
Evolution of Molecular Biomarkers and Precision Molecular Therapeutic Strategies in Glioblastoma


Simple Summary
Abstract
1. Introduction
2. Current Nomenclature for Classification of Tumors
3. Histopathological Features of Glioblastoma
4. Radiographic Presentation of Glioblastoma
4.1. Criteria for Assessment of Imaging in Brain Tumors
4.2. Standard and Advanced MRI Imaging of Brain Tumors
4.3. Radiogenomics in Tumors of the Brain
5. Molecular Features of Glioblastoma and Prognostic Implications
5.1. IDH Mutation Status
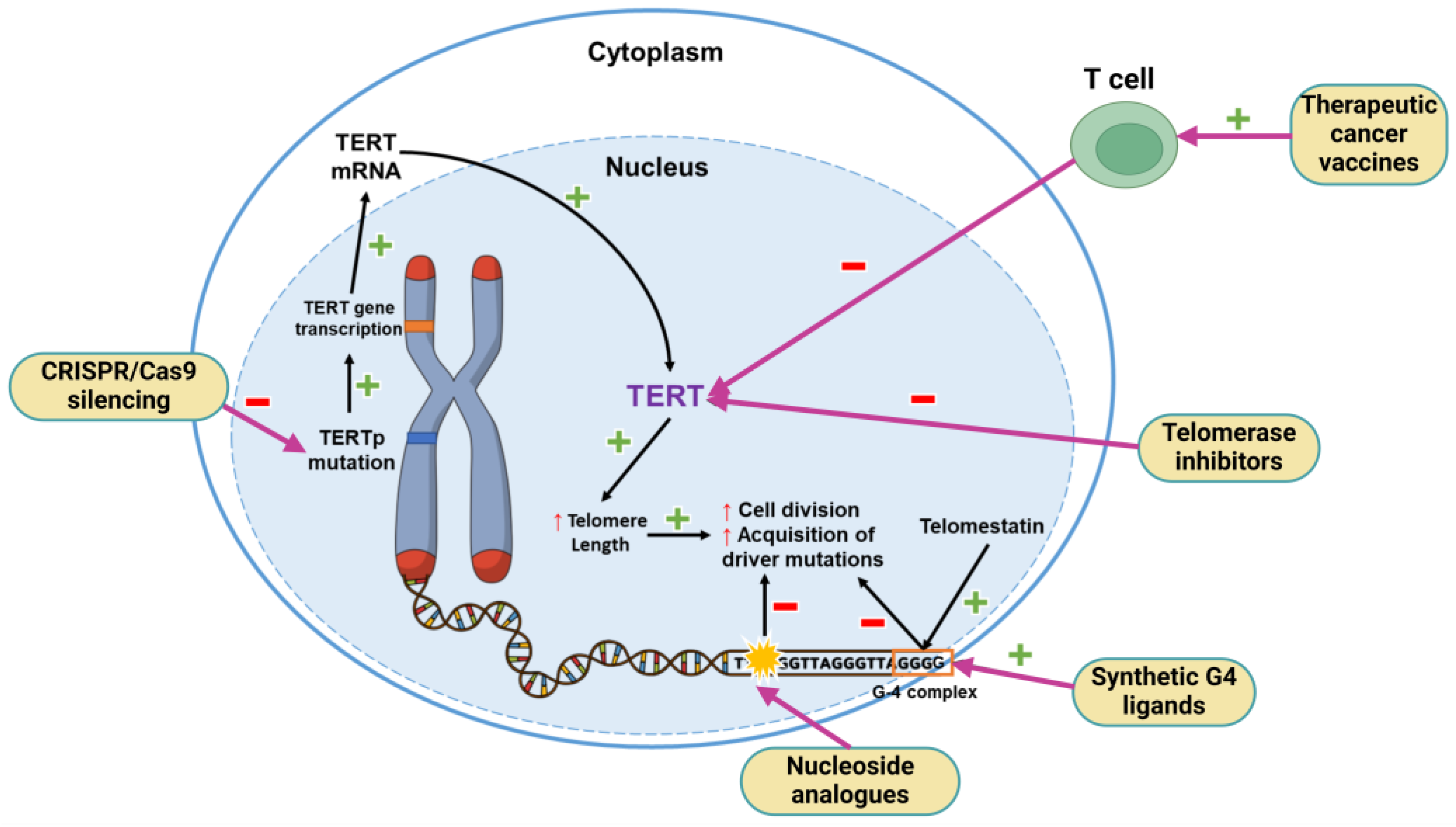
5.2. TERT Promoter Mutation
5.3. EGFR Gene Amplification
5.4. Concomitant Chromosome 10 Loss and Chromosome 7 Gain
5.5. MGMT Promoter Methylation Status
5.6. Other Potential Targets for Therapy
6. Discussion and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Worldwide Cancer Data. Available online: https://www.wcrf.org/cancer-trends/worldwide-cancer-data/ (accessed on 16 March 2024).
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2023, 25, iv1–iv99. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Senhaji, N.; Houssaini, A.S.; Lamrabet, S.; Louati, S.; Bennis, S. Molecular and Circulating Biomarkers in Patients with Glioblastoma. Int. J. Mol. Sci. 2022, 23, 7474. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E. Updates on the WHO Diagnosis of IDH-Mutant Glioma. J. Neurooncol. 2023, 162, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Akyerli, C.B.; Yüksel, Ş.; Can, Ö.; Erson-Omay, E.Z.; Oktay, Y.; Coşgun, E.; Ülgen, E.; Erdemgil, Y.; Sav, A.; von Deimling, A.; et al. Use of Telomerase Promoter Mutations to Mark Specific Molecular Subsets with Reciprocal Clinical Behavior in IDH Mutant and IDH Wild-Type Diffuse Gliomas. J. Neurosurg. 2018, 128, 1102–1114. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. CIMPACT-NOW Update 3: Recommended Diagnostic Criteria for “Diffuse Astrocytic Glioma, IDH-Wildtype, with Molecular Features of Glioblastoma, WHO Grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Figrarella-Branger, D.; Fuller, G.N.; Giannini, C.; Holland, E.C.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Komori, T.; et al. CIMPACT-NOW Update 5: Recommended Grading Criteria and Terminologies for IDH-Mutant Astrocytomas. Acta Neuropathol. 2020, 139, 603–608. [Google Scholar] [CrossRef]
- Tesileanu, C.M.S.; Dirven, L.; Wijnenga, M.M.J.; Koekkoek, J.A.F.; Vincent, A.J.P.E.; Dubbink, H.J.; Atmodimedjo, P.N.; Kros, J.M.; Van Duinen, S.G.; Smits, M.; et al. Survival of Diffuse Astrocytic Glioma, IDH1/2 Wildtype, with Molecular Features of Glioblastoma, WHO Grade IV: A Confirmation of the CIMPACT-NOW Criteria. Neuro Oncol. 2020, 22, 515–523. [Google Scholar] [CrossRef]
- Guo, X.; Gu, L.; Li, Y.; Zheng, Z.; Chen, W.; Wang, Y.; Wang, Y.; Xing, H.; Shi, Y.; Liu, D.; et al. Histological and Molecular Glioblastoma, IDH-Wildtype: A Real-World Landscape Using the 2021 WHO Classification of Central Nervous System Tumors. Front. Oncol. 2023, 13, 1200815. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Rahim, S.; Abdul-Ghafar, J.; Chundriger, Q.; Din, N.U. Events in CNS Tumor Pathology Post-2016 WHO CNS: CIMPACT-NOW Updates and Other Advancements: A Comprehensive Review Plus a Summary of the Salient Features of 2021 WHO CNS 5. Int. J. Gen. Med. 2023, 16, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Shan, F.Y.; Zhao, D.; Tirado, C.A.; Fonkem, E.; Zhang, Y.; Feng, D.; Huang, J.H. Glioblastomas: Molecular Diagnosis and Pathology. In Glioblastoma-Current Evidence; IntechOpen: London, UK, 2023. [Google Scholar]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Beiriger, J.; Habib, A.; Jovanovich, N.; Kodavali, C.V.; Edwards, L.; Amankulor, N.; Zinn, P.O. The Subventricular Zone in Glioblastoma: Genesis, Maintenance, and Modeling. Front. Oncol. 2022, 12, 790976. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; Al-Shammari, A.R.; Soares, L.; Tomlinson, I.; O’Neill, E.; Szele, F.G. The Role of Inflammation in Subventricular Zone Cancer. Prog. Neurobiol. 2018, 170, 37–52. [Google Scholar] [CrossRef]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of Origin for Malignant Gliomas and Its Implication in Therapeutic Development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Ah-Pine, F.; Khettab, M.; Bedoui, Y.; Slama, Y.; Daniel, M.; Doray, B.; Gasque, P. On the Origin and Development of Glioblastoma: Multifaceted Role of Perivascular Mesenchymal Stromal Cells. Acta Neuropathol. Commun. 2023, 11, 104. [Google Scholar] [CrossRef]
- Zheng, Y.; Carrillo-Perez, F.; Pizurica, M.; Heiland, D.H.; Gevaert, O. Spatial Cellular Architecture Predicts Prognosis in Glioblastoma. Nat. Commun. 2023, 14, 4122. [Google Scholar] [CrossRef]
- Cheung, E.Y.W.; Chu, E.S.M.; Li, A.S.M.; Tang, F.; Wu, R.W. 408P Machine Learning for Glioblastoma Screening from Histopathology Whole Slide Imaging. Ann. Oncol. 2022, 33, S1602. [Google Scholar] [CrossRef]
- Ortega, S.; Halicek, M.; Fabelo, H.; Camacho, R.; Plaza, M.d.l.L.; Godtliebsen, F.; Callicó, G.M.; Fei, B. Hyperspectral Imaging for the Detection of Glioblastoma Tumor Cells in H&E Slides Using Convolutional Neural Networks. Sensors 2020, 20, 1911. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G.S.; Petkova, L.; Dzhenkov, D.L. A Practical Approach to the Differential Diagnosis of Intracranial Tumors: Gross, Histology, and Immunoprofile-Based Algorithm. Cureus 2019, 11, e6384. [Google Scholar] [CrossRef]
- Mikkelsen, V.E.; Solheim, O.; Salvesen, Ø.; Torp, S.H. The Histological Representativeness of Glioblastoma Tissue Samples. Acta Neurochir. 2021, 163, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Unruh, D.; Schwarze, S.R.; Khoury, L.; Thomas, C.; Wu, M.; Chen, L.; Chen, R.; Liu, Y.; Schwartz, M.A.; Amidei, C.; et al. Mutant IDH1 and Thrombosis in Gliomas. Acta Neuropathol. 2016, 132, 917–930. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T. Molecular Mechanisms Involved in Gliomagenesis. Brain Tumor Pathol. 2017, 34, 1–7. [Google Scholar] [CrossRef]
- Becker, A.; Sells, B.; Haque, S.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Skjulsvik, A.J.; Mørk, J.N.; Torp, M.O.; Torp, S.H. Ki-67/MIB-1 Immunostaining in a Cohort of Human Gliomas. Int. J. Clin. Exp. Pathol. 2014, 7, 8905–8910. [Google Scholar]
- Priambada, D.; Thohar Arifin, M.; Saputro, A.; Muzakka, A.; Karlowee, V.; Sadhana, U.; Bakhtiar, Y.; Prihastomo, K.T.; Risdianto, A.; Brotoarianto, H.K.; et al. Immunohistochemical Expression of IDH1, ATRX, Ki67, GFAP, and Prognosis in Indonesian Glioma Patients. Int. J. Gen. Med. 2023, 16, 393–403. [Google Scholar] [CrossRef]
- Alkhaibary, A.; Alassiri, A.H.; AlSufiani, F.; Alharbi, M.A. Ki-67 Labeling Index in Glioblastoma; Does It Really Matter? Hematol. Oncol. Stem Cell Ther. 2019, 12, 82–88. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Dong, X.; Liu, L.; Huo, L.; Chen, H. High Expression of Vimentin Is Associated With Progression and a Poor Outcome in Glioblastoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Lee, M.K.; Dent, A.; Fiala, C.; Portante, A.; Rabindranath, M.; Alsafwani, N.; Gao, A.; Djuric, U.; Diamandis, P. Integrating Morphologic and Molecular Histopathological Features through Whole Slide Image Registration and Deep Learning. Neurooncol. Adv. 2022, 4, vdac001. [Google Scholar] [CrossRef] [PubMed]
- Lonjon, M.; Mondot, L.; Lonjon, N.; Chanalet, S. Chemins Cliniques Des Glioblastomes et Neuroradiologie. Neurochirurgie 2010, 56, 449–454. [Google Scholar] [CrossRef]
- Khandwala, K.; Mubarak, F.; Minhas, K. The Many Faces of Glioblastoma: Pictorial Review of Atypical Imaging Features. Neuroradiol. J. 2021, 34, 33–41. [Google Scholar] [CrossRef]
- Ellingson, B.M.; Wen, P.Y.; van den Bent, M.J.; Cloughesy, T.F. Pros and Cons of Current Brain Tumor Imaging. Neuro Oncol. 2014, 16, vii2–vii11. [Google Scholar] [CrossRef]
- Ellingson, B.M.; Bendszus, M.; Boxerman, J.; Barboriak, D.; Erickson, B.J.; Smits, M.; Nelson, S.J.; Gerstner, E.; Alexander, B.; Goldmacher, G.; et al. Consensus Recommendations for a Standardized Brain Tumor Imaging Protocol in Clinical Trials. Neuro Oncol. 2015, 17, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Meyer, J.E.; Mabray, M.C.; Cha, S. Current Clinical Brain Tumor Imaging. Neurosurgery 2017, 81, 397–415. [Google Scholar] [CrossRef]
- Sanvito, F.; Kaufmann, T.J.; Cloughesy, T.F.; Wen, P.Y.; Ellingson, B.M. Standardized Brain Tumor Imaging Protocols for Clinical Trials: Current Recommendations and Tips for Integration. Front. Radiol. 2023, 3, 1267615. [Google Scholar] [CrossRef]
- Treister, D.; Kingston, S.; Hoque, K.E.; Law, M.; Shiroishi, M.S. Multimodal Magnetic Resonance Imaging Evaluation of Primary Brain Tumors. Semin. Oncol. 2014, 41, 478–495. [Google Scholar] [CrossRef]
- Wen, P.Y.; Macdonald, D.R.; Reardon, D.A.; Cloughesy, T.F.; Sorensen, A.G.; Galanis, E.; DeGroot, J.; Wick, W.; Gilbert, M.R.; Lassman, A.B.; et al. Updated Response Assessment Criteria for High-Grade Gliomas: Response Assessment in Neuro-Oncology Working Group. J. Clin. Oncol. 2010, 28, 1963–1972. [Google Scholar] [CrossRef]
- Macdonald, D.R.; Cascino, T.L.; Schold, S.C.; Cairncross, J.G. Response Criteria for Phase II Studies of Supratentorial Malignant Glioma. J. Clin. Oncol. 1990, 8, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, K.; Kamimura, Y.; Nakano, T.; Hasegawa, T.; Nakajo, M.; Yamada, C.; Akune, K.; Ejima, F.; Ayukawa, T.; Ito, S.; et al. Differentiating Brain Metastasis from Glioblastoma by Time-Dependent Diffusion MRI. Cancer Imaging 2023, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Thust, S.C.; Hassanein, S.; Bisdas, S.; Rees, J.H.; Hyare, H.; Maynard, J.A.; Brandner, S.; Tur, C.; Jäger, H.R.; Yousry, T.A.; et al. Apparent Diffusion Coefficient for Molecular Subtyping of Non-Gadolinium-Enhancing WHO Grade II/III Glioma: Volumetric Segmentation versus Two-Dimensional Region of Interest Analysis. Eur. Radiol. 2018, 28, 3779–3788. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.; Okuchi, S.; Wastling, S.; Al Busaidi, A.; Almossawi, O.; Mbatha, W.; Brandner, S.; Jaunmuktane, Z.; Koc, A.M.; Mancini, L.; et al. World Health Organization Grade II/III Glioma Molecular Status: Prediction by MRI Morphologic Features and Apparent Diffusion Coefficient. Radiology 2020, 296, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shu, X.; He, P.; Cai, Y.; Geng, Y.; Hu, X.; Sun, Y.; Xiao, H.; Zheng, W.; Song, Y.; et al. Ultra-High b-Value DWI Accurately Identifies Isocitrate Dehydrogenase Genotypes and Tumor Subtypes of Adult-Type Diffuse Gliomas. Eur. Radiol. 2024, 34, 6751–6762. [Google Scholar] [CrossRef]
- Ellingson, B.M.; Kim, E.; Woodworth, D.C.; Marques, H.; Boxerman, J.L.; Safriel, Y.; McKinstry, R.C.; Bokstein, F.; Jain, R.; Chi, T.L.; et al. Diffusion MRI Quality Control and Functional Diffusion Map Results in ACRIN 6677/RTOG 0625: A Multicenter, Randomized, Phase II Trial of Bevacizumab and Chemotherapy in Recurrent Glioblastoma. Int. J. Oncol. 2015, 46, 1883–1892. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Branzoli, F.; Di Stefano, A.L.; Capelle, L.; Ottolenghi, C.; Valabrègue, R.; Deelchand, D.K.; Bielle, F.; Villa, C.; Baussart, B.; Lehéricy, S.; et al. Highly Specific Determination of IDH Status Using Edited in Vivo Magnetic Resonance Spectroscopy. Neuro Oncol. 2018, 20, 907–916. [Google Scholar] [CrossRef]
- Pope, W.B.; Prins, R.M.; Albert Thomas, M.; Nagarajan, R.; Yen, K.E.; Bittinger, M.A.; Salamon, N.; Chou, A.P.; Yong, W.H.; Soto, H.; et al. Non-Invasive Detection of 2-Hydroxyglutarate and Other Metabolites in IDH1 Mutant Glioma Patients Using Magnetic Resonance Spectroscopy. J. Neurooncol. 2012, 107, 197–205. [Google Scholar] [CrossRef]
- Verma, G.; Chawla, S.; Mohan, S.; Wang, S.; Nasrallah, M.; Sheriff, S.; Desai, A.; Brem, S.; O’Rourke, D.M.; Wolf, R.L.; et al. Three-dimensional Echo Planar Spectroscopic Imaging for Differentiation of True Progression from Pseudoprogression in Patients with Glioblastoma. NMR Biomed. 2019, 32, e4042. [Google Scholar] [CrossRef]
- Booth, T.C.; Wiegers, E.C.; Warnert, E.A.H.; Schmainda, K.M.; Riemer, F.; Nechifor, R.E.; Keil, V.C.; Hangel, G.; Figueiredo, P.; Álvarez-Torres, M.D.M.; et al. High-Grade Glioma Treatment Response Monitoring Biomarkers: A Position Statement on the Evidence Supporting the Use of Advanced MRI Techniques in the Clinic, and the Latest Bench-to-Bedside Developments. Part 2: Spectroscopy, Chemical Exchange Saturation, Multiparametric Imaging, and Radiomics. Front. Oncol. 2022, 11, 811425. [Google Scholar] [CrossRef]
- Yuan, Y.; Yu, Y.; Chang, J.; Chu, Y.-H.; Yu, W.; Hsu, Y.-C.; Patrick, L.A.; Liu, M.; Yue, Q. Convolutional Neural Network to Predict IDH Mutation Status in Glioma from Chemical Exchange Saturation Transfer Imaging at 7 Tesla. Front. Oncol. 2023, 13, 1134626. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, C.; Barritault, M.; Poncet, D.; Cartalat, S.; Joubert, B.; Bruna, J.; Jouanneau, E.; Guyotat, J.; Vasiljevic, A.; Fenouil, T.; et al. Radiological Characteristics and Natural History of Adult IDH-Wildtype Astrocytomas with TERT Promoter Mutations. Neurosurgery 2019, 85, E448–E456. [Google Scholar] [CrossRef]
- Fathi Kazerooni, A.; Bakas, S.; Saligheh Rad, H.; Davatzikos, C. Imaging Signatures of Glioblastoma Molecular Characteristics: A Radiogenomics Review. J. Magn. Reson. Imaging 2020, 52, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Lasocki, A.; Rosenthal, M.A.; Roberts-Thomson, S.J.; Neal, A.; Drummond, K.J. Neuro-Oncology and Radiogenomics: Time to Integrate? Am. J. Neuroradiol. 2020, 41, 1982–1988. [Google Scholar] [CrossRef]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images Are More than Pictures, They Are Data. Radiology 2016, 278, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Auer, T.A. Advanced MR Techniques in Glioblastoma Imaging—Upcoming Challenges and How to Face Them. Eur. Radiol. 2021, 31, 6652–6654. [Google Scholar] [CrossRef]
- Choi, Y.S.; Bae, S.; Chang, J.H.; Kang, S.-G.; Kim, S.H.; Kim, J.; Rim, T.H.; Choi, S.H.; Jain, R.; Lee, S.-K. Fully Automated Hybrid Approach to Predict the IDH Mutation Status of Gliomas via Deep Learning and Radiomics. Neuro Oncol. 2021, 23, 304–313. [Google Scholar] [CrossRef]
- Lotan, E.; Jain, R.; Razavian, N.; Fatterpekar, G.M.; Lui, Y.W. State of the Art: Machine Learning Applications in Glioma Imaging. Am. J. Roentgenol. 2019, 212, 26–37. [Google Scholar] [CrossRef]
- Sanvito, F.; Castellano, A.; Falini, A. Advancements in Neuroimaging to Unravel Biological and Molecular Features of Brain Tumors. Cancers 2021, 13, 424. [Google Scholar] [CrossRef]
- Díaz-Pernas, F.J.; Martínez-Zarzuela, M.; Antón-Rodríguez, M.; González-Ortega, D. A Deep Learning Approach for Brain Tumor Classification and Segmentation Using a Multiscale Convolutional Neural Network. Healthcare 2021, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.M.; Zakariah, M.; Aldakheel, E.A. Brain Tumor Segmentation Using Deep Learning on MRI Images. Diagnostics 2023, 13, 1562. [Google Scholar] [CrossRef] [PubMed]
- Ben naceur, M.; Akil, M.; Saouli, R.; Kachouri, R. Fully Automatic Brain Tumor Segmentation with Deep Learning-Based Selective Attention Using Overlapping Patches and Multi-Class Weighted Cross-Entropy. Med. Image Anal. 2020, 63, 101692. [Google Scholar] [CrossRef] [PubMed]
- Munir, K.; Frezza, F.; Rizzi, A. Deep Learning Hybrid Techniques for Brain Tumor Segmentation. Sensors 2022, 22, 8201. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Grinband, J.; Weinberg, B.D.; Bardis, M.; Khy, M.; Cadena, G.; Su, M.-Y.; Cha, S.; Filippi, C.G.; Bota, D.; et al. Deep-Learning Convolutional Neural Networks Accurately Classify Genetic Mutations in Gliomas. Am. J. Neuroradiol. 2018, 39, 1201–1207. [Google Scholar] [CrossRef]
- Luckett, P.H.; Olufawo, M.; Lamichhane, B.; Park, K.Y.; Dierker, D.; Verastegui, G.T.; Yang, P.; Kim, A.H.; Chheda, M.G.; Snyder, A.Z.; et al. Predicting Survival in Glioblastoma with Multimodal Neuroimaging and Machine Learning. J. Neurooncol. 2023, 164, 309–320. [Google Scholar] [CrossRef]
- Karami, G.; Pascuzzo, R.; Figini, M.; Del Gratta, C.; Zhang, H.; Bizzi, A. Combining Multi-Shell Diffusion with Conventional MRI Improves Molecular Diagnosis of Diffuse Gliomas with Deep Learning. Cancers 2023, 15, 482. [Google Scholar] [CrossRef]
- Han, Y.; Yan, L.-F.; Wang, X.-B.; Sun, Y.-Z.; Zhang, X.; Liu, Z.-C.; Nan, H.-Y.; Hu, Y.-C.; Yang, Y.; Zhang, J.; et al. Structural and Advanced Imaging in Predicting MGMT Promoter Methylation of Primary Glioblastoma: A Region of Interest Based Analysis. BMC Cancer 2018, 18, 215. [Google Scholar] [CrossRef]
- Ladenhauf, V.K.; Galijasevic, M.; Kerschbaumer, J.; Freyschlag, C.F.; Nowosielski, M.; Birkl-Toeglhofer, A.M.; Haybaeck, J.; Gizewski, E.R.; Mangesius, S.; Grams, A.E. Peritumoral ADC Values Correlate with the MGMT Methylation Status in Patients with Glioblastoma. Cancers 2023, 15, 1384. [Google Scholar] [CrossRef]
- Ahn, S.S.; Shin, N.-Y.; Chang, J.H.; Kim, S.H.; Kim, E.H.; Kim, D.W.; Lee, S.-K. Prediction of Methylguanine Methyltransferase Promoter Methylation in Glioblastoma Using Dynamic Contrast-Enhanced Magnetic Resonance and Diffusion Tensor Imaging. J. Neurosurg. 2014, 121, 367–373. [Google Scholar] [CrossRef]
- Rundle-Thiele, D.; Day, B.; Stringer, B.; Fay, M.; Martin, J.; Jeffree, R.L.; Thomas, P.; Bell, C.; Salvado, O.; Gal, Y.; et al. Using the Apparent Diffusion Coefficient to Identifying MGMT Promoter Methylation Status Early in Glioblastoma: Importance of Analytical Method. J. Med. Radiat. Sci. 2015, 62, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-C.; Bai, H.; Sun, Q.; Li, Q.; Liu, L.; Zou, Y.; Chen, Y.; Liang, C.; Zheng, H. Multiregional Radiomics Features from Multiparametric MRI for Prediction of MGMT Methylation Status in Glioblastoma Multiforme: A Multicentre Study. Eur. Radiol. 2018, 28, 3640–3650. [Google Scholar] [CrossRef] [PubMed]
- Korfiatis, P.; Kline, T.L.; Coufalova, L.; Lachance, D.H.; Parney, I.F.; Carter, R.E.; Buckner, J.C.; Erickson, B.J. MRI Texture Features as Biomarkers to Predict MGMT Methylation Status in Glioblastomas. Med. Phys. 2016, 43, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Yogananda, C.G.B.; Shah, B.R.; Nalawade, S.S.; Murugesan, G.K.; Yu, F.F.; Pinho, M.C.; Wagner, B.C.; Mickey, B.; Patel, T.R.; Fei, B.; et al. MRI-Based Deep-Learning Method for Determining Glioma MGMT Promoter Methylation Status. Am. J. Neuroradiol. 2021, 42, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Jian, A.; Jang, K.; Manuguerra, M.; Liu, S.; Magnussen, J.; Di Ieva, A. Machine Learning for the Prediction of Molecular Markers in Glioma on Magnetic Resonance Imaging: A Systematic Review and Meta-Analysis. Neurosurgery 2021, 89, 31–44. [Google Scholar] [CrossRef]
- Choi, Y.; Jang, J.; Kim, B.; Ahn, K.-J. Pretreatment MR-Based Radiomics in Patients with Glioblastoma: A Systematic Review and Meta-Analysis of Prognostic Endpoints. Eur. J. Radiol. 2023, 168, 111130. [Google Scholar] [CrossRef]
- Jiang, S.; Wen, Z.; Ahn, S.S.; Cai, K.; Paech, D.; Eberhart, C.G.; Zhou, J. Applications of Chemical Exchange Saturation Transfer Magnetic Resonance Imaging in Identifying Genetic Markers in Gliomas. NMR Biomed. 2023, 36, e4731. [Google Scholar] [CrossRef]
- Park, Y.W.; Ahn, S.S.; Park, C.J.; Han, K.; Kim, E.H.; Kang, S.-G.; Chang, J.H.; Kim, S.H.; Lee, S.-K. Diffusion and Perfusion MRI May Predict EGFR Amplification and the TERT Promoter Mutation Status of IDH-Wildtype Lower-Grade Gliomas. Eur. Radiol. 2020, 30, 6475–6484. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.; Zhang, Y.; Zhou, B.; Wu, L.; Lei, Y.; Huang, B. Deep Learning Radiomics for the Assessment of Telomerase Reverse Transcriptase Promoter Mutation Status in Patients With Glioblastoma Using Multiparametric MRI. J. Magn. Reson. Imaging 2023, 58, 1441–1451. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Li, T.; Huang, L.; Tang, C.; Li, Y.; Zeng, Z. MRI Radiomics Model for Predicting TERT Promoter Mutation Status in Glioblastoma. Brain Behav. 2023, 13, e3324. [Google Scholar] [CrossRef]
- Buz-Yalug, B.; Turhan, G.; Cetin, A.I.; Dindar, S.S.; Danyeli, A.E.; Yakicier, C.; Pamir, M.N.; Özduman, K.; Dincer, A.; Ozturk-Isik, E. Identification of IDH and TERTp Mutations Using Dynamic Susceptibility Contrast MRI with Deep Learning in 162 Gliomas. Eur. J. Radiol. 2024, 170, 111257. [Google Scholar] [CrossRef] [PubMed]
- Jovanovich, N.; Habib, A.; Chilukuri, A.; Hameed, N.U.F.; Deng, H.; Shanahan, R.; Head, J.R.; Zinn, P.O. Sex-Specific Molecular Differences in Glioblastoma: Assessing the Clinical Significance of Genetic Variants. Front. Oncol. 2024, 13, 1340386. [Google Scholar] [CrossRef] [PubMed]
- Shireman, J.M.; Ammanuel, S.; Eickhoff, J.C.; Dey, M. Sexual Dimorphism of the Immune System Predicts Clinical Outcomes in Glioblastoma Immunotherapy: A Systematic Review and Meta-Analysis. Neurooncol. Adv. 2022, 4, vdac082. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.E.; Ozair, A.; Bamashmos, A.S.; Li, H.; Bosler, D.S.; Yeaney, G.; Ali, A.; Peereboom, D.M.; Lathia, J.D.; Ahluwalia, M.S. MGMT Methylation and Differential Survival Impact by Sex in Glioblastoma. Cancers 2024, 16, 1374. [Google Scholar] [CrossRef]
- Sarhadi, V.K.; Armengol, G. Molecular Biomarkers in Cancer. Biomolecules 2022, 12, 1021. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science (1979) 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Pandey, M.; Anoosha, P.; Yesudhas, D.; Gromiha, M.M. Identification of Potential Driver Mutations in Glioblastoma Using Machine Learning. Brief Bioinform. 2022, 23, bbac451. [Google Scholar] [CrossRef]
- Herrera-Oropeza, G.E.; Angulo-Rojo, C.; Gástelum-López, S.A.; Varela-Echavarría, A.; Hernández-Rosales, M.; Aviña-Padilla, K. Glioblastoma Multiforme: A Multi-Omics Analysis of Driver Genes and Tumour Heterogeneity. Interface Focus 2021, 11, 20200072. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Alzial, G.; Renoult, O.; Paris, F.; Gratas, C.; Clavreul, A.; Pecqueur, C. Wild-Type Isocitrate Dehydrogenase under the Spotlight in Glioblastoma. Oncogene 2022, 41, 613–621. [Google Scholar] [CrossRef]
- Nakhate, V.; Lasica, A.B.; Wen, P.Y. The Role of Mutant IDH Inhibitors in the Treatment of Glioma. Curr. Neurol. Neurosci. Rep. 2024. Online early access. Available online: https://link.springer.com/article/10.1007/s11910-024-01378-3 (accessed on 20 September 2024). [CrossRef]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Jakob, C.G.; Upadhyay, A.K.; Donner, P.L.; Nicholl, E.; Addo, S.N.; Qiu, W.; Ling, C.; Gopalakrishnan, S.M.; Torrent, M.; Cepa, S.P.; et al. Novel Modes of Inhibition of Wild-Type Isocitrate Dehydrogenase 1 (IDH1): Direct Covalent Modification of His315. J. Med. Chem. 2018, 61, 6647–6657. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Dresser, J.; Wilder-Romans, K.; Parsels, J.D.; Zhao, S.G.; Davis, M.; Zhao, L.; Kachman, M.; Wernisch, S.; Burant, C.F.; et al. Glioblastoma Therapy Can Be Augmented by Targeting IDH1-Mediated NADPH Biosynthesis. Cancer Res. 2017, 77, 960–970. [Google Scholar] [CrossRef] [PubMed]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. IDH3α Regulates One-Carbon Metabolism in Glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef]
- Pierini, T.; Nardelli, C.; Lema Fernandez, A.G.; Pierini, V.; Pellanera, F.; Nofrini, V.; Gorello, P.; Moretti, M.; Arniani, S.; Roti, G.; et al. New Somatic TERT Promoter Variants Enhance the Telomerase Activity in Glioblastoma. Acta Neuropathol. Commun. 2020, 8, 145. [Google Scholar] [CrossRef]
- Bollam, S.R.; Berens, M.E.; Dhruv, H.D. When the Ends Are Really the Beginnings: Targeting Telomerase for Treatment of GBM. Curr. Neurol. Neurosci. Rep. 2018, 18, 15. [Google Scholar] [CrossRef]
- Di Nunno, V.; Aprile, M.; Bartolini, S.; Gatto, L.; Tosoni, A.; Ranieri, L.; De Biase, D.; Asioli, S.; Franceschi, E. The Biological and Clinical Role of the Telomerase Reverse Transcriptase Gene in Glioblastoma: A Potential Therapeutic Target? Cells 2023, 13, 44. [Google Scholar] [CrossRef]
- Giunco, S.; Padovan, M.; Angelini, C.; Cavallin, F.; Cerretti, G.; Morello, M.; Caccese, M.; Rizzo, B.; d’Avella, D.; Della Puppa, A.; et al. Prognostic Role and Interaction of TERT Promoter Status, Telomere Length and MGMT Promoter Methylation in Newly Diagnosed IDH Wild-Type Glioblastoma Patients. ESMO Open 2023, 8, 101570. [Google Scholar] [CrossRef]
- Cappelli, L.; Khan, M.M.; Kayne, A.; Poiset, S.; Miller, R.; Ali, A.; Niazi, M.; Shi, W.; Alnahhas, I. Differences in Clinical Outcomes Based on Molecular Markers in Glioblastoma Patients Treated with Concurrent Tumor-Treating Fields and Chemoradiation: Exploratory Analysis of the SPARE Trial. Chin. Clin. Oncol. 2023, 12, 23. [Google Scholar] [CrossRef]
- Salloum, R.; Hummel, T.R.; Kumar, S.S.; Dorris, K.; Li, S.; Lin, T.; Daryani, V.M.; Stewart, C.F.; Miles, L.; Poussaint, T.Y.; et al. A Molecular Biology and Phase II Study of Imetelstat (GRN163L) in Children with Recurrent or Refractory Central Nervous System Malignancies: A Pediatric Brain Tumor Consortium Study. J. Neurooncol. 2016, 129, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, E.B.; Mangsbo, S.M.; Hovig, E.; Gaudernack, G. Telomerase as a Target for Therapeutic Cancer Vaccines and Considerations for Optimizing Their Clinical Potential. Front. Immunol. 2021, 12, 682492. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brem, S.; Desai, A.S.; Bagley, S.J.; Kurz, S.C.; De La Fuente, M.I.; Nagpal, S.; Welch, M.R.; Hormigo, A.; Forsyth, P.A.J.; et al. Intramuscular (IM) INO-5401 + INO-9012 with Electroporation (EP) in Combination with Cemiplimab (REGN2810) in Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2022, 40, 2004. [Google Scholar] [CrossRef]
- Carpentier, A.F.; Verlut, C.; Ghiringhelli, F.; Bronnimann, C.; Ursu, R.; Fumet, J.D.; Gherga, E.; Lefort, F.; Belin, C.; Vernerey, D.; et al. Anti-Telomerase Vaccine in Patients with Newly Diagnosed, Unmethylated MGMT Glioblastoma: A Phase II Study. J. Clin. Oncol. 2023, 41, 2005. [Google Scholar] [CrossRef]
- Xiong, Z.; Raphael, I.; Olin, M.; Okada, H.; Li, X.; Kohanbash, G. Glioblastoma Vaccines: Past, Present, and Opportunities. EBioMedicine 2024, 100, 104963. [Google Scholar] [CrossRef]
- Mender, I.; Zhang, A.; Ren, Z.; Han, C.; Deng, Y.; Siteni, S.; Li, H.; Zhu, J.; Vemula, A.; Shay, J.W.; et al. Telomere Stress Potentiates STING-Dependent Anti-Tumor Immunity. Cancer Cell 2020, 38, 400–411.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Yan, L.; You, Y.; Zhao, F.; Cheng, J.; Yang, L.; Sun, Y.; Chang, Q.; Liu, R.; et al. Zeolitic Imidazolate Framework-8 (ZIF-8) as a Drug Delivery Vehicle for the Transport and Release of Telomerase Inhibitor BIBR 1532. Nanomaterials 2023, 13, 1779. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, X.; Li, Y.; Xu, S.; Ma, C.; Wu, X.; Cheng, Y.; Yu, Z.; Zhao, G.; Chen, Y. Telomere Targeting with a Novel G-Quadruplex-Interactive Ligand BRACO-19 Induces T-Loop Disassembly and Telomerase Displacement in Human Glioblastoma Cells. Oncotarget 2016, 7, 14925–14939. [Google Scholar] [CrossRef]
- Nakamura, T.; Okabe, S.; Yoshida, H.; Iida, K.; Ma, Y.; Sasaki, S.; Yamori, T.; Shin-ya, K.; Nakano, I.; Nagasawa, K.; et al. Targeting Glioma Stem Cells in Vivo by a G-Quadruplex-Stabilizing Synthetic Macrocyclic Hexaoxazole. Sci. Rep. 2017, 7, 3605. [Google Scholar] [CrossRef]
- Berardinelli, F.; Tanori, M.; Muoio, D.; Buccarelli, M.; di Masi, A.; Leone, S.; Ricci-Vitiani, L.; Pallini, R.; Mancuso, M.; Antoccia, A. G-Quadruplex Ligand RHPS4 Radiosensitizes Glioblastoma Xenograft in Vivo through a Differential Targeting of Bulky Differentiated- and Stem-Cancer Cells. J. Exp. Clin. Cancer Res. 2019, 38, 311. [Google Scholar] [CrossRef]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable Base Editing of Mutated TERT Promoter Inhibits Brain Tumour Growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Amen, A.M.; Fellmann, C.; Soczek, K.M.; Ren, S.M.; Lew, R.J.; Knott, G.J.; Park, J.E.; McKinney, A.M.; Mancini, A.; Doudna, J.A.; et al. Cancer-Specific Loss of TERT Activation Sensitizes Glioblastoma to DNA Damage. Proc. Natl. Acad. Sci. USA 2021, 118, e2008772118. [Google Scholar] [CrossRef]
- Mender, I.; Gryaznov, S.; Dikmen, Z.G.; Wright, W.E.; Shay, J.W. Induction of Telomere Dysfunction Mediated by the Telomerase Substrate Precursor 6-Thio-2′-Deoxyguanosine. Cancer Discov. 2015, 5, 82–95. [Google Scholar] [CrossRef]
- Zeng, X.; Hernandez-Sanchez, W.; Xu, M.; Whited, T.L.; Baus, D.; Zhang, J.; Berdis, A.J.; Taylor, D.J. Administration of a Nucleoside Analog Promotes Cancer Cell Death in a Telomerase-Dependent Manner. Cell Rep. 2018, 23, 3031–3041. [Google Scholar] [CrossRef]
- Lavanya, C.; Venkataswamy, M.M.; Sibin, M.K.; Srinivas Bharath, M.M.; Chetan, G.K. Down Regulation of Human Telomerase Reverse Transcriptase (HTERT) Expression by BIBR1532 in Human Glioblastoma LN18 Cells. Cytotechnology 2018, 70, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, D.; Okabe, S.; Okamoto, K.; Nakano, I.; Shin-ya, K.; Seimiya, H. G-Quadruplex Ligand-Induced DNA Damage Response Coupled with Telomere Dysfunction and Replication Stress in Glioma Stem Cells. Biochem. Biophys. Res. Commun. 2016, 471, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zong, H.; Ma, C.; Ming, X.; Shang, M.; Li, K.; He, X.; Du, H.; Cao, L. Epidermal Growth Factor Receptor in Glioblastoma. Oncol. Lett. 2017, 14, 512–516. [Google Scholar] [CrossRef]
- Maire, C.L.; Ligon, K.L. Molecular Pathologic Diagnosis of Epidermal Growth Factor Receptor. Neuro Oncol. 2014, 16, viii1–viii6. [Google Scholar] [CrossRef]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic Significance of Epidermal Growth Factor Receptor Expression in Glioma Patients. Onco Targets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting Cellular Pathways in Glioblastoma Multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef]
- Ezzati, S.; Salib, S.; Balasubramaniam, M.; Aboud, O. Epidermal Growth Factor Receptor Inhibitors in Glioblastoma: Current Status and Future Possibilities. Int. J. Mol. Sci. 2024, 25, 2316. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A Phase II Study of Bevacizumab and Erlotinib after Radiation and Temozolomide in MGMT Unmethylated GBM Patients. J. Neurooncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.F.; Jaramillo-Velásquez, D.; Ruiz-Patiño, A.; Polo, C.; Jiménez, E.; Hakim, F.; Gómez, D.; Ramón, J.F.; Cifuentes, H.; Mejía, J.A.; et al. Efficacy of Osimertinib plus Bevacizumab in Glioblastoma Patients with Simultaneous EGFR Amplification and EGFRvIII Mutation. J. Neurooncol. 2021, 154, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Oguchi, K.; Araki, H.; Tsuji, S.; Nakamura, M.; Miura, A.; Funabashi, K.; Osada, A.; Tanaka, S.; Suzuki, T.; Kobayashi, S.S.; et al. TAS2940, a Novel Brain-penetrable Pan-ERBB Inhibitor, for Tumors with HER2 and EGFR Aberrations. Cancer Sci. 2023, 114, 654–664. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; Vogelbaum, M.A.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab Mafodotin in EGFR-Amplified Newly Diagnosed Glioblastoma: A Phase III Randomized Clinical Trial. Neuro Oncol. 2023, 25, 339–350. [Google Scholar] [CrossRef]
- Choi, S.W.; Jung, H.A.; Cho, H.; Kim, T.M.; Park, C.; Nam, D.; Lee, S. A Multicenter, Phase II Trial of GC1118, a Novel Anti-EGFR Antibody, for Recurrent Glioblastoma Patients with EGFR Amplification. Cancer Med. 2023, 12, 15788–15796. [Google Scholar] [CrossRef] [PubMed]
- Spiekman, I.A.C.; Geurts, B.S.; Zeverijn, L.J.; de Wit, G.F.; van der Noort, V.; Roepman, P.; de Leng, W.W.J.; Jansen, A.M.L.; Kusters, B.; Beerepoot, L.V.; et al. Efficacy and Safety of Panitumumab in Patients With RAF/RAS-Wild-Type Glioblastoma: Results From the Drug Rediscovery Protocol. Oncologist 2024, 29, 431–440. [Google Scholar] [CrossRef]
- Du, X.-J.; Li, X.-M.; Cai, L.-B.; Sun, J.-C.; Wang, S.-Y.; Wang, X.-C.; Pang, X.-L.; Deng, M.-L.; Chen, F.-F.; Wang, Z.-Q.; et al. Efficacy and Safety of Nimotuzumab in Addition to Radiotherapy and Temozolomide for Cerebral Glioblastoma: A Phase II Multicenter Clinical Trial. J. Cancer 2019, 10, 3214–3223. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, Y.; Wanggou, S.; Lin, D.; Su, J.; Li, X.; Tao, E. A Natural Compound Melatonin Enhances the Effects of Nimotuzumab via Inhibiting EGFR in Glioblastoma. Cancer Lett. 2024, 592, 216920. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Appolloni, I.; Alessandrini, F.; Menotti, L.; Avitabile, E.; Marubbi, D.; Piga, N.; Ceresa, D.; Piaggio, F.; Campadelli-Fiume, G.; Malatesta, P. Specificity, Safety, Efficacy of EGFRvIII-Retargeted Oncolytic HSV for Xenotransplanted Human Glioblastoma. Viruses 2021, 13, 1677. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Xu, B.; Chen, Y.; Li, Z.; Wang, J.; Zhang, J.; Ma, R.; Cao, S.; Hu, W.; Chiocca, E.A.; et al. Specific Targeting of Glioblastoma with an Oncolytic Virus Expressing a Cetuximab-CCL5 Fusion Protein via Innate and Adaptive Immunity. Nat. Cancer 2022, 3, 1318–1335. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.-Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J.; et al. An Oncolytic Virus Expressing IL15/IL15Rα Combined with Off-the-Shelf EGFR-CAR NK Cells Targets Glioblastoma. Cancer Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of Glioblastoma by Immuno-Virotherapy with a Retargeted Oncolytic HSV in a Preclinical Model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–Transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.P.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G.; et al. Repeated Peripheral Infusions of Anti-EGFRvIII CAR T Cells in Combination with Pembrolizumab Show No Efficacy in Glioblastoma: A Phase 1 Trial. Nat. Cancer 2024, 5, 517–531. [Google Scholar] [CrossRef]
- Bagley, S.J.; Logun, M.; Fraietta, J.A.; Wang, X.; Desai, A.S.; Bagley, L.J.; Nabavizadeh, A.; Jarocha, D.; Martins, R.; Maloney, E.; et al. Intrathecal Bivalent CAR T Cells Targeting EGFR and IL13Rα2 in Recurrent Glioblastoma: Phase 1 Trial Interim Results. Nat. Med. 2024, 30, 1320–1329. [Google Scholar] [CrossRef]
- Luwor, R.B.; Johns, T.G.; Murone, C.; Huang, H.J.; Cavenee, W.K.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. Monoclonal Antibody 806 Inhibits the Growth of Tumor Xenografts Expressing Either the De2-7 or Amplified Epidermal Growth Factor Receptor (EGFR) but Not Wild-Type EGFR. Cancer Res. 2001, 61, 5355–5361. [Google Scholar]
- Greenall, S.A.; McKenzie, M.; Seminova, E.; Dolezal, O.; Pearce, L.; Bentley, J.; Kuchibhotla, M.; Chen, S.C.; McDonald, K.L.; Kornblum, H.I.; et al. Most Clinical Anti-EGFR Antibodies Do Not Neutralize Both WtEGFR and EGFRvIII Activation in Glioma. Neuro Oncol. 2019, 21, 1016–1027. [Google Scholar] [CrossRef]
- van der Velden, D.L.; Hoes, L.R.; van der Wijngaart, H.; van Berge Henegouwen, J.M.; van Werkhoven, E.; Roepman, P.; Schilsky, R.L.; de Leng, W.W.J.; Huitema, A.D.R.; Nuijen, B.; et al. The Drug Rediscovery Protocol Facilitates the Expanded Use of Existing Anticancer Drugs. Nature 2019, 574, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Medikonda, R.; Pant, A.; Lim, M. Immunotherapy as a New Therapeutic Approach for Brain and Spinal Cord Tumors. Adv. Exp. Med. Biol. 2023, 1394, 73–84. [Google Scholar]
- Agosti, E.; Zeppieri, M.; De Maria, L.; Tedeschi, C.; Fontanella, M.M.; Panciani, P.P.; Ius, T. Glioblastoma Immunotherapy: A Systematic Review of the Present Strategies and Prospects for Advancements. Int. J. Mol. Sci. 2023, 24, 15037. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR Amplification, Combined Chromosome 7 Gain and Chromosome 10 Loss, and TERT Promoter Mutation in Brain Tumors and Their Potential for the Reclassification of IDHwt Astrocytoma to Glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Körber, V.; Yang, J.; Barah, P.; Wu, Y.; Stichel, D.; Gu, Z.; Fletcher, M.N.C.; Jones, D.; Hentschel, B.; Lamszus, K.; et al. Evolutionary Trajectories of IDHWT Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years Ahead of Initial Diagnosis. Cancer Cell 2019, 35, 692–704.e12. [Google Scholar] [CrossRef]
- Koshiyama, D.B.; Trevisan, P.; Graziadio, C.; Rosa, R.F.M.; Cunegatto, B.; Scholl, J.; Provenzi, V.O.; de Sá, A.P.; Soares, F.P.; Velho, M.C.; et al. Frequency and Clinical Significance of Chromosome 7 and 10 Aneuploidies, Amplification of the EGFR Gene, Deletion of PTEN and TP53 Genes, and 1p/19q Deficiency in a Sample of Adult Patients Diagnosed with Glioblastoma from Southern Brazil. J. Neurooncol. 2017, 135, 465–472. [Google Scholar] [CrossRef]
- Yang, H.; Han, F.; Hu, R.; Liu, J.; Sui, J.; Xiang, X.; Wang, F.; Chu, L.; Song, S. PTEN Gene Mutations Correlate to Poor Prognosis in Glioma Patients: A Meta-Analysis. OncoTargets Ther. 2016, 2016, 3485–3492. [Google Scholar] [CrossRef]
- Srividya, M.R.; Thota, B.; Shailaja, B.C.; Arivazhagan, A.; Thennarasu, K.; Chandramouli, B.A.; Hegde, A.S.; Santosh, V. Homozygous 10q23/PTEN Deletion and Its Impact on Outcome in Glioblastoma: A Prospective Translational Study on a Uniformly Treated Cohort of Adult Patients. Neuropathology 2011, 31, 376–383. [Google Scholar] [CrossRef]
- Thuy, M.N.T.; Kam, J.K.T.; Lee, G.C.Y.; Tao, P.L.; Ling, D.Q.; Cheng, M.; Goh, S.K.; Papachristos, A.J.; Shukla, L.; Wall, K.-L.; et al. A Novel Literature-Based Approach to Identify Genetic and Molecular Predictors of Survival in Glioblastoma Multiforme: Analysis of 14,678 Patients Using Systematic Review and Meta-Analytical Tools. J. Clin. Neurosci. 2015, 22, 785–799. [Google Scholar] [CrossRef]
- Al-Ghabkari, A.; Huang, B.; Park, M. Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions. Cells 2024, 13, 218. [Google Scholar] [CrossRef]
- Nair, N.U.; Schäffer, A.A.; Gertz, E.M.; Cheng, K.; Zerbib, J.; Das Sahu, A.; Leor, G.; Shulman, E.D.; Aldape, K.D.; Ben-David, U.; et al. Chromosome 7 to the Rescue: Overcoming Chromosome 10 Loss in Gliomas. bioRxiv 2024. [Google Scholar] [CrossRef]
- Maroto, P.; Porta, C.; Capdevila, J.; Apolo, A.B.; Viteri, S.; Rodriguez-Antona, C.; Martin, L.; Castellano, D. Cabozantinib for the Treatment of Solid Tumors: A Systematic Review. Ther. Adv. Med. Oncol. 2022, 14, 175883592211071. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Holland, J.; et al. Phase II Study of Cabozantinib in Patients with Progressive Glioblastoma: Subset Analysis of Patients Naive to Antiangiogenic Therapy. Neuro Oncol. 2018, 20, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.; Su, W.; Schuler, M.; Nam, D.; Lim, W.T.; Bauer, T.M.; Azaro, A.; Poon, R.T.P.; Hong, D.; Lin, C.; et al. Phase 1 Study of Capmatinib in MET-positive Solid Tumor Patients: Dose Escalation and Expansion of Selected Cohorts. Cancer Sci. 2020, 111, 536–547. [Google Scholar] [CrossRef]
- Martínez-García, M.; Velasco, G.; Pineda, E.; Gil-Gil, M.; Alameda, F.; Capellades, J.; Martín-Soberón, M.C.; López-Valero, I.; Tovar Ambel, E.; Foro, P.; et al. Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers 2022, 14, 2393. [Google Scholar] [CrossRef]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine–DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; Ponte de Albuquerque, C.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 504–518.e7. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Guo, L.; Song, Y.; Wang, L.; Yu, K.; Huang, Q.; Zhong, Y. Combinatorial Therapy with Adenoviral-Mediated PTEN and a PI3K Inhibitor Suppresses Malignant Glioma Cell Growth in Vitro and in Vivo by Regulating the PI3K/AKT Signaling Pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 1477–1487. [Google Scholar] [CrossRef]
- Tiu, C.; Yau, W.H.; Welsh, L.C.; Jones, T.L.; Zachariou, A.; Prout, T.; Parmar, M.; Turner, A.J.; Daly, R.W.; Yap, C.; et al. Abstract CT093: Preliminary Evidence of Antitumor Activity of Ipatasertib (Ipat) and Atezolizumab (A) in Glioblastoma (GBM) Patients (Pts) with PTEN Loss in the Phase 1 Ice-CAP Trial (NCT03673787). Cancer Res. 2023, 83, CT093. [Google Scholar] [CrossRef]
- Tiu, C.; Welsh, L.; Jones, T.; Zachariou, A.; Prout, T.; Turner, A.; Daly, R.; Tunariu, N.; Riisnaes, R.; Gurel, B.; et al. Preliminary Evidence of Antitumour Activity of Ipatasertib (Ipat) and Atezolizumab (ATZ) in Glioblastoma Patients (Pts) with PTEN Loss from the Phase 1 Ice-CAP Trial (NCT03673787). Neuro Oncol. 2021, 23, iv10. [Google Scholar] [CrossRef]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.; Clement, P.M.; Campone, M.; Gil-Gil, M.J.; DeGroot, J.; Chinot, O.; Idbaih, A.; Gan, H.; Raizer, J.; Wen, P.Y.; et al. Buparlisib plus Carboplatin or Lomustine in Patients with Recurrent Glioblastoma: A Phase Ib/II, Open-Label, Multicentre, Randomised Study. ESMO Open 2020, 5, e000672. [Google Scholar] [CrossRef]
- van den Bent, M.; Azaro, A.; De Vos, F.; Sepulveda, J.; Yung, W.K.A.; Wen, P.Y.; Lassman, A.B.; Joerger, M.; Tabatabai, G.; Rodon, J.; et al. A Phase Ib/II, Open-Label, Multicenter Study of INC280 (Capmatinib) Alone and in Combination with Buparlisib (BKM120) in Adult Patients with Recurrent Glioblastoma. J. Neurooncol. 2020, 146, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K.; et al. Phase I/II Study of Bevacizumab with BKM120, an Oral PI3K Inhibitor, in Patients with Refractory Solid Tumors (Phase I) and Relapsed/Refractory Glioblastoma (Phase II). J. Neurooncol. 2019, 144, 303–311. [Google Scholar] [CrossRef]
- Wen, P.Y.; Rodon, J.A.; Mason, W.; Beck, J.T.; DeGroot, J.; Donnet, V.; Mills, D.; El-Hashimy, M.; Rosenthal, M. Phase I, Open-Label, Multicentre Study of Buparlisib in Combination with Temozolomide or with Concomitant Radiation Therapy and Temozolomide in Patients with Newly Diagnosed Glioblastoma. ESMO Open 2020, 5, e000673. [Google Scholar] [CrossRef]
- Noch, E.K.; Palma, L.N.; Yim, I.; Bullen, N.; Qiu, Y.; Ravichandran, H.; Kim, J.; Rendeiro, A.; Davis, M.B.; Elemento, O.; et al. Insulin Feedback Is a Targetable Resistance Mechanism of PI3K Inhibition in Glioblastoma. Neuro Oncol. 2023, 25, 2165–2176. [Google Scholar] [CrossRef]
- Guo, T.; Wu, C.; Zhang, J.; Yu, J.; Li, G.; Jiang, H.; Zhang, X.; Yu, R.; Liu, X. Dual Blockade of EGFR and PI3K Signaling Pathways Offers a Therapeutic Strategy for Glioblastoma. Cell Commun. Signal. 2023, 21, 363. [Google Scholar] [CrossRef]
- Bao, L.; Li, X.; Lin, Z. PTEN Overexpression Promotes Glioblastoma Death through Triggering Mitochondrial Division and Inactivating the Akt Pathway. J. Recept. Signal Transduct. 2019, 39, 215–225. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Mizoguchi, M.; Hata, N.; Murata, H.; Hatae, R.; Amano, T.; Nakamizo, A.; Sasaki, T. Complex DNA Repair Pathways as Possible Therapeutic Targets to Overcome Temozolomide Resistance in Glioblastoma. Front. Oncol. 2012, 2, 186. [Google Scholar] [CrossRef]
- Storey, K.; Leder, K.; Hawkins-Daarud, A.; Swanson, K.; Ahmed, A.U.; Rockne, R.C.; Foo, J. Glioblastoma Recurrence and the Role of O6–Methylguanine–DNA Methyltransferase Promoter Methylation. JCO Clin. Cancer Inform. 2019, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Håvik, A.B.; Brandal, P.; Honne, H.; Dahlback, H.-S.S.; Scheie, D.; Hektoen, M.; Meling, T.R.; Helseth, E.; Heim, S.; Lothe, R.A.; et al. MGMT Promoter Methylation in Gliomas-Assessment by Pyrosequencing and Quantitative Methylation-Specific PCR. J. Transl. Med. 2012, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Salvati, M.; Arcella, A.; Buttarelli, F.; D’Elia, A.; Lanzetta, G.; Esposito, V.; Scarpino, S.; Maurizi Enrici, R.; Giangaspero, F. Correlation between O6-Methylguanine-DNA Methyltransferase and Survival in Elderly Patients with Glioblastoma Treated with Radiotherapy plus Concomitant and Adjuvant Temozolomide. J. Neurooncol. 2011, 102, 311–316. [Google Scholar] [CrossRef]
- Zapanta Rinonos, S.; Li, T.; Pianka, S.T.; Prins, T.J.; Eldred, B.S.C.; Kevan, B.M.; Liau, L.M.; Nghiemphu, P.L.; Cloughesy, T.F.; Lai, A. DCas9/CRISPR-Based Methylation of O-6-Methylguanine-DNA Methyltransferase Enhances Chemosensitivity to Temozolomide in Malignant Glioma. J. Neurooncol. 2024, 166, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Abdallah, M.O.E.; Breuer, P.; Stahl, F.; Bakhit, Y.; Potthoff, A.-L.; Pregler, B.E.F.; Schneider, M.; Waha, A.; Wüllner, U.; et al. Downregulation of MGMT Expression by Targeted Editing of DNA Methylation Enhances Temozolomide Sensitivity in Glioblastoma. Neoplasia 2023, 44, 100929. [Google Scholar] [CrossRef]
- Frenel, J.-S.; Cartron, P.-F.; Gourmelon, C.; Campion, L.; Aumont, M.; Augereau, P.; Ducray, F.; Loussouarn, D.; Lallier, L.; Robert, M.; et al. 370MO FOLAGLI: A Phase I Study of Folinic Acid Combined with Temozolomide and Radiotherapy to Modulate MGMT Gene Promoter Methylation in Newly Diagnosed MGMT Non-Methytated Glioblastoma. Ann. Oncol. 2020, 31, S400. [Google Scholar] [CrossRef]
- Salas, L.A.; Stewart, T.G.; Mobley, B.C.; Peng, C.; Liu, J.; Loganathan, S.N.; Wang, J.; Ma, Y.; Berger, M.S.; Absher, D.; et al. Phase I Study of High-Dose L-Methylfolate in Combination with Temozolomide and Bevacizumab in Recurrent IDH Wild-Type High-Grade Glioma. Cancer Res. Commun. 2022, 2, 1–9. [Google Scholar] [CrossRef]
- Kirstein, A.; Schilling, D.; Combs, S.E.; Schmid, T.E. Lomeguatrib Increases the Radiosensitivity of MGMT Unmethylated Human Glioblastoma Multiforme Cell Lines. Int. J. Mol. Sci. 2021, 22, 6781. [Google Scholar] [CrossRef]
- Wu, Q.; Berglund, A.E.; Macaulay, R.J.; Etame, A.B. Epigenetic Activation of TUSC3 Sensitizes Glioblastoma to Temozolomide Independent of MGMT Promoter Methylation Status. Int. J. Mol. Sci. 2023, 24, 15179. [Google Scholar] [CrossRef]
- Rahman, M.A.; Gras Navarro, A.; Brekke, J.; Engelsen, A.; Bindesbøll, C.; Sarowar, S.; Bahador, M.; Bifulco, E.; Goplen, D.; Waha, A.; et al. Bortezomib Administered Prior to Temozolomide Depletes MGMT, Chemosensitizes Glioblastoma with Unmethylated MGMT Promoter and Prolongs Animal Survival. Br. J. Cancer 2019, 121, 545–555. [Google Scholar] [CrossRef]
- Roth, P.; Gorlia, T.; Reijneveld, J.C.; de Vos, F.; Idbaih, A.; Frenel, J.-S.; Le Rhun, E.; Sepulveda, J.M.; Perry, J.; Masucci, G.L.; et al. Marizomib for Patients with Newly Diagnosed Glioblastoma: A Randomized Phase 3 Trial. Neuro Oncol. 2024, 26, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Sun, X.; Yang, Q.; Zheng, M.; Shimoni, O.; Ruan, W.; Wang, Y.; Zhang, D.; Yin, J.; Huang, X.; et al. Blood-Brain Barrier–Penetrating Single CRISPR-Cas9 Nanocapsules for Effective and Safe Glioblastoma Gene Therapy. Sci. Adv. 2022, 8, eabm8011. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.S.; Kramp, T.R.; Palanichamy, K.; Tofilon, P.J.; Camphausen, K. MGMT Inhibition Regulates Radioresponse in GBM, GSC, and Melanoma. Sci. Rep. 2024, 14, 12363. [Google Scholar] [CrossRef]
- Rahman, M.A.; Brekke, J.; Arnesen, V.; Hannisdal, M.H.; Navarro, A.G.; Waha, A.; Herfindal, L.; Rygh, C.B.; Bratland, E.; Brandal, P.; et al. Sequential Bortezomib and Temozolomide Treatment Promotes Immunological Responses in Glioblastoma Patients with Positive Clinical Outcomes: A Phase 1B Study. Immun. Inflamm. Dis. 2020, 8, 342–359. [Google Scholar] [CrossRef]
- Roth, P.; Mason, W.P.; Richardson, P.G.; Weller, M. Proteasome Inhibition for the Treatment of Glioblastoma. Expert Opin. Investig. Drugs 2020, 29, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib Alone or in Combination with Bevacizumab in Patients with Recurrent Glioblastoma: Phase I/II Clinical Trial Data. Neurooncol. Adv. 2021, 3, vdab142. [Google Scholar] [CrossRef]
- Mason, W.P.; Kesari, S.; Stupp, R.; Aregawi, D.G.; Piccioni, D.E.; Roth, P.; Desjardins, A.; Reich, S.D.; Casadebaig, M.-L.; Elias, I.; et al. Full Enrollment Results from an Extended Phase I, Multicenter, Open Label Study of Marizomib (MRZ) with Temozolomide (TMZ) and Radiotherapy (RT) in Newly Diagnosed Glioblastoma (GBM). J. Clin. Oncol. 2019, 37, 2021. [Google Scholar] [CrossRef]
- Kesari, S.; Juarez, T.; Carrillo, J.; Truong, J.; Nguyen, M.; Heng, A.; Gill, J.; Nguyen, H.; Nomura, N.; Grigorian, B.; et al. RBTT-01. A phase 2 trial with ABI-009 (nab-sirolimus) as single-agent and combinations in recurrent high-grade glioma (rhgg) and in newly diagnosed glioblastoma (Ndgbm). Neuro Oncol. 2019, 21, vi218–vi219. [Google Scholar] [CrossRef]
- Dewdney, B.; Jenkins, M.R.; Best, S.A.; Freytag, S.; Prasad, K.; Holst, J.; Endersby, R.; Johns, T.G. From Signalling Pathways to Targeted Therapies: Unravelling Glioblastoma’s Secrets and Harnessing Two Decades of Progress. Signal Transduct. Target. Ther. 2023, 8, 400. [Google Scholar] [CrossRef]
- Fusco, M.J.; Piña, Y.; Macaulay, R.J.; Sahebjam, S.; Forsyth, P.A.; Peguero, E.; Walko, C.M. Durable Progression-Free Survival With the Use of BRAF and MEK Inhibitors in Four Cases With BRAF V600E-Mutated Gliomas. Cancer Control 2021, 28, 107327482110400. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, G.; Liu, Y.; Feng, L.; Wang, M.; Liu, J.; Chen, Y.; Ouyang, L. Development of Small-Molecule Tropomyosin Receptor Kinase (TRK) Inhibitors for NTRK Fusion Cancers. Acta Pharm. Sin. B 2021, 11, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Long, P.; Wang, Y.; Ma, W. NTRK Fusions and TRK Inhibitors: Potential Targeted Therapies for Adult Glioblastoma. Front. Oncol. 2020, 10, 593578. [Google Scholar] [CrossRef] [PubMed]
- König, D.; Hench, J.; Frank, S.; Dima, L.; Bratic Hench, I.; Läubli, H. Larotrectinib Response in NTRK3 Fusion-Driven Diffuse High-Grade Glioma. Pharmacology 2022, 107, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.-F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M.; et al. Molecular and Clinicopathologic Features of Gliomas Harboring NTRK Fusions. Acta Neuropathol. Commun. 2020, 8, 107. [Google Scholar] [CrossRef]
- Kim, E.E.; Park, C.-K.; Kim, S.-K.; Phi, J.H.; Paek, S.H.; Choi, J.Y.; Kang, H.J.; Lee, J.H.; Won, J.K.; Yun, H.; et al. NTRK-Fused Central Nervous System Tumours: Clinicopathological and Genetic Insights and Response to TRK Inhibitors. Acta Neuropathol. Commun. 2024, 12, 118. [Google Scholar] [CrossRef]
- Grogan, P.T.; Deming, D.A.; Helgager, J.; Ruszkiewicz, T.; Baskaya, M.K.; Howard, S.P.; Robins, H.I. Entrectinib Demonstrates Prolonged Efficacy in an Adult Case of Radiation-Refractory NTRK Fusion Glioblastoma. Neurooncol. Adv. 2022, 4, vdac046. [Google Scholar] [CrossRef]
- Pattwell, S.S.; Arora, S.; Cimino, P.J.; Ozawa, T.; Szulzewsky, F.; Hoellerbauer, P.; Bonifert, T.; Hoffstrom, B.G.; Boiani, N.E.; Bolouri, H.; et al. A Kinase-Deficient NTRK2 Splice Variant Predominates in Glioma and Amplifies Several Oncogenic Signaling Pathways. Nat. Commun. 2020, 11, 2977. [Google Scholar] [CrossRef]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical Trial Links Oncolytic Immunoactivation to Survival in Glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef]
- Scott, R. OncLive. Available online: https://www.onclive.com/view/fda-grants-fast-track-designation-to-can-3110-for-recurrent-high-grade-glioma (accessed on 11 September 2024).
- Farooq, M.; Scalia, G.; Umana, G.; Parekh, U.; Naeem, F.; Abid, S.; Khan, M.; Zahra, S.; Sarkar, H.; Chaurasia, B. A Systematic Review of Nanomedicine in Glioblastoma Treatment: Clinical Efficacy, Safety, and Future Directions. Brain Sci. 2023, 13, 1727. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| IDH-Wildtype and H3-Wildtype Positive Gliomas Plus Either: | |
|---|---|
| Histopathological features | Molecular features |
| -Microvascular proliferation -Necrosis | -TERT promoter mutation -EGFR amplification -Combined gain of chromosome 7 and loss of chromosome 10 |
| Therapy | Mechanism of Action | Results | Ref. |
|---|---|---|---|
| Imetelstat | Telomerase inhibitor | Phase II clinical trial needed to be terminated due to two patients dying of intratumoral hemorrhage secondary to thrombocytopenia. | [103] |
| INO-5401 + INO-9012 + cemiplimab | Vaccine containing a DNA plasmid encoding human TERT, WT-1 and PSMA + a DNA plasmid encoding IL-12 + PD-1 inhibitor | Phase I/II study in newly diagnosed glioblastoma patients administered a DNA vaccine concomitantly with standard chemoradiation, demonstrating safety and a robust immune response and enhanced survival. Median OS in cohorts A (unmethylated MGMT) and B (methylated MGMT) was 17.9 and 32.5 months, respectively. | [105] |
| UCPVax | CD4 helper peptides derived from TERT | In a phase IIa trial in glioblastoma, IDH-wildtype, MGMT-unmethylated patients, the therapy was shown to be highly immunogenic and safe, improving OS. | [106] |
| 6-thio-2-deoxyguanosine (6-thio-dG) | Nucleoside analogue that targets newly synthesized telomeres | This therapy caused telomere stress and DNA damage, whichactivated innate and adaptative immune-dependent responses. Not tested in glioblastoma cells. | [108] |
| BIBR 1532 + ZIF-8 | Telomerase inhibitor in combination with zeolitic imidazole framework-8, a drug delivery vehicle | Reduced TERT mRNA expression with cell cycle arrest and cellular senescence with improved transportation and delivery of BIBR1532 to the nucleus. Not tested in glioblastoma cells. | [109] |
| BRACO-19 | Synthetic G4 ligand | Decreased viability of human glioblastoma cells while sparing normal surrounding cells in a highly specific manner. | [110] |
| Y2H2-6M(4)-oxazole telomestatin derivative (6OTD) | Synthetic G4 ligand | Inhibition of the growth of GSCs and DNA damage, preferentially in telomeres of GSCs. | [111] |
| RHPS4 + ionizing radiation | Synthetic G4 ligand + ionizing radiation | Inhibition of growth in both differentiated glioblastoma and GSCs with synergistic radiosensitization in the differentiated cells but not in GSCs, possibly because effects on GSC growth inhibition are not mediated by telomeric dysfunction. | [112] |
| Adenoviruses expressing sgRNA-guided CjABE | sgRNA-guided and catalytically impaired Campylobacter jejuni CRISPR-associated protein 9-fused adenine base editor (CjABE) in an adeno-associated virus vector | Correction of TERTp mutation, reducing TERT transcription and TERT protein expression in human glioblastoma cell lines, which ultimately inhibited growth | [113] |
| shRNA + temozolomide | shRNAs targeting GABPB1L + alkylating agent chemotherapy | Reduction in the respective mRNA and protein levels, leading to reduced TERT mRNA and telomerase activity exclusively in TERTp-mutant glioblastoma cells. Chemotherapy sensitization resulted in increasing survival in a synergistic manner. | [114] |
| Therapy | Mechanism of Action | Results | Ref. |
|---|---|---|---|
| Erlotinib + Bevacizumab | First-generation EGFR inhibitor + monoclonal antibody against VEGF | Phase II clinical trial showed no improvement in OS in unmethylated MGMT glioblastoma patients. | [125] |
| Osimertinib + Bevacizumab | Third-generation EGFR inhibitor + monoclonal antibody against VEGF | Retrospective cohort study showed that the osimertinib/bevacizumab combination was marginally effective in most patients with simultaneous EGFR amplification plus EGFRvIII mutation, and a meaningful benefit was seen in a patient subgroup. | [126] |
| TAS2940 | Irreversible pan-ErbB inhibitor | Improved brain penetration in in vitro and in vivo mouse xenograft models, inhibition of tumor growth against cells with HER2 amplification and EGFRvIII mutation, improving survival in mice. Ongoing clinical trials: NCT04982926, https://clinicaltrials.gov/study/NCT04982926, accessed on 27 October 2024. | [127] |
| Depatuxizumab mafodotin | Monoclonal antibody against EGFRvIII | Phase III clinical trial showed increased PFS but no improvement in OS versus placebo in newly diagnosed glioblastoma patients with EGFR. Corneal epitheliopathy occurred as an adverse effect in 94% of treated patients. | [128] |
| GC1118 | Monoclonal antibody against EGFR | A multicenter, open-label, single-arm phase II trial demonstrated good tolerance and improved immune signatures in tumors but did not show benefit in terms of PFS or OS in patients with recurrent glioblastoma and EGFR amplification. | [129] |
| Panitumumab | Monoclonal antibody against EGFR | A cohort under the DRUP trial demonstrated safety of use but limited clinical benefit in only 21% of patients. Ongoing clinical trials: NCT03510208, https://clinicaltrials.gov/study/NCT03510208, accessed on 27 October 2024. | [130] |
| Nimotuzumab | Monoclonal antibody against extracellular region of EGFR | A phase II, single-arm, multicenter clinical trial showed increased OS in patients with newly diagnosed EGFR-expressed glioblastoma when added to standard chemoradiation. Importantly, MGMT status showed no correlation with these results. | [131] |
| Nimotuzumab + Melatonin | Monoclonal antibody against extracellular region of EGFR + hormone-blocking ATP binding to the kinase domain of EGFR | Synergistic increase in cytotoxicity and apoptosis of cancer cells in vitro and in xenograft mouse glioblastoma models. | [132] |
| Rindopepimut + Bevacizumab | Peptide-based vaccine targeting EGFRvIII + monoclonal antibody against VEGF | A double-blind, randomized phase II trial in recurrent EGFRvIII-expressing glioblastoma patients showed that concurrent administration of rindopepimut with Bevacizumab increased PFS at 6 months and OS compared to bevacizumab alone. | [133] |
| R-613 | Oncolytic herpes simplex virus (oHSV) retargeted to EGFRvIII | The therapy delayed the development of tumor masses and increased OS in orthotopically transplanted mice when given as an early treatment. | [134] |
| OV-Cmab-CCL5 | oHSV containing an IgG1 form of cetuximab and chemokine C-C motif ligand 5, a chemotactic chemokine | The therapy upregulated immune cell trafficking in the tumor microenvironment, with enhanced migration and activation of natural killer cells, macrophages and T cells; inhibition of tumor EGFR signaling; a subsequent reduction in tumor size; and increased survival in mouse models. | [135] |
| OV-IL15C | oHSV-expressing IL-15/IL-15Rα + CAR NK cells | Synergistic inhibition of tumor growth and increased survival in mouse models. | [136] |
| R-115 | Fully virulent oHSV retargeted to human ErbB-2 | A single injection showed significant improvement in the OS of mice and resistance to recurrence, with an unprecedented complete eradication of tumor in 30% of subjects. | [137] |
| CAR T-cell therapy | Chimeric antigen receptor T cells against EGFRvIII | A phase I trial demonstrated patient safety but no clinically significant change in PFS. | [138] |
| CAR T cells + Pembrolizumab | CAR T cells against EGFRvIII + monoclonal antibody against PD1 | A phase I trial showed upregulation of the tumor microenvironment but no improvement in terms of PFS or OS. | [139] |
| Intrathecal CAR T cells | CAR T cells against EGFR and IL13Rα2 | A phase I trial demonstrated safety, with only early-onset neurotoxicity and moderate efficacy and reductions in enhancement and tumor size detected by MRI in all patients. | [140] |
| Therapy | Mechanism of Action | Results | Ref. |
|---|---|---|---|
| Cabozantinib | TKI against VEGFR2 and MET | A multicenter, open-label, single-agent phase II trial enrolled recurrent glioblastoma patients, with a PFS at 6 months of 27.8% and an OS of 10.4 months, failing to meet the statistical target for success. Ongoing trials: NCT05039281 (https://clinicaltrials.gov/study/NCT05039281, accessed on 27 October 2024) (+ atezolizumab) | [156] |
| Capmatinib (INC280) | MET inhibitor | A multicenter, open-label, non-randomized, two-part study comprising a dose-escalation and expansion phase I trial including patients with various MET-positive solid tumors, including glioblastoma, showed that the drug was well tolerated and exhibited antitumor activity. Ongoing trials: NCT02386826 (https://clinicaltrials.gov/study/NCT02386826, accessed on 27 October 2024) (+ bevacizumab) | [157] |
| Crizotinib | ALK, ROS1 and c-MET inhibitor | A multicenter, open-label, single-arm phase Ib trial demonstrated a median PFS of 10.7 months and OS of 22.6 months, showing a possible synergistic effect of crizotinib when was added to standard chemoradiation in newly diagnosed glioblastoma patients | [158] |
| Onartuzumab + bevacizumab | Monoclonal antibody against MET + monoclonal antibody against VEGF | A multicenter, randomized, double-blind, placebo-controlled phase II trial showed no clinical benefit of adding onartuzumab to bevacizumab when compared to placebo in recurrent glioblastoma patients. | [159] |
| Therapy | Mechanism of Action | Results | Ref. |
|---|---|---|---|
| Ad-PTEN + LY294002 | Oncolytic adenovirus retargeted to upregulate PTEN + PI3K inhibitor | The combination of Ad-PTEN and LY294002 inhibited the PI3K/AKT pathway more effectively than either therapy alone, suppressing tumor growth in vitro and in in vivo glioblastoma xenograft mouse models. | [161] |
| Ipatasertib + atezolizumab | Akt inhibitor + monoclonal antibody against PD-L1 | A single-center, open-label phase I/II trial showed the combination to be well tolerated, with clinical benefit in 32% of all patients and in 28.6% of patients with PTEN loss, making it a promising predictive biomarker for response to the combination. | [162,163] |
| Buparlisib (BK120) | PI3K inhibitor | A multicenter, open-label, multi-arm phase II trial in patients with recurrent glioblastoma showed significant brain penetration but incomplete blockade of the PI3K pathway and minimal efficacy. | [164] |
| Buparlisib + either carboplatin or lomustine | PI3K inhibitor + either platinum-based chemo or alkylating agent | A multicenter, open-label, randomized phase Ib/II trial in patients with recurrent glioblastoma showed insufficient antitumor activity compared with data on the use of single-agent carboplatin or lomustine. | [165] |
| Capmatinib + buparlisib | MET inhibitor + PI3K inhibitor | A multicenter, open-label phase Ib/II trial in patients with recurrent glioblastoma showed no clear activity using the combination. | [166] |
| Buparlisib + bevacizumab | PI3K inhibitor + monoclonal antibody against VEGF | A multicenter, phase I/II study in patients with recurrent glioblastoma showed poor tolerability of the combination, with 57% of patients experiencing at least one serious treatment-related toxicity and similar efficacy to that of single-agent bevacizumab. | [167] |
| Buparlisib + standard chemoradiation | PI3K inhibitor + alkylating agent + radiation therapy | A two-stage, multicenter, open-label phase I trial in newly diagnosed glioblastoma patients was not able to determine the maximum tolerated dose due to dose-limiting toxicities. Subsequently, Novartis decided not to pursue the development of buparlisib in newly diagnosed glioblastoma. | [168] |
| Paxalisib + metformin | PI3K/mTOR inhibitor + biguanide | The therapy increased the efficacy of PI3K inhibitors when combined with metformin and a ketogenic diet to reduce insulin feedback and hyperglycemia in orthotopic glioblastoma mouse models. Ongoing trials: NCT05183204 (https://clinicaltrials.gov/study/NCT05183204, accessed on 27 October 2024) | [169] |
| AZD-9291 + GDC-0084 | EGFR/MEK/ERK pathway inhibitor + PI3K/AKT/mTOR inhibitor | Synergistic inhibition of proliferation and survival in in vitro and in vivo glioblastoma mice models was seen with this combination. | [170] |
| Therapy | Mechanism of Action | Results | Ref. |
|---|---|---|---|
| d3A + temozolomide | Chimeric fusion protein (CRISPR/dCas9 + DNA methyltransferase 3A catalytic domain) + alkylating agent | Targeted MGMT methylation in specific CpG clusters in the vicinity of the promoter, with consequent MGMT downregulation and enhanced chemosensitivity to temozolomide of glioblastoma cells in vitro. | [176,177] |
| Folic acid + temozolomide | Water-soluble vitamin + alkylating agent | A phase I trial demonstrated the safety of adding folic acid and restored methylation of the promoter in samples of circulating tumor DNA of 8 glioblastoma patients. | [178] |
| L-methylfolate + temozolomide + bevacizumab | Water-soluble vitamin + alkylating agent + monoclonal antibody against VEGF | A phase I trial showed the safety of L-methylfolate and DNA methylome reprogramming of recurrent IDH-wild-type glioblastomas. However, no significant impact on survival could be demonstrated due to a lack of statistical power. | [179] |
| Lomeguatrib | MGMT inhibitor | Inactivation of MGMT protein in glioblastoma cells in vitro, with increased radiosensitization at lower concentrations and radioprotective effects at higher doses. | [180] |
| 5-Azacitidine (5-Aza) + lomeguatrib | Demethylating agent + MGMT inhibitor | Epigenetic reactivation of TUSC3, which increased GSC sensitivity to temozolomide in MGMT-unmethylated orthotopic GSC mouse models. | [181] |
| Bortezomib + temozolomide | 26S proteasome inhibitor + alkylating agent | Depletion of MGMT mRNA and protein in glioblastoma cells in vitro and diminished proteasome activity in orthotopic mouse models, with increased survival. Ongoing trials: NCT03643549 (https://clinicaltrials.gov/study/NCT03643549, accessed on 27 October 2024) | [182] |
| Marizomib + standard treatment | Pan-proteasome inhibitor ± alkylating agent and radiotherapy | A multicenter, randomized, controlled, open-label phase III trial in patients with newly diagnosed glioblastoma showed increased toxicity and no statistically significant difference in OS or PFS between patients receiving marizomib in addition to standard treatment (RT/Temozolomide) compared with patients receiving standard treatment alone, either in the MGMT-methylated or unmethylated subgroup. | [183] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacome, M.A.; Wu, Q.; Piña, Y.; Etame, A.B. Evolution of Molecular Biomarkers and Precision Molecular Therapeutic Strategies in Glioblastoma. Cancers 2024, 16, 3635. https://doi.org/10.3390/cancers16213635
Jacome MA, Wu Q, Piña Y, Etame AB. Evolution of Molecular Biomarkers and Precision Molecular Therapeutic Strategies in Glioblastoma. Cancers. 2024; 16(21):3635. https://doi.org/10.3390/cancers16213635
Chicago/Turabian StyleJacome, Maria A., Qiong Wu, Yolanda Piña, and Arnold B. Etame. 2024. "Evolution of Molecular Biomarkers and Precision Molecular Therapeutic Strategies in Glioblastoma" Cancers 16, no. 21: 3635. https://doi.org/10.3390/cancers16213635
APA StyleJacome, M. A., Wu, Q., Piña, Y., & Etame, A. B. (2024). Evolution of Molecular Biomarkers and Precision Molecular Therapeutic Strategies in Glioblastoma. Cancers, 16(21), 3635. https://doi.org/10.3390/cancers16213635