol. Res. 2023, 276, 127470. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Sato, H.; Mizusawa, T.; Tominaga, K.; Ikarashi, S.; Hayashi, K.; Mizuno, K.I.; Hashimoto, S.; Yokoyama, J.; Terai, S. Comparison of Oral and Esophageal Microbiota in Patients with Achalasia Before and After Peroral Endoscopic Myotomy. Turk. J. Gastroenterol. 2021, 32, 42–52. [Google Scholar] [CrossRef]
- Fleming, S.E.; Yeo, S. Production and Absorption of Short-Chain Fatty Acids. In Dietary Fiber: Chemistry, Physiology, and Health Effects; Kritchevsky, D., Bonfield, C., Anderson, J.W., Eds.; Springer: Boston, MA, USA, 1990; pp. 301–315. [Google Scholar]
- Tomás-Pejó, E.; González-Fernández, C.; Greses, S.; Kennes, C.; Otero-Logilde, N.; Veiga, M.C.; Bolzonella, D.; Müller, B.; Passoth, V. Production of short-chain fatty acids (SCFAs) as chemicals or substrates for microbes to obtain biochemicals. Biotechnol. Biofuels Bioprod. 2023, 16, 96. [Google Scholar] [CrossRef]
- Peterson, C.T.; Perez Santiago, J.; Iablokov, S.N.; Chopra, D.; Rodionov, D.A.; Peterson, S.N. Short-Chain Fatty Acids Modulate Healthy Gut Microbiota Composition and Functional Potential. Curr. Microbiol. 2022, 79, 128. [Google Scholar] [CrossRef]
- Ghosh, S.; Pramanik, S. Structural diversity, functional aspects and future therapeutic applications of human gut microbiome. Arch. Microbiol. 2021, 203, 5281–5308. [Google Scholar] [CrossRef]
- Guan, X.; Li, W.; Meng, H. A double-edged sword: Role of butyrate in the oral cavity and the gut. Mol. Oral. Microbiol. 2021, 36, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Tang, D.; Hou, P.; Shen, W.; Li, H.; Wang, T.; Liu, R. Dysbiosis of gut microbiota in patients with esophageal cancer. Microb. Pathog. 2021, 150, 104709. [Google Scholar] [CrossRef] [PubMed]
- Tagaino, R.; Washio, J.; Abiko, Y.; Tanda, N.; Sasaki, K.; Takahashi, N. Metabolic property of acetaldehyde production from ethanol and glucose by oral Streptococcus and Neisseria. Sci. Rep. 2019, 9, 10446. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Helicobacter pylori: A ROS-inducing bacterial species in the stomach. Inflamm. Res. 2010, 59, 997–1003. [Google Scholar] [CrossRef]
- Charoensaensuk, V.; Chen, Y.-C.; Lin, Y.-H.; Ou, K.-L.; Yang, L.-Y.; Lu, D.-Y. Porphyromonas gingivalis Induces Proinflammatory Cytokine Expression Leading to Apoptotic Death through the Oxidative Stress/NF-κB Pathway in Brain Endothelial Cells. Cells 2021, 10, 3033. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef]
- Abranches, J.; Zeng, L.; Kajfasz, J.K.; Palmer, S.R.; Chakraborty, B.; Wen, Z.T.; Richards, V.P.; Brady, L.J.; Lemos, J.A. Biology of Oral Streptococci. Microbiol. Spectr. 2018, 6, 426. [Google Scholar] [CrossRef]
- Uitto, V.J.; Baillie, D.; Wu, Q.; Gendron, R.; Grenier, D.; Putnins, E.E.; Kanervo, A.; Firth, J.D. Fusobacterium nucleatum increases collagenase 3 production and migration of epithelial cells. Infect. Immun. 2005, 73, 1171–1179. [Google Scholar] [CrossRef]
- Wu, D.-D.; Ngowi, E.E.; Zhai, Y.-K.; Wang, Y.-Z.; Khan, N.H.; Kombo, A.F.; Khattak, S.; Li, T.; Ji, X.-Y. Role of Hydrogen Sulfide in Oral Disease. Oxidative Med. Cell. Longev. 2022, 2022, 1886277. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 2006, 4, 9–14. [Google Scholar] [CrossRef]
- Karpiński, T.M.; Szkaradkiewicz, A.K. Characteristic of bacteriocines and their application. Pol. J. Microbiol. 2013, 62, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef]
- Nieminen, M.T.; Salaspuro, M. Local Acetaldehyde-An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis. Cancers 2018, 10, 11. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef]
- Byun, J.-K. Tumor lactic acid: A potential target for cancer therapy. Arch. Pharmacal Res. 2023, 46, 90–110. [Google Scholar] [CrossRef]
- Lunt, S.J.; Chaudary, N.; Hill, R.P. The tumor microenvironment and metastatic disease. Clin. Exp. Metastasis 2009, 26, 19–34. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Ranjbar, M.; Salehi, R.; Haghjooy Javanmard, S.; Rafiee, L.; Faraji, H.; Jafarpor, S.; Ferns, G.A.; Ghayour-Mobarhan, M.; Manian, M.; Nedaeinia, R. The dysbiosis signature of Fusobacterium nucleatum in colorectal cancer-cause or consequences? A systematic review. Cancer Cell Int. 2021, 21, 194. [Google Scholar] [CrossRef]
- McIlvanna, E.; Linden, G.J.; Craig, S.G.; Lundy, F.T.; James, J.A. Fusobacterium nucleatum and oral cancer: A critical review. BMC Cancer 2021, 21, 1212. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Park, Y.; Hasegawa, Y.; Tribble, G.D.; James, C.E.; Handfield, M.; Stavropoulos, M.F.; Yilmaz, O.; Lamont, R.J. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007, 9, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.K. Porphyromonas gingivalis in oral squamous cell carcinoma: A review. Microbes Infect. 2022, 24, 104925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, L. Fusobacterium nucleatum carcinogenesis and drug delivery interventions. Adv. Drug Deliv. Rev. 2024, 209, 115319. [Google Scholar] [CrossRef]
- Li, Y.; Xing, S.; Chen, F.; Li, Q.; Dou, S.; Huang, Y.; An, J.; Liu, W.; Zhang, G. Intracellular Fusobacterium nucleatum infection attenuates antitumor immunity in esophageal squamous cell carcinoma. Nat. Commun. 2023, 14, 5788. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e516. [Google Scholar] [CrossRef]
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 227. [Google Scholar] [CrossRef]
- Reddy, M.G.S.; Kakodkar, P.; Nayanar, G. Capacity of Candida species to produce acetaldehyde at various concentrations of alcohol. J. Oral. Maxillofac. Pathol. 2022, 26, 161–165. [Google Scholar] [CrossRef]
- Muto, M.; Hitomi, Y.; Ohtsu, A.; Shimada, H.; Kashiwase, Y.; Sasaki, H.; Yoshida, S.; Esumi, H. Acetaldehyde production by non-pathogenic Neisseria in human oral microflora: Implications for carcinogenesis in upper aerodigestive tract. Int. J. Cancer 2000, 88, 342–350. [Google Scholar] [CrossRef]
- Huhta, H.; Helminen, O.; Lehenkari, P.P.; Saarnio, J.; Karttunen, T.J.; Kauppila, J.H. Toll-like receptors 1, 2, 4 and 6 in esophageal epithelium, Barrett’s esophagus, dysplasia and adenocarcinoma. Oncotarget 2016, 7, 23658–23667. [Google Scholar] [CrossRef]
- Verbeek, R.E.; Siersema, P.D.; Ten Kate, F.J.; Fluiter, K.; Souza, R.F.; Vleggaar, F.P.; Bus, P.; van Baal, J.W. Toll-like receptor 4 activation in Barrett’s esophagus results in a strong increase in COX-2 expression. J. Gastroenterol. 2014, 49, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, Y.; Yin, Y.; Wang, L.; Yang, H.; Luo, S.; Zheng, Q.; Pan, Y.; Zhang, D. Potential role of epithelial-mesenchymal transition induced by periodontal pathogens in oral cancer. J. Cell Mol. Med. 2024, 28, e18064. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Jungas, T.; Verbeke, P.; Ojcius, D.M. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 2004, 72, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Jermanus, C.; Barbetta, B.; Choi, C.; Verbeke, P.; Ojcius, D.M.; Yilmaz, O. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol. Oral. Microbiol. 2010, 25, 89–101. [Google Scholar] [CrossRef]
- Nakhjiri, S.F.; Park, Y.; Yilmaz, O.; Chung, W.O.; Watanabe, K.; El-Sabaeny, A.; Park, K.; Lamont, R.J. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol. Lett. 2001, 200, 145–149. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Liu, S.; Zhang, S.; Pan, Y. The Role of Porphyromonas gingivalis Outer Membrane Vesicles in Periodontal Disease and Related Systemic Diseases. Front. Cell. Infect. Microbiol. 2021, 10, 585917. [Google Scholar] [CrossRef]
- Huang, S.; Cao, G.; Dai, D.; Xu, Q.; Ruiz, S.; Shindo, S.; Nakamura, S.; Kawai, T.; Lin, J.; Han, X. Porphyromonas gingivalis outer membrane vesicles exacerbate retinal microvascular endothelial cell dysfunction in diabetic retinopathy. Front. Microbiol. 2023, 14, 1167160. [Google Scholar] [CrossRef]
- Ye, C.; Liu, X.; Liu, Z.; Pan, C.; Zhang, X.; Zhao, Z.; Sun, H. Fusobacterium nucleatum in tumors: From tumorigenesis to tumor metastasis and tumor resistance. Cancer Biol. Ther. 2024, 25, 2306676. [Google Scholar] [CrossRef]
- Ergun, P.; Kipcak, S.; Bor, S. Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7817. [Google Scholar] [CrossRef]
- Liang, G.; Wang, H.; Shi, H.; Zhu, M.; An, J.; Qi, Y.; Du, J.; Li, Y.; Gao, S. Porphyromonas gingivalis Promotes the Proliferation and Migration of Esophageal Squamous Cell Carcinoma through the miR-194/GRHL3/PTEN/Akt Axis. ACS Infect. Dis. 2020, 6, 871–881. [Google Scholar] [CrossRef]
- Park, H.E.; Kim, J.H.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Intratumoral Fusobacterium nucleatum abundance correlates with macrophage infiltration and CDKN2A methylation in microsatellite-unstable colorectal carcinoma. Virchows Arch. 2017, 471, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, Y.; Yu, J.; Chen, T.; Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Fu, X. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 2017, 8, 31802–31814. [Google Scholar] [CrossRef]
- Fan, S.; Xing, J.; Jiang, Z.; Zhang, Z.; Zhang, H.; Wang, D.; Tang, D. Effects of Long Non-Coding RNAs Induced by the Gut Microbiome on Regulating the Development of Colorectal Cancer. Cancers 2022, 14, 5813. [Google Scholar] [CrossRef]
- Nikolaieva, N.; Sevcikova, A.; Omelka, R.; Martiniakova, M.; Mego, M.; Ciernikova, S. Gut Microbiota-MicroRNA Interactions in Intestinal Homeostasis and Cancer Development. Microorganisms 2022, 11, 107. [Google Scholar] [CrossRef]
- Olsen, I.; Yilmaz, Ö. Possible role of Porphyromonas gingivalis in orodigestive cancers. J. Oral. Microbiol. 2019, 11, 1563410. [Google Scholar] [CrossRef]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef]
- Lv, J.; Guo, L.; Liu, J.J.; Zhao, H.P.; Zhang, J.; Wang, J.H. Alteration of the esophageal microbiota in Barrett’s esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 2019, 25, 2149–2161. [Google Scholar] [CrossRef]
- Ajayi, T.A.; Cantrell, S.; Spann, A.; Garman, K.S. Barrett’s esophagus and esophageal cancer: Links to microbes and the microbiome. PLoS Pathog. 2018, 14, e1007384. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.; Yang, Y.; Han, B.; Wang, Q.; Du, S. Investigating the causal relationship of gut microbiota with GERD and BE: A bidirectional mendelian randomization. BMC Genom. 2024, 25, 471. [Google Scholar] [CrossRef]
- Bakhti, S.Z.; Latifi-Navid, S. Oral microbiota and Helicobacter pylori in gastric carcinogenesis: What do we know and where next? BMC Microbiol. 2021, 21, 71. [Google Scholar] [CrossRef]
- Asili, P.; Mirahmad, M.; Rezaei, P.; Mahdavi, M.; Larijani, B.; Tavangar, S.M. The Association of Oral Microbiome Dysbiosis with Gastrointestinal Cancers and Its Diagnostic Efficacy. J. Gastrointest. Cancer 2023, 54, 1082–1101. [Google Scholar] [CrossRef] [PubMed]
- Canzi Almada de Souza, R.; Hermênio Cavalcante Lima, J. Helicobacter pylori and gastroesophageal reflux disease: A review of this intriguing relationship. Dis. Esophagus 2009, 22, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Jalanka, J.; Gunn, D.; Singh, G.; Krishnasamy, S.; Lingaya, M.; Crispie, F.; Finnegan, L.; Cotter, P.; James, L.; Nowak, A.; et al. Postinfective bowel dysfunction following Campylobacter enteritis is characterised by reduced microbiota diversity and impaired microbiota recovery. Gut 2023, 72, 451. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Danhof, H.A.; Ruan, W.; Engevik, A.C.; Chang-Graham, A.L.; Engevik, K.A.; Shi, Z.; Zhao, Y.; Brand, C.K.; Krystofiak, E.S.; et al. Fusobacterium nucleatum Secretes Outer Membrane Vesicles and Promotes Intestinal Inflammation. mBio 2021, 12, e02706-20. [Google Scholar] [CrossRef]
- Kawar, N.; Park, S.G.; Schwartz, J.L.; Callahan, N.; Obrez, A.; Yang, B.; Chen, Z.; Adami, G.R. Salivary microbiome with gastroesophageal reflux disease and treatment. Sci. Rep. 2021, 11, 188. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef]
- Lory, S. The Family Leptotrichiaceae. In The Prokaryotes: Firmicutes and Tenericutes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 213–214. [Google Scholar]
- Zeng, R.; Gou, H.; Lau, H.C.H.; Yu, J. Stomach microbiota in gastric cancer development and clinical implications. Gut 2024, 1–12. [Google Scholar] [CrossRef]
- Rigauts, C.; Aizawa, J.; Taylor, S.L.; Rogers, G.B.; Govaerts, M.; Cos, P.; Ostyn, L.; Sims, S.; Vandeplassche, E.; Sze, M.; et al. R othia mucilaginosa is an anti-inflammatory bacterium in the respiratory tract of patients with chronic lung disease. Eur. Respir. J. 2022, 59, 2101293. [Google Scholar] [CrossRef]
- Chesdachai, S.; Tai, D.B.G.; Yetmar, Z.A.; Misra, A.; Ough, N.; Abu Saleh, O. The Characteristics of Capnocytophaga Infection: 10 Years of Experience. Open Forum Infect. Dis. 2021, 8, ofab175. [Google Scholar] [CrossRef]
- Abdel-Latif, M.M.; Kelleher, D.; Reynolds, J.V. Potential role of NF-kappaB in esophageal adenocarcinoma: As an emerging molecular target. J. Surg. Res. 2009, 153, 172–180. [Google Scholar] [CrossRef]
- Rai, A.K.; Panda, M.; Das, A.K.; Rahman, T.; Das, R.; Das, K.; Sarma, A.; Kataki, A.C.; Chattopadhyay, I. Dysbiosis of salivary microbiome and cytokines influence oral squamous cell carcinoma through inflammation. Arch. Microbiol. 2021, 203, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Lee, S.K. Exploring Esophageal Microbiomes in Esophageal Diseases: A Systematic Review. J. Neurogastroenterol. Motil. 2020, 26, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Tateda, M.; Saijo, S.; Hori, T.; Sato, I.; Tateno, H.; Matsuura, K.; Takasaka, T.; Miyagi, T. Presence of Streptococcus infection in extra-oropharyngeal head and neck squamous cell carcinoma and its implication in carcinogenesis. Oncol. Rep. 2001, 8, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quinn, P.J. Endotoxins: Lipopolysaccharides of Gram-Negative Bacteria. In Endotoxins: Structure, Function and Recognition; Wang, X., Quinn, P.J., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 3–25. [Google Scholar]
- Li, Q.; von Ehrlich-Treuenstätt, V.; Schardey, J.; Wirth, U.; Zimmermann, P.; Andrassy, J.; Bazhin, A.V.; Werner, J.; Kühn, F. Gut Barrier Dysfunction and Bacterial Lipopolysaccharides in Colorectal Cancer. J. Gastrointest. Surg. 2023, 27, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: A review. Appl. Biol. Chem. 2017, 60, 327–338. [Google Scholar] [CrossRef]
- Bertona, S.; Monrabal Lezama, M.; Patti, M.G.; Herbella, F.A.M.; Schlottmann, F. Gastroesophageal Reflux Disease in Obese Patients. In Gastroesophageal Reflux Disease: From Pathophysiology to Treatment; Schlottmann, F., Herbella, F.A.M., Patti, M.G., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 117–125. [Google Scholar]
- Lee, K.X.; Quek, K.F.; Ramadas, A. Dietary and Lifestyle Risk Factors of Obesity Among Young Adults: A Scoping Review of Observational Studies. Curr. Nutr. Rep. 2023, 12, 733–743. [Google Scholar] [CrossRef]
- Scida, S.; Russo, M.; Miraglia, C.; Leandro, G.; Franzoni, L.; Meschi, T.; De’ Angelis, G.L.; Di Mario, F. Relationship between Helicobacter pylori infection and GERD. Acta Biomed. 2018, 89, 40–43. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Medhat, A.; Spector, I. Short-chain fatty acids in cancer pathogenesis. Cancer Metastasis Rev. 2023, 42, 677–698. [Google Scholar] [CrossRef]
- Xie, F.J.; Zhang, Y.P.; Zheng, Q.Q.; Jin, H.C.; Wang, F.L.; Chen, M.; Shao, L.; Zou, D.H.; Yu, X.M.; Mao, W.M. Helicobacter pylori infection and esophageal cancer risk: An updated meta-analysis. World J. Gastroenterol. 2013, 19, 6098–6107. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Beltrán-Velasco, A.I.; Redondo-Flórez, L.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Global Impacts of Western Diet and Its Effects on Metabolism and Health: A Narrative Review. Nutrients 2023, 15, 2749. [Google Scholar] [CrossRef]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sah, D.K.; Arjunan, A.; Lee, B.; Jung, Y.D. Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development. Antioxidants 2023, 12, 1712. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Valderrama, M.; Cerda-Opazo, P.; Backert, S.; González, M.F.; Carrasco-Véliz, N.; Jorquera-Cordero, C.; Wehinger, S.; Canales, J.; Bravo, D.; Quest, A.F.G. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1α in Gastric Cells. Cancers 2019, 11, 799. [Google Scholar] [CrossRef]
- Aral, K.; Milward, M.R.; Gupta, D.; Cooper, P.R. Effects of Porphyromonas gingivalis and Fusobacterium nucleatum on inflammasomes and their regulators in H400 cells. Mol. Oral. Microbiol. 2020, 35, 158–167. [Google Scholar] [CrossRef]
- Yao, Y.; Shen, X.; Zhou, M.; Tang, B. Periodontal Pathogens Promote Oral Squamous Cell Carcinoma by Regulating ATR and NLRP3 Inflammasome. Front. Oncol. 2021, 11, 722797. [Google Scholar] [CrossRef]
- Brouwer, S.; Rivera-Hernandez, T.; Curren, B.F.; Harbison-Price, N.; De Oliveira, D.M.P.; Jespersen, M.G.; Davies, M.R.; Walker, M.J. Pathogenesis, epidemiology and control of Group A Streptococcus infection. Nat. Rev. Microbiol. 2023, 21, 431–447. [Google Scholar] [CrossRef]
- Shaw, M.H.; Kamada, N.; Kim, Y.G.; Núñez, G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258. [Google Scholar] [CrossRef]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. [Google Scholar] [CrossRef]
- Shimada, H.; Takeda, A.; Nabeya, Y.; Okazumi, S.I.; Matsubara, H.; Funami, Y.; Hayashi, H.; Gunji, Y.; Kobayashi, S.; Suzuki, T.; et al. Clinical significance of serum vascular endothelial growth factor in esophageal squamous cell carcinoma. Cancer 2001, 92, 663–669. [Google Scholar] [CrossRef]
- Malhotra, R.; Tyson, D.W.; Rosevear, H.M.; Brosius, F.C. Hypoxia-inducible factor-1alpha is a critical mediator of hypoxia induced apoptosis in cardiac H9c2 and kidney epithelial HK-2 cells. BMC Cardiovasc. Disord. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Sun, F.; Meng, Q.; Zhou, M.; Yu, J.; Hu, L. Investigating the predictive value of vascular endothelial growth factor in the evaluation of treatment efficacy and prognosis for patients with non-surgical esophageal squamous cell carcinoma. Front. Oncol. 2022, 12, 843250. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Peng, C.; Ouyang, Y.; Lu, N.; Li, N. The NF-κB Signaling Pathway, the Microbiota, and Gastrointestinal Tumorigenesis: Recent Advances. Front. Immunol. 2020, 11, 1387. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Newman, A.C.; Hughes, C.C.W. Macrophages and angiogenesis: A role for Wnt signaling. Vasc. Cell 2012, 4, 13. [Google Scholar] [CrossRef]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef]
- Malkova, A.M.; Gubal, A.R.; Petrova, A.L.; Voronov, E.; Apte, R.N.; Semenov, K.N.; Sharoyko, V.V. Pathogenetic role and clinical significance of interleukin-1β in cancer. Immunology 2023, 168, 203–216. [Google Scholar] [CrossRef]
- El-Omar, E.M. The importance of interleukin 1β in Helicobacter pylori associated disease. Gut 2001, 48, 743. [Google Scholar] [CrossRef]
- Yoshida, S.; Ono, M.; Shono, T.; Izumi, H.; Ishibashi, T.; Suzuki, H.; Kuwano, M. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol. Cell Biol. 1997, 17, 4015–4023. [Google Scholar] [CrossRef]
- Naserian, S.; Abdelgawad, M.E.; Afshar Bakshloo, M.; Ha, G.; Arouche, N.; Cohen, J.L.; Salomon, B.L.; Uzan, G. The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Commun. Signal. 2020, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Basic, A.; Blomqvist, M.; Dahlén, G.; Svensäter, G. The proteins of Fusobacterium spp. involved in hydrogen sulfide production from L-cysteine. BMC Microbiol. 2017, 17, 61. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Microbes Increased in ESCC | Microbes Decreased in ESCC or Increased in Control Samples | References |
|---|---|---|---|
| 67 paired samples (ESCC tissue vs. non-tumor tissue) | Fusobacteria phylum Fusobacterium genus | Firmicutes phylum Streptococcus genus | [41] |
| 32 ESCC samples vs. 21 healthy controls | Streptococcus genus Actinobacillus genus Peptostreptococcus genus Fusobacterium genus Prevotella genus | Fusobacteria phylum Faecalibacterium genus Bacteroides genus Curvibacter genus Blautia genus | [42] |
| 32 ESCC samples vs. 15 esophagitis samples | Streptococcus genus | Bacteroidetes genus Faecalibacterium genus Bacteroides genus Blautia genus | [42] |
| 17 ESCC samples vs. 16 healthy control samples | Fusobacteria phylum Prevotella genus Pseudomonas genus | Actinobacteria phylum Ralstonia genus Burkholderia-Caballeronia-Paraburkholderia genus | [43] |
| 17 ESCC samples vs. 15 post-op ESCC samples | Fusobacteria phylum Bacteroidetes phylum Prevotella genus | Pseudomonas genus | [43] |
| 100 ESCC samples vs. 100 adjacent tissue samples or 30 normal esophagus samples | P. gingivalis | [44] | |
| 18 ESCC samples vs. 11 normal esophagus samples | Fusobacteria phylum Bacteroidetes phylum Spirochaetes phylum T. amylovorum, S. infantis, P. nigrescens, P. endodontalis, V. dispar, A. segnis, P. melaninogenica, P. intermedia P. tannerae, P. nanceiensis, S. anginosus | Proteobacteria phylum Thermi Phylum | [45] |
| 120 ESCC samples vs. adjacent tissue sample from same subjects | R. mucilaginosa, P. endodontalis unclassified species in the genus Leptotrichia unclassified species in the genus Phyllobacterium unclassified species in the genus Sphingomonas | class Bacilli N. subflava H. pylori A. parahaemolyticus A. rhizosphaerae, unclassified species in the genus Campylobacter unclassified species in the genus Haemophilus | [46] |
| 60 ESCC samples vs. paired adjacent normal tissue samples | F. nucleatum | [47] | |
| 54 ESCC samples vs. 4 normal esophageal tissues | Proteus genus Firmicutes genus Bacteroides genus Fusobacterium genus | [48] | |
| 7 ESCC samples vs. 70 normal control samples (together with 70 esophagitis, 70 low-grade intraepithelial neoplasia and 19 high-grade intraepithelial neoplasia) | Streptococcus genus Haemophilus genus Neisseria genus Porphyromonas genus | [49] | |
| 48 ESCC samples vs. matched control samples | Staphylococcus genus | [50] | |
| 111 ESCC samples vs. 41 normal samples | Bacteroidetes phylum Fusobacteria phylum Spirochaetae phylum Streptococcus genus F. nucleatum | Butyrivibrio genus Lactobacillus genus | [51] |
| 31 ESCC samples vs. matched controls | Peptostreptococcaceae, Leptotrichia, Peptostreptococcus, Anaerovoracaceae, Filifactor, Anaerovoracaceae-Eubacterium_ brachygroup, Lachnoanaerobaculum, Dethiosulfatibacteraceae, Solobacterium, Johnsonella, Prevotellaceae UCG_001, and Tannerella (higher in N0 stage) Treponema and Brevibacillus (higher in N1 and N2 stages) Acinetobacter (higher in T3 stage) Corynebacterium, Aggregatibacter, Saccharimonadaceae-TM7x, and Cupriavidus (higher in T4 stage) | [52] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis | Activates ERK1/2–Ets1 and PAR2/NF-κB pathways | Increased secretion of pro-inflammatory cytokines and chemokines reprogramming TME | [53,54,55] |
| Interacts with T cells and macrophages | Disrupts epithelial barrier, induces DNA damage, triggers pro-oncogenic signals | [56] | |
| LPS activates TLR-4 leading to NF-κB activation | Promotes cell proliferation, inhibits apoptosis, induces angiogenesis through VEGF expression | [57] | |
| Inhibits HDACs through SCFAs modulating Treg cell function | Supports tumor growth, metastasis, and resistance to therapy | [58,59,60] | |
| F. nucleatum | Activates NOD1/RIPK2/NF-κB and NLRP3 inflammasome pathways | Induces high levels of IL-6 and IL-8, driving inflammation-related carcinogenesis | [53,61] |
| LPS activates TLR-4 leading to NF-κB activation | Recruits and reprograms immune cells within TME, supporting tumor progression and immune evasion | [62] | |
| Interacts with T cells and macrophages | Promotes cell proliferation, inhibits apoptosis, induces angiogenesis through VEGF expression | [57] | |
| E. coli | Upregulates TLRs 1–3, 6, 7, and 9 | Induces early carcinogenic molecular changes through TLR signaling pathway activation | [63] |
| A. actinomycetemcomitans | Produces virulence factors such as leukotoxin and cytotoxic distension toxin | Exacerbates inflammation and cancer risk | [64] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis |
|
| [53,54,55] |
|
| [44] | |
| F. nucleatum |
|
| [53,61] |
|
| [47] | |
|
| [70] | |
| T. denticola, S. anginosus |
|
| [71] |
| E. coli |
|
| [63] |
| Prevotella |
|
| [74] |
| Neisseria |
|
| [75,76] |
| Eikenella |
|
| [77] |
| A. segnis, T. amylovorum, P. endodontalis, S. infantis, V. dispar, S. anginosus, P. intermedia, P. melaninogenica |
|
| [45] |
| Campylobacter |
|
| [78,79] |
| Parvimonas |
|
| [77] |
| Leptotrichia |
|
| [80] |
| Lautropia, Bulleidia, Catonella, Corynebacterium, Moryella, Peptococcus, Cardiobacterium |
|
| [81] |
| Tannerella forsythia |
|
| [7] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| Bacteroides, Clostridium, Faecalibacterium, Ruminococcus | Produce SCFAs like butyrate, acetate, and propionate through dietary fiber fermentation | Reduced SCFA production contributes to a pro-inflammatory environment and weakened intestinal barrier, promoting carcinogenesis | [92] |
| Neisseria, Streptococcus, Candida | Metabolize alcohol into acetaldehyde, a highly toxic and carcinogenic substance | Causes DNA damage, mutagenesis, and gut microbiota disruption, increasing EC risk | [95] |
| P. gingivalis, H. pylori, E. coli | Produce ROS | Leads to DNA damage, cellular transformation, tumor survival, invasion, angiogenesis, and metastasis | [96,97] |
| S. oralis, S. mitis, S. sanguinis, S. gordonii, L. fermentum, L. jensenii, L. acidophilus, B. adolescentis | Produce RNS | Contribute to DNA damage and cancer progression through nitrosative stress | [98,99] |
| P. gingivalis, F. nucleatum | Overexpress MMPs; P. gingivalis produces gingipains to activate MMP-9; F. nucleatum stimulates MMP-9 and MMP-13 through p38 signaling | Degrade extracellular matrix, disrupt cell adhesion, facilitating cancer cell invasion and metastasis, critical in tumor progression | [55,100] |
| P. gingivalis, Prevotella intermedia, A. actinomycetemcomitans, F. nucleatum | Produce H2S, a genotoxic volatile sulfur compound | Induces genomic instability and cumulative mutations, promoting tumor growth and spread by activating various signaling pathways | [101,102] |
| Lactobacillus, Lactococcus, Bifidobacterium, Streptococcus, Leuconostoc, Pediococcus | Produce lactic acid through fermentation | Overproduction creates an acidic and hypoxic tumor microenvironment, suppressing immune responses and enhancing metastatic efficiency | [103] |
| E. coli | Secretes colibactin, a metabolic genetic toxic substance | Induces DNA double-strand breaks, leading to genomic instability and contributing significantly to carcinogenesis | [104] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis | Activates ERK1/2–Ets1 and PAR2/NF-κB pathways | Promotes proliferation, migration, and invasion of epithelial cells | [53,54] |
| Induces antiapoptotic activity via JAK1/AKT/STAT3 pathway | Reduces apoptotic activity of epithelial cells | [114] | |
| Secretes NDK | Enhances BCL2 to BAX ratio | [106] | |
| Accelerates S-phase progression by manipulating CDK activity | Promotes cancer cell proliferation | [115] | |
| F. nucleatum | Activates NOD1/RIPK2/NF-κB pathway | Enhances ESCC cell growth and migration | [53,61] |
| Influences TME through chemokine activation | Associated with shorter survival times and aggressive tumor behavior | [116,117] | |
| Activates TLR-4 | Promotes β-catenin signaling leading to oncogene activation | [70,118] | |
| Binds to E-cadherin on carcinoma cells | Facilitates cancer cell proliferation | [70] | |
| Campylobacter, Leptotrichia, Rothia, Capnocytophaga | Enriched in GERD and BE | Contributes to chronic inflammation and epithelial cell transformation | [79,119] |
| A. actinomycetemcomitans | Produces virulence factors that interact with epithelial cells | Promotes cell transformation and carcinogenesis | [64] |
| T. denticola, S. mitis, S. anginosus | Dominates microbiota in cancerous esophageal tissues | Suggests direct interaction with epithelial cells contributing to disease progression | [71] |
| Candida, Neisseria | Metabolizes alcohol into acetaldehyde | Causes DNA damage, mutagenesis, and disrupts gut microbiota | [120,121] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| P. gingivalis |
|
| [58,60] |
|
| [132] | |
| F. nucleatum |
|
| [133] |
|
| [70,134] | |
| Microbiota in General |
|
| [60] |
|
| [135,136] | |
| Microbiota in BE and EAC |
|
| [123,131] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| Veillonella, Prevotella, Neisseria | Produces LPS, activates TLR-4 leading to NF-κB activation | Creates a pro-inflammatory environment, contributing to carcinogenesis | [142,143] |
| Streptococcus | Increases prevalence with age, producing pro-inflammatory cytokines | Influences chronic inflammation and increases the risk of EC | [119] |
| H. pylori |
|
| [144] |
| Campylobacter |
|
| [145] |
| F. nucleatum |
|
| [146] |
| Prevotella |
|
| [147] |
| S. anginosus |
|
| [71,148] |
| Leptotrichia |
|
| [149,150] |
| Rothia |
|
| [151] |
| Capnocytophaga |
|
| [152] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| Bacteroides, Clostridium, Faecalibacterium, Ruminococcus | Produce SCFAs, modulate inflammation | Maintain gut health; reduced SCFA production leads to a pro-inflammatory environment and cancer risk | [89,92] |
| H. pylori | Induces chronic gastritis, alters gastric acid secretion | Promotes GERD, BE, and EAC | [164] |
| Campylobacter | Induces inflammatory responses | Promotes chronic inflammation and progression to BE and EAC | [46] |
| Lactobacillus, Streptococcus, Bifidobacterium, Leuconostoc | Produce lactic acid, create low pH hypoxic environment, induce Warburg effect | Immunosuppression, enhanced tumor metastasis, support cancer cell survival and proliferation | [80] |
| F. nucleatum | Produces LPS, activates β-catenin signaling, enhances oncogene expression (C-myc, cyclin D1) | Promotes cancer cell proliferation, chronic inflammation, and carcinogenesis | [134] |
| P. gingivalis | Modulates ATP/P2X7 signaling, affects ROS and antioxidant responses | Contributes to cancer development through ROS-mediated DNA damage and inflammatory responses | [106] |
| Streptococci, Candida yeasts | Metabolize alcohol to acetaldehyde via ADH activity | Causes DNA damage, increases carcinogenesis risk | [95] |
| Bacteria | Mechanism | Impact on EC | References |
|---|---|---|---|
| H. pylori | Increases ROS production through virulence factors | Activates angiogenesis and cancer development | [168] |
| Promotes hypoxic conditions stabilizing HIF-1α | Upregulates pro-angiogenic genes such as VEGF, contributing to tumor progression and poor prognosis | [169] | |
| F. nucleatum | Influences IL-8 production | Enhances angiogenesis and tumor invasiveness | [113] |
| Enhances IL-1β production | Creates a pro-inflammatory and pro-angiogenic microenvironment | [170] | |
| Increases TNF-α levels | Contributes to angiogenesis and tumor progression | [66] | |
| Activates β-catenin signaling, enhancing β-catenin, C-myc, and cyclin D1 expression | Enhances cancer cell proliferation and tumor growth | [70] | |
| P. gingivalis | Modulates inflammatory responses and cytokine production | Enhances tumor angiogenesis | [170] |
| Increases TNF-α levels | Promotes cancer cell proliferation and metastasis | [171] | |
| Produces H2S, activating proliferation, migration, and invasive signaling pathways | Contributes to a hypoxic, pro-angiogenic microenvironment | [101] | |
| Streptococcus species | Stimulates the production of angiogenic factors such as IL-8, VEGF, and bFGF | Promotes angiogenesis and cancer cell growth | [172] |
| General oral microbiota | Produces IL-1β, which activates endothelial cells to produce VEGF and other pro-angiogenic factors | Provides an inflammatory microenvironment conducive to angiogenesis and tumor progression | [173,174] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moe, K.T.; Tan, K.S.-W. Mechanistic Insights on Microbiota-Mediated Development and Progression of Esophageal Cancer. Cancers 2024, 16, 3305. https://doi.org/10.3390/cancers16193305
Moe KT, Tan KS-W. Mechanistic Insights on Microbiota-Mediated Development and Progression of Esophageal Cancer. Cancers. 2024; 16(19):3305. https://doi.org/10.3390/cancers16193305
Chicago/Turabian StyleMoe, Kyaw Thu, and Kevin Shyong-Wei Tan. 2024. "Mechanistic Insights on Microbiota-Mediated Development and Progression of Esophageal Cancer" Cancers 16, no. 19: 3305. https://doi.org/10.3390/cancers16193305
APA StyleMoe, K. T., & Tan, K. S.-W. (2024). Mechanistic Insights on Microbiota-Mediated Development and Progression of Esophageal Cancer. Cancers, 16(19), 3305. https://doi.org/10.3390/cancers16193305