
The Emerging Roles of the Stress Epigenetic Reader LEDGF/p75 in Cancer Biology and Therapy Resistance: Mechanisms and Targeting Opportunities

 ,
,
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. LEDGF/p75 Discovery
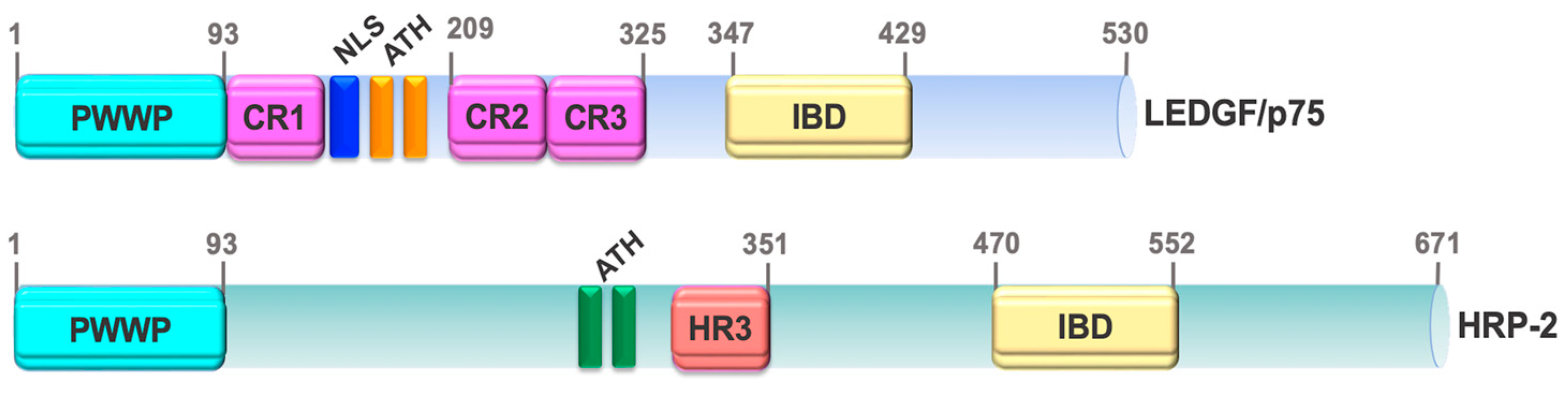
3. LEDGF/p75 Structural Domains and Functions
3.1. N-Terminal PWWP Domain
3.2. C-Terminal IBD Domain
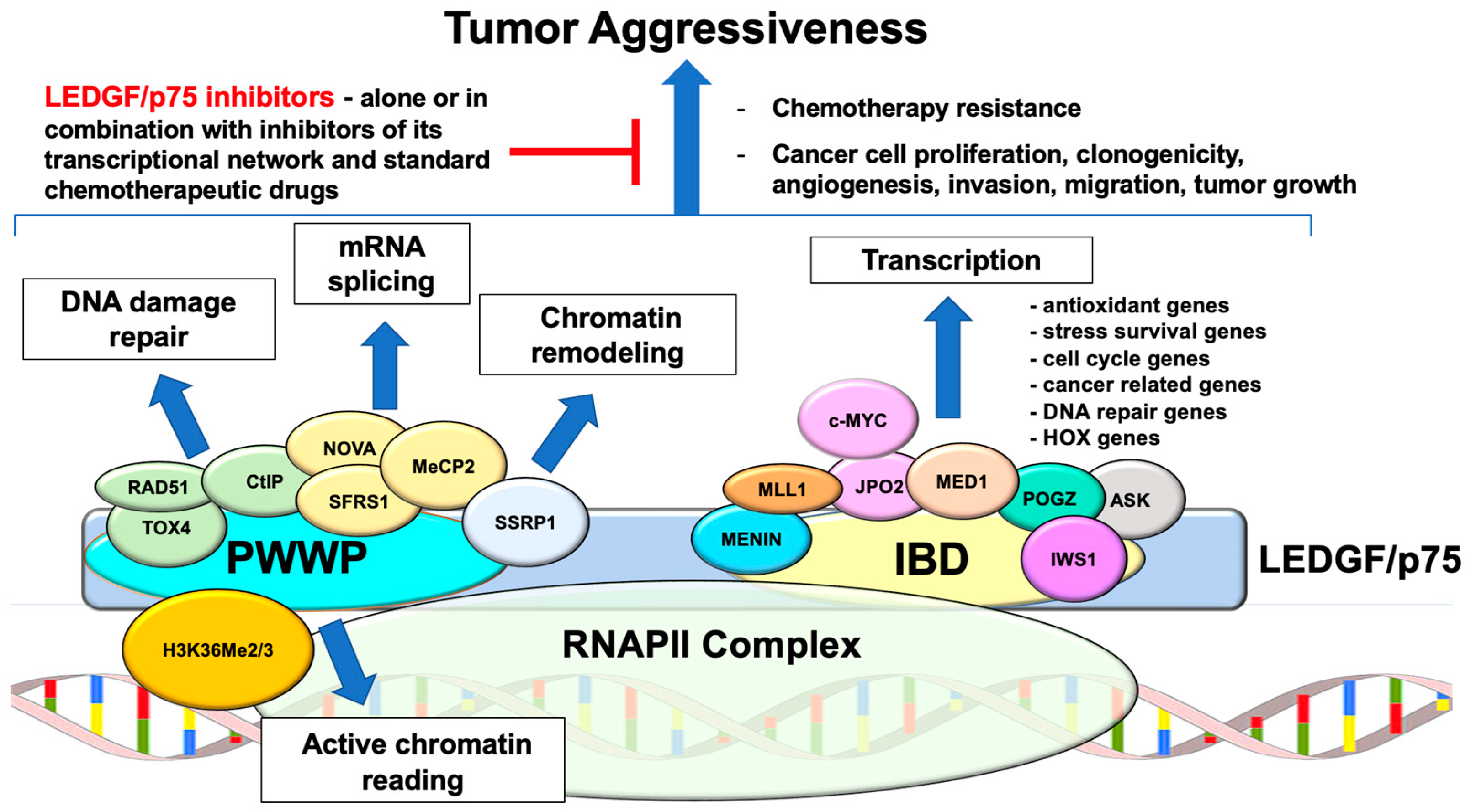
4. Stress Survival Functions of LEDGF/p75
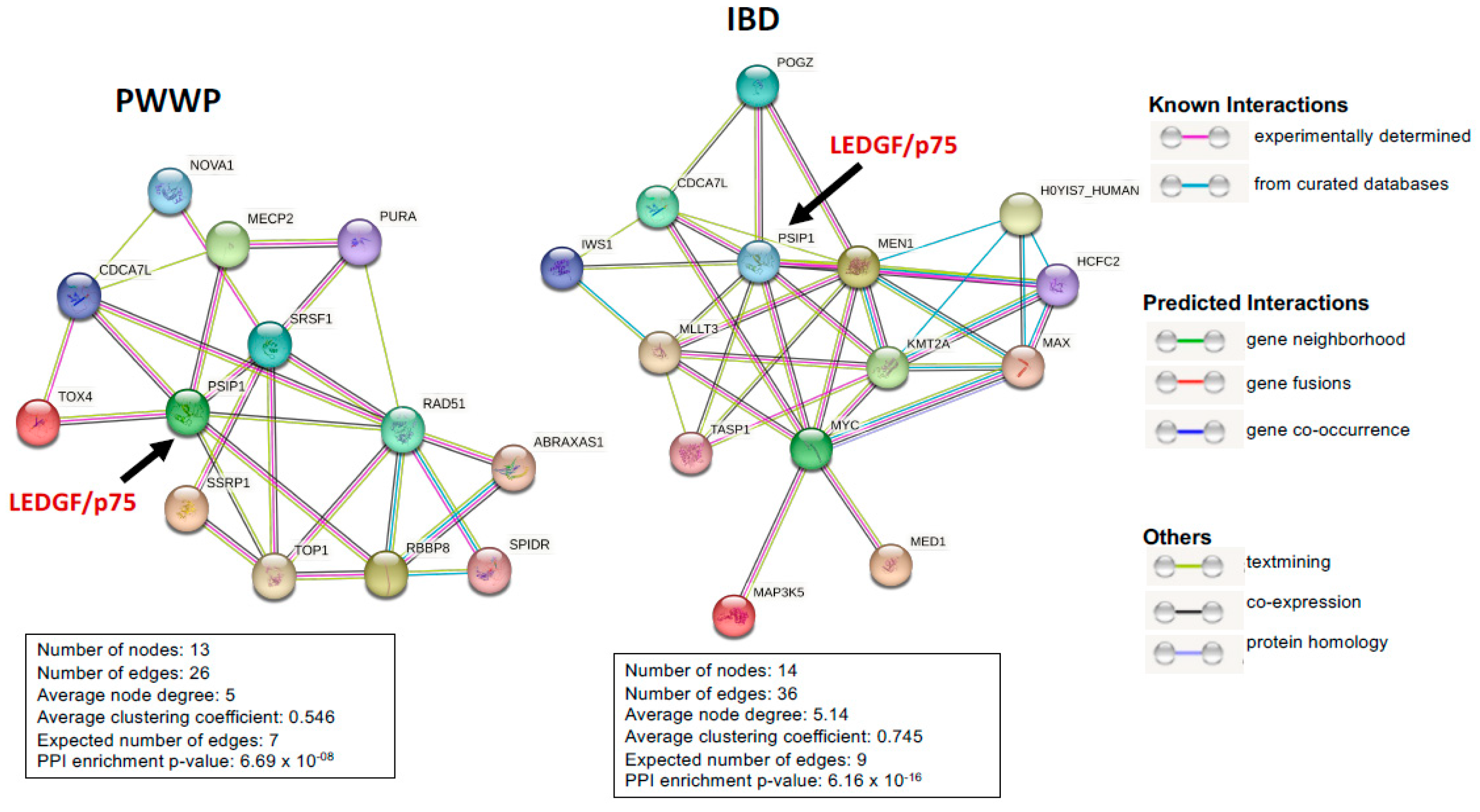
5. The LEDGF/p75 Interactome
5.1. PWWP Domain Interacting Partners
5.2. IBD Domain Interacting Partners
6. LEDGF/p75 Roles in Cancer Aggressiveness and Chemoresistance
6.1. Leukemia
6.2. Prostate Cancer
6.3. Breast Cancer
6.4. Ovarian Cancer
6.5. Cervical Cancer
6.6. Pediatric Brain Tumors
6.7. Esophageal Squamous Cell Carcinoma
6.8. Renal Carcinoma and Other Cancers
7. LEDGF/p75 Potential as Oncotherapeutic Target
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palacio, M.; Taatjes, D.J. Merging Established Mechanisms with New Insights: Condensates, Hubs, and the Regulation of RNA Polymerase II Transcription. J. Mol. Biol. 2022, 434, 167216. [Google Scholar] [CrossRef] [PubMed]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef]
- Vervoort, S.J.; Devlin, J.R.; Kwiatkowski, N.; Teng, M.; Gray, N.S.; Johnstone, R.W. Targeting transcription cycles in cancer. Nat. Rev. Cancer 2022, 22, 5–24. [Google Scholar] [CrossRef]
- Ahmad, K.; Brahma, S.; Henikoff, S. Epigenetic pioneering by SWI/SNF family remodelers. Mol. Cell 2024, 84, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Vijayakumar, U.; Zhang, Y.; Fullwood, M.J. Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer. Cancers 2022, 14, 2866. [Google Scholar] [CrossRef]
- Ramanand, S.G.; Chen, Y.; Yuan, J.; Daescu, K.; Lambros, M.B.; Houlahan, K.E.; Carreira, S.; Yuan, W.; Baek, G.; Sharp, A.; et al. The landscape of RNA polymerase II-associated chromatin interactions in prostate cancer. J. Clin. Investig. 2020, 3, 3987–4005. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Feng, Q. Androgen receptor signaling and spatial chromatin organization in castration-resistant prostate cancer. Front. Med. 2022, 9, 924087. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef]
- Sakellakis, M.; Flores, L.J. Is the glucocorticoid receptor a key player in prostate cancer?: A literature review. Medicine 2022, 101, e29716. [Google Scholar] [CrossRef]
- Chen, Y.; Lan, T. Molecular Origin, Expression Regulation, and Biological Function of Androgen Receptor Splicing Variant 7 in Prostate Cancer. Urol. Int. 2021, 105, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Luo, J. Regulation of androgen receptor variants in prostate cancer. Asian J. Urol. 2020, 7, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, J.; Liu, Y.; Shi, Z.; Xuan, Z.; Yang, K.; Xu, C.; Bai, Y.; Fu, M.; Xiao, Q.; et al. The Crucial Role of AR-V7 in Enzalutamide-Resistance of Castration-Resistant Prostate Cancer. Cancers 2022, 14, 4877. [Google Scholar] [CrossRef]
- Shim, M.; Kim, Y.; Park, Y.; Ahn, H. Taxane-based Chemotherapy Induced Androgen Receptor Splice Variant 7 in Patients with Castration-Resistant Prostate Cancer: A Tissue-based Analysis. Sci. Rep. 2019, 9, 16794. [Google Scholar] [CrossRef] [PubMed]
- Ehsani, M.; David, F.O.; Baniahmad, A. Androgen Receptor-Dependent Mechanisms Mediating Drug Resistance in Prostate Cancer. Cancers 2021, 13, 1534. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Ge, H.; Si, Y.; Roeder, R.G. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 1998, 17, 6723–6729. [Google Scholar] [CrossRef]
- Singh, D.P.; Ohguro, N.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Increased resistance to thermal and oxidative stresses. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1444–1451. [Google Scholar]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T., Jr.; Shinohara, T. Lens epithelium-derived growth factor: Effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar] [CrossRef]
- Chen, Z.Z.; Bowden, P.; Dufresne, J.; Miao, M.; Marshall, J.G. LEDGF is a new growth factor in fetal serum. Anal. Biochem. 2022, 655, 114845. [Google Scholar] [CrossRef]
- Blokken, J.; De Rijck, J.; Christ, F.; Debyser, Z. Protein–protein and protein–chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 2017, 24, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Tesina, P.; Cermakova, K.; Horejsi, M.; Prochazkova, K.; Fabry, M.; Sharma, S.; Christ, F.; Demeulemeester, J.; Debyser, Z.; Rijck, J.; et al. Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 2015, 6, 7968. [Google Scholar] [CrossRef]
- Nakamura, M.; Singh, D.P.; Kubo, E.; Chylack, L.T., Jr.; Shinohara, T. LEDGF: Survival of embryonic chick retinal photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1168–1175. [Google Scholar]
- Sharma, P.; Singh, D.P.; Fatma, L.T.; Chylack, L.T., Jr.; Shinohara, T. Activation of LEDGF gene by thermal- and oxidative-stresses. Biochem. Biophys. Res. Commun. 2000, 276, 1320–1324. [Google Scholar] [CrossRef]
- Fatma, N.; Singh, D.P.; Shinohara, T.; Chylack, L.T., Jr. Transcriptional regulation of the AOP2 gene, a thiol-specific antioxidant, by LEDGF to protect cells from oxidative stress. J. Biol. Chem. 2001, 276, 48899–48907. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Fatma, N.; Kimura, A.; Chylack, L.T., Jr.; Shinohara, T. LEDGF binds to heat shock and stress-related element to activate the expression of stress-related genes. Biochem. Biophys. Res. Commun. 2001, 283, 943–955. [Google Scholar] [CrossRef]
- Shinohara, T.; Singh, D.P.; Fatma, N. LEDGF, a survival factor, activates stress-related genes. Prog. Retin. Ocular Res. 2002, 21, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Lin, L.R.; Singh, D.P.; Shinohara, T.; Reddy, V.N. Lens epithelium-derived growth factor: Increased survival and decreased DNA breakage of human RPE cells induced by oxidative stress. Investig. Ophthalmol. Vis. Sci. 2002, 42, 2935–2941. [Google Scholar]
- Kubo, E.; Fatma, N.; Sharma, P.; Shinohara, T.; Chylack, L.T., Jr.; Akagi, Y.; Singh, D.P. Transactivation of involucrin, a marker of differentiation in keratinocytes, by lens epithelium-derived growth factor (LEDGF). J. Mol. Biol. 2002, 320, 1053–1063. [Google Scholar] [CrossRef]
- Inomata, Y.; Hirata, A.; Koga, T.; Kimura, A.; Singh, D.P.; Shinohara, T.; Tanihara, H. Lens epithelium-derived growth factor: Neuroprotection on rat retinal damage induced by N-methyl-D-aspartate. Brain Res. 2003, 991, 163–170. [Google Scholar] [CrossRef]
- Fatma, N.; Kubo, E.; Chylack, L.T., Jr.; Shinohara, T.; Akagi, Y.; Singh, D.P. LEDGF regulation of alcohol and aldehyde dehydrogenases in lens epithelial cells: Stimulation of retinoic acid production and protection from ethanol toxicity. Am. J. Physiol. Cell Physiol. 2004, 287, C508–C516. [Google Scholar] [CrossRef] [PubMed]
- Takamura, Y.; Fatma, N.; Kubo, E.; Singh, D.P. Regulation of heavy subunit chain of gamma-glutamylcysteine synthetase by tumor necrosis factor-alpha in lens epithelial cells: Role of LEDGF/p75. Am. J. Physiol. Cell Physiol. 2006, 290, C554–C566. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Piao, C.S.; Lim, C.M.; Lee, J.K. LEDGF binding to stress response element increases alphaB-crystallin expression in astrocytes with oxidative stress. Neurosci. Lett. 2008, 435, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Drame, A.; Muñoz, R.; Gijsbers, R.; Debyser, Z.; De Leon, M.; Casiano, C.A. Pathway specific gene expression profiling reveals oxidative stress genes potentially regulated by transcription co-activator LEDGF/p75 in prostate cancer cells. Prostate 2012, 72, 597–611. [Google Scholar] [CrossRef]
- Basu, A.; Cajigas-Du Ross, C.; Rios-Colon, L.; Mediavilla-Varela, M.; Daniels-Wells, T.R.; Leoh, L.S.; Rojas, H.; Banerjee, H.; Martinez, S.R.; Acevedo-Martinez, S.; et al. LEDGF/p75 overexpression attenuates oxidative stress-induced necrosis and upregulates ERp57/PDIA3/GRp58 in prostate cancer. PLoS ONE 2016, 11, e0146549. [Google Scholar] [CrossRef]
- Leitz, J.; Reuschenbach, M.; Lohrey, C.; Honegger, A.; Accardi, R.; Tommasino, M.; Llano, M.; von Knebel Doeberitz, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Oncogenic human papillomaviruses activate the tumor-associated lens epithelial-derived growth factor (LEDGF) gene. PLoS Pathog. 2014, 10, 1003957. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Addadi, Y.; Sapoznik, S.; Meir, G.; Kalchenko, V.; Harmelin, A.; Ben-Dor, S.; Neeman, M. Transcriptional regulation of vascular endothelial growth factor C by oxidative and thermal stress is mediated by lens epithelium-derived growth factor/p75. Neoplasia 2009, 11, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Daniels, T.; Molinaro, C.; Lilly, M.B.; Casiano, C.A. Caspase cleavage of the nuclear autoantigen LEDGF/p75 abrogates its pro-survival function: Implications for autoimmunity in atopic disorders. Cell Death Differ. 2002, 9, 915–925. [Google Scholar] [CrossRef]
- Daniels, T.; Zhang, J.; Gutierrez, I.; Elliot, M.L.; Yamada, B.; Heeb, M.J.; Sheets, S.M.; Wu, X.; Casiano, C.A. Antinuclear autoantibodies in PCa: Immunity to LEDGF/p75, a survival protein highly expressed in prostate tumors and cleaved during apoptosis. Prostate 2005, 62, 14–26. [Google Scholar] [CrossRef]
- Huang, T.S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, O.; Doskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef]
- Daugaard, M.; Kirkegaard-Sorensen, T.; Ostenfeld, M.S.; Aaboe, M.; Hoyer-Hansen, M.; Orntoft, T.F.; Rohde, M.; Jaattela, M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef] [PubMed]
- Sapoznik, S.; Cohen, B.; Tzuman, Y.; Meir, G.; Ben-Dor, S.; Harmelin, A.; Neeman, M. Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res. 2009, 69, 9306–9314. [Google Scholar] [CrossRef]
- Bhargavan, B.; Fatma, N.; Chhunchha, B.; Singh, V.; Kubo, E.; Singh, D.P. LEDGF gene silencing impairs the tumorigenicity of prostate cancer DU145 cells by abating the expression of Hsp27 and activation of the Akt/ERK signaling pathway. Cell Death Dis. 2012, 3, e316. [Google Scholar] [CrossRef]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; De Leon, M.; Casiano, C.A. Expression of the stress response oncoprotein LEDGF/p75 in human cancer: A study of 21 tumor types. PLoS ONE 2012, 7, e30132. [Google Scholar] [CrossRef]
- Chan, T.S.; Hawkins, C.; Krieger, J.R.; McGlade, C.J.; Huang, A. JPO2/CDCA7L and LEDGF/p75 Are Novel Mediators of PI3K/AKT Signaling and Aggressive Phenotypes in Medulloblastoma. Cancer Res. 2016, 76, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.S.; Padilla, A.; Wall, N.R.; Lilly, M.B.; et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer 2009, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sørensen, C.S.; Petersen, N.H.; Sorensen, P.H.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef]
- Liedtke, V.; Schroder, C.; Roggenbuck, D.; Weiss, R.; Stohwasser, R.; Schierack, P.; Rodiger, S.; Schenk, L. LEDGF/p75 Is Required for an Efficient DNA Damage Response. Int. J. Mol. Sci. 2021, 22, 5866. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Hernandez, E.S.; Ochoa, P.T.; Suzuki, T.; Ortiz-Hernandez, G.L.; Unternaehrer, J.J.; Alkashgari, H.R.; Diaz Osterman, C.J.; Martinez, S.R.; Chen, Z.; Kremsky, I.; et al. Glucocorticoid Receptor Regulates and Interacts with LEDGF/p75 to Promote Docetaxel Resistance in Prostate Cancer Cells. Cells 2023, 12, 2046. [Google Scholar] [CrossRef]
- Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Ochoa, P.T.; Elix, C.C.; Alkashgari, H.R.; McMullen, J.R.W.; Soto, U.; Martinez, S.R.; Diaz Osterman, C.J.; Mahler, M.; et al. The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells. Cells 2021, 10, 2723. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, W.; Feng, Y.; Yang, L.; Lin, H.; Zhou, P.; Zhao, K.; Jiang, L.; Yao, B.; Feng, N. Identification of the H3K36me3 reader LEDGF/p75 in the pancancer landscape and functional exploration in clear cell renal cell carcinoma. Comput. Struct. Biotechnol. J. 2023, 21, 4134–4148. [Google Scholar] [CrossRef] [PubMed]
- Ochs, R.L.; Mahler, M.; Basu, A.; Rios-Colon, L.; Sanchez, T.W.; Andrade, L.E.; Fritzler, M.J.; Casiano, C.A. The significance of autoantibodies to DFS70/LEDGF in health and disease: Integrating basic science with clinical understanding. Clin. Exp. Med. 2016, 16, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.; Röber, N.; Andrade, L.E.; Mahler, M. The Clinical Relevance of Anti-DFS70 Autoantibodies. Clin. Rev. Allergy Immunol. 2017, 52, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Andrade, L.E.; Casiano, C.A.; Malyavantham, K.; Fritzler, M.J. Anti-DFS70 antibodies: An update on our current understanding and their clinical usefulness. Expert Rev. Clin. Immunol. 2019, 15, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Casiano, C.A. Twenty years of research on the DFS70/LEDGF autoantibody-autoantigen system: Many lessons learned but still many questions. Autoimmun. Highlights 2020, 11, 3. [Google Scholar] [CrossRef]
- Bossuyt, X. DFS70 Autoantibodies: Clinical Utility in Antinuclear Antibody Testing. Clin. Chem. 2024, 70, 374–381. [Google Scholar] [CrossRef]
- Llano, M.; Morrison, J.; Poeschla, E.M. Virological and cellular roles of the transcriptional coactivator LEDGF/p75. Curr. Top. Microbiol. Immunol. 2009, 339, 125–146. [Google Scholar]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host factors for retroviral integration site selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef]
- Engelman, A.N.; Singh, P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018, 75, 2491–2507. [Google Scholar] [CrossRef]
- Ochs, R.L.; Muro, Y.; Si, Y.; Ge, H.; Chan, E.K.; Tan, E.M. Autoantibodies to DFS 70 kd/transcription coactivator p75 in atopic dermatitis and other conditions. J. Allergy Clin. Immunol. 2000, 105, 1211–1220. [Google Scholar] [CrossRef]
- Singh, D.P.; Kimura, A.; Chylack, L.T.; and Shinohara, T. Lens epithelium-derived growth factor (LEDGF/p75) and p52 are derived from a single gene by alternative splicing. Gene 2000, 242, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Debyser, Z.; Engelborghs, Y.; Engelman, A. Identification and characterization of a functional nuclear localization signal in the HIV-1 integrase interactor LEDGF/p75. J. Biol. Chem. 2004, 279, 33421–33429. [Google Scholar] [CrossRef]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef]
- Janssens, J.; Bruggemans, A.; Christ, F.; Debyser, Z. Towards a Functional Cure of HIV-1: Insight Into the Chromatin Landscape of the Provirus. Front. Microbiol. 2021, 12, 636642. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.G.; Newton, K.; Brownstein, D.G.; Holmes, M.C.; Kress, C.; Semple, C.A.; Bickmore, W.A. Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol. Cell. Biol. 2006, 26, 7201–7210. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Usukura, J.; Singh, D.P.; Chylack, L.T., Jr.; Shinohara, T. Spatial and temporal dynamics of two alternatively spliced regulatory factors, lens epithelium-derived growth factor (ledgf/p75) and p52, in the nucleus. Cell Tissue Res. 2001, 305, 107–114. [Google Scholar] [CrossRef]
- Brown-Bryan, T.A.; Leoh, L.S.; Ganapathy, V.; Pacheco, F.J.; Mediavilla-Varela, M.; Filippova, M.; Linkhart, T.A.; Gijsbers, R.; Debyser, Z.; Casiano, C.A. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/p75. Mol. Cancer Res. 2008, 6, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Dietz, F.; Franken, S.; Yoshida, K.; Nakamura, H.; Kappler, J.; Gieselmann, V. The family of hepatoma-derived growth factor proteins: Characterization of a new member HRP-4 and classification of its subfamilies. Biochem. J. 2002, 366, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, Y.; Zhou, M.; Dong, A.; Yan, X.; Loppnau, P.; Min, J.; Liu, Y. Histone and DNA binding ability studies of the NSD subfamily of PWWP domains. Biochem. Biophys. Res. Commun. 2021, 569, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lei, M.; Qin, S.; Dong, A.; Yang, A.; Li, Y.; Loppnau, P.; Hughes, T.R.; Min, J.; Liu, Y. Crystal structure of the BRPF2 PWWP domain in complex with DNA reveals a different binding mode than the HDGF family of PWWP domains. Biochim. Biophys. Acta Gene Regul. Mech. 2021, 1864, 194688. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Y.; Min, J. Advances in inhibitor development targeting the PWWP domain. Trends Pharmacol. Sci. 2024, 45, 193–196. [Google Scholar] [CrossRef]
- Tajima, S.; Suetake, I.; Takeshita, K.; Nakagawa, A.; Kimura, H.; Song, J. Domain Structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA Methyltransferases. Adv. Exp. Med. Biol. 2022, 1389, 45–68. [Google Scholar]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Q.; Wong, S.H.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef]
- Yu, J.R.; LeRoy, G.; Bready, D.; Frenster, J.D.; Saldaña-Meyer, R.; Jin, Y.; Descostes, N.; Stafford, J.M.; Placantonakis, D.G.; Reinberg, D. The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci. Adv. 2021, 7, eabg7444. [Google Scholar] [CrossRef] [PubMed]
- Sundarraj, J.; Taylor, G.C.A.; von Kriegsheim, A.; Pradeepa, M.M. H3K36me3 and PSIP1/LEDGF associate with several DNA repair proteins, suggesting their role in efficient DNA repair at actively transcribing loci. Wellcome Open Res. 2021, 2, 83. [Google Scholar] [CrossRef]
- Acke, A.; Van Belle, S.; Louis, B.; Vitale, R.; Rocha, S.; Voet, T.; Debyser, Z.; Hofkens, J. Expansion microscopy allows high resolution single cell analysis of epigenetic readers. Nucleic Acids Res. 2022, 50, e100. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.N.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K.N.; Horth, C.; McGuire, J.T.; Xu, X.; Nikbakht, H.; et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019, 573, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, S.; Patel, M.; Boulet, F.; Aziz, H.; Brooke, G.N.; Tummala, H.; Pradeepa, M.M. PSIP1/LEDGF reduces R-loops at transcription sites to maintain genome integrity. Nat. Commun. 2024, 15, 361. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Lan, L.; Zou, L. Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat. Rev. Mol. Cell Biol. 2022, 23, 521–540. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Sharma, S.; Čermáková, K.; De Rijck, J.; Demeulemeester, J.; Fábry, M.; El Ashkar, S.; Van Belle, S.; Lepšík, M.; Tesina, P.; Duchoslav, V.; et al. Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E7053–E7062. [Google Scholar] [CrossRef]
- Vanegas, M.; Llano, M.; Delgado, S.; Thompson, D.; Peretz, M.; Poeschla, E. Identification of the LEDGF/p75 HIV-1 integrase-interaction domain and NLS reveals NLS-independent chromatin tethering. J. Cell Sci. 2005, 118, 1733–1743. [Google Scholar] [CrossRef]
- Thakar, K.; Votteler, I.; Kelkar, D.; Shidore, T.; Gupta, S.; Kelm, S.; Dietz, F. Interaction of HRP-2 isoforms with HDGF: Chromatin binding of a specific heteromer. FEBS J. 2012, 279, 737–751. [Google Scholar] [CrossRef]
- Vandegraaff, N.; Devroe, E.; Turlure, F.; Silver, P.A.; Engelman, A. Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP-2) in preintegration complex function and HIV-1 replication. Virology 2006, 346, 415–426. [Google Scholar] [CrossRef]
- Wang, H.; Jurado, K.A.; Wu, X.; Shun, M.C.; Li, X.; Ferris, A.L.; Smith, S.J.; Patel, P.A.; Fuchs, J.R.; Cherepanov, P.; et al. HRP-2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [Google Scholar] [CrossRef]
- Schrijvers, R.; Vets, S.; De Rijck, J.; Malani, N.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 2012, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Xu, C.; Jin, X.; Wumaier, R.; Ma, J.; Peng, J.; Wang, Y.; Tang, Y.; Yu, L.; Zhang, P. HDGF-related protein-2 (HRP-2) acts as an oncogene to promote cell growth in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2015, 458, 849–855. [Google Scholar] [CrossRef]
- Van Belle, S.; El Ashkar, S.; Cermakova, K.; Matthijssens, F.; Goossens, S.; Canella, A.; Hodges, C.H.; Christ, F.; De Rijck, J.; Van Vlierberghe, P.; et al. Unlike Its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival. Cells 2021, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Oksuz, O.; Descostes, N.; Aoi, Y.; Ganai, R.A.; Kara, H.O.; Yu, J.R.; Lee, C.H.; Stafford, J.; Shilatifard, A.; et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019, 5, eaay3068. [Google Scholar] [CrossRef]
- Bhargavan, B.; Chhunchha, B.; Kubo, E.; Singh, D.P. DNA methylation as an epigenetic mechanism in the regulation of LEDGF expression and biological response in aging and oxidative stress. Cell Death Discov. 2024, 10, 296. [Google Scholar] [CrossRef]
- Leoh, L.S.; van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.R.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar] [CrossRef]
- Lampros, M.; Vlachos, N.; Voulgaris, S.; Alexiou, G.A. The Role of Hsp27 in Chemotherapy Resistance. Biomedicines 2022, 10, 897. [Google Scholar] [CrossRef]
- Rizvi, S.F.; Hasan, A.; Parveen, S.; Mir, S.S. Untangling the complexity of heat shock protein 27 in cancer and metastasis. Arch. Biochem. Biophys. 2023, 736, 109537. [Google Scholar] [CrossRef]
- Bueno, M.T.; Garcia-Rivera, J.A.; Kugelman, J.R.; Morales, E.; Rosas-Acosta, G.; Llano, M. SUMOylation of the lens epithelium-derived growth factor/p75 attenuates its transcriptional activity on the heat shock protein 27 promoter. J. Mol. Biol. 2010, 399, 221–239. [Google Scholar] [CrossRef]
- Ishihara, K.; Fatma, N.; Bhargavan, B.; Chhunchha, B.; Kubo, E.; Dey, S.; Takamura, Y.; Kumar, A.; Singh, D.P. Lens epithelium-derived growth factor deSumoylation by Sumo-specific protease-1 regulates its transcriptional activation of small heat shock protein and the cellular response. FEBS J. 2012, 279, 3048–3070. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Li, X.L.; Lin, Y.C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/p75 promotes tumorigenicity in breast cancer cells by promoting the transcription of cell cycle genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Plumb, M.R.; Ferris, A.L.; Iben, J.R.; Wu, X.; Fadel, H.J.; Luke, B.T.; Esnault, C.; Poeschla, E.M.; Hughes, S.H.; et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015, 29, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Morchikh, M.; Naughtin, M.; Di Nunzio, F.; Xavier, J.; Charneau, P.; Jacob, Y.; Lavigne, M. TOX4 and NOVA1 proteins are partners of the LEDGF PWWP domain and affect HIV-1 replication. PLoS ONE 2013, 8, e81217. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.R.; Wang, X.; Mort, M.; Vanduyn, N.; Cooper, D.N.; Mooney, S.D.; Edenberg, H.J.; Liu, Y. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009, 19, 381–394. [Google Scholar] [CrossRef]
- Li, R.; Dong, Q.; Yuan, X.; Zeng, X.; Gao, Y.; Chiao, C.; Li, H.; Zhao, X.; Keles, S.; Wang, Z.; et al. Misregulation of Alternative Splicing in a Mouse Model of Rett Syndrome. PLoS Genet. 2016, 12, e1006129. [Google Scholar] [CrossRef]
- Nejati-Koshki, K.; Roberts, C.T.; Babaei, G.; Rastegar, M. The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers 2023, 15, 2683. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Grimes, G.R.; Taylor, G.C.; Sutherland, H.G.; Bickmore, W.A. Psip1/Ledgf p75 restrains Hox gene expression by recruiting both trithorax and polycomb group proteins. Nucleic Acids Res. 2014, 42, 9021–9032. [Google Scholar] [CrossRef]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451. [Google Scholar] [CrossRef]
- Mac, M.; DeVico, B.M.; Raspanti, S.M.; Moody, C.A. The SETD2 Methyltransferase Supports Productive HPV31 Replication through the LEDGF/CtIP/Rad51 Pathway. J. Virol. 2023, 97, e0020123. [Google Scholar] [CrossRef]
- Lopez, A.P.; Kugelman, J.R.; Garcia-Rivera, J.; Urias, E.; Salinas, S.A.; Fernandez-Zapico, M.E.; Llano, M. The Structure-Specific Recognition Protein 1 Associates with Lens Epithelium-Derived Growth Factor Proteins and Modulates HIV-1 Replication. J. Mol. Biol. 2016, 428, 2814–2831. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cao, R.; Tao, B.; Wu, P.; Peng, C.; Gao, H.; Liang, J.; Yang, W. Pyruvate Facilitates FACT-Mediated γH2AX Loading to Chromatin and Promotes the Radiation Resistance of Glioblastoma. Adv. Sci. 2022, 9, e2104055. [Google Scholar] [CrossRef] [PubMed]
- Hannon, C.; Cruz-Migoni, A.; Platonova, O.; Owen, R.L.; Nettleship, J.E.; Miller, A.; Carr, S.B.; Harris, G.; Rabbitts, T.H.; Phillips, S.E.V. Cloning, purification and structure determination of the HIV integrase-binding domain of lens epithelium-derived growth factor. Acta Crystallogr. F Struct. Biol. Commun. 2018, 74, 143–149. [Google Scholar] [CrossRef]
- Lux, V.; Brouns, T.; Čermáková, K.; Srb, P.; Fábry, M.; Mádlíková, M.; Hořejší, M.; Kukačka, Z.; Novák, P.; Kugler, M.; et al. Molecular Mechanism of LEDGF/p75 Dimerization. Structure 2020, 28, 1288–1299. [Google Scholar] [CrossRef]
- Brouns, T.; Lux, V.; Van Belle, S.; Christ, F.; Veverka, V.; Debyser, Z. The Impact of Lens Epithelium-Derived Growth Factor p75 Dimerization on Its Tethering Function. Cells 2024, 13, 227. [Google Scholar] [CrossRef]
- Wistner, S.C.; MacDonald, I.A.; Stanley, K.A.; Hathaway, N.A. Characterization of Hepatoma-Derived Growth Factor-Related Protein 2 Interactions with Heterochromatin. Cells 2023, 12, 325. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, X.; Dang, L.; Jiang, H.; Xie, Y.; Li, X.; Guo, J.; Wang, Y.; Peng, Z.; Wang, M.; et al. Epigenomic reprogramming via HRP-2-MINA dictates response to proteasome inhibitors in multiple myeloma with t(4;14) translocation. J. Clin. Investig. 2022, 132, e149526. [Google Scholar] [CrossRef]
- Gérard, A.; Ségéral, E.; Naughtin, M.; Abdouni, A.; Charmeteau, B.; Cheynier, R.; Rain, J.C.; Emiliani, S. The integrase cofactor LEDGF/p75 associates with Iws1 and Spt6 for postintegration silencing of HIV-1 gene expression in latently infected cells. Cell Host Microbe 2015, 17, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.P.; Hu, W.; Chen, C.W.; Chu, S.H.; Brien, G.L.; Park, C.Y.; et al. NUP98 Fusion Proteins Interact with the NSL and MLL1 Complexes to Drive Leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Isaksson, M.; Billström, R.; Strömbeck, B.; Mitelman, F.; Johansson, B. Fusion of the NUP98 gene and the homeobox gene HOXC13 in acute myeloid leukemia with t(11;12)(p15;q13). Genes Chromosomes Cancer 2003, 36, 107–112. [Google Scholar] [CrossRef]
- Ahuja, H.G.; Hong, J.; Aplan, P.D.; Tcheurekdjian, L.; Forman, S.J.; Slovak, M.L. t(9;11)(p22;p15) in acute myeloid leukemia results in a fusion between NUP98 and the gene encoding transcriptional coactivators p52 and p75-lens epithelium-derived growth factor (LEDGF). Cancer Res. 2000, 60, 6227–6229. [Google Scholar] [PubMed]
- Hussey, D.J.; Moore, S.; Nicola, M.; Dobrovic, A. Fusion of the NUP98 gene with the LEDGF/p52 gene defines a recurrent acute myeloid leukemia translocation. BMC Genet. 2001, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; Horvat, A.; Karlsson, K.; Olofsson, T.; Paulsson, K.; Johansson, B. t(9;11)(p22;p15) [NUP98/PSIP1] is a poor prognostic marker associated with de novo acute myeloid leukaemia expressing both mature and immature surface antigens. Leuk. Res. 2011, 35, e75–e76. [Google Scholar] [CrossRef]
- Ha, S.Y.; Chan, L.C. Biphenotypic leukemia with t(9;11)(p22;p15). Cancer Genet. Cytogenet. 1994, 76, 116–117. [Google Scholar] [CrossRef]
- Grand, F.H.; Koduru, P.; Cross, N.C.; Allen, S.L. NUP98-LEDGF fusion and t(9;11) in transformed chronic myeloid leukemia. Leuk. Res. 2005, 29, 1469–1472. [Google Scholar] [CrossRef]
- Morerio, C.; Acquila, M.; Rosanda, C.; Rapella, A.; Tassano, E.; Micalizzi, C.; Panarello, C. t(9;11)(p22;p15) with NUP98-LEDGF fusion gene in pediatric acute myeloid leukemia. Leuk. Res. 2005, 9, 467–740. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nakamachi, Y.; Yakushijin, K.; Funakoshi, Y.; Okamura, A.; Kawano, S.; Matsuoka, H.; Minami, H. Expression of the novel NUP98/PSIP1 fusion transcripts in myelodysplastic syndrome with t(9;11)(p22;p15). Eur. J. Haematol. 2012, 88, 244–248. [Google Scholar]
- Xue, S.; Chen, J.Q.; Wang, T.; Zhang, L.N.; Chen, M.; Sun, H.P.; Cao, X.Y. Case report: NUP98::LEDGF fusion gene drives malignant hematological tumor with mixed immunological phenotype. Front. Oncol. 2024, 14, 1396655. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Kubo, E.; Takamura, Y.; Shinohara, T.; Kumar, A.; Chylack, L.T., Jr.; Fatma, N. DNA binding domains and nuclear localization signal of LEDGF: Contribution of two helix-turn-helix (HTH)-like domains and a stretch of 58 amino acids of the N-terminal to the trans-activation potential of LEDGF. J. Mol. Biol. 2006, 355, 379–394. [Google Scholar] [CrossRef]
- Gallego Hernanz, M.P.; Torregrosa Diaz, J.M.; Sorel, N.; Bobin, A.; Dindinaud, E.; Bouyer, S.; Desmier, D.; Brizard, F.; Leleu, X.; Maillard, N.; et al. Long-term molecular remission in a patient with acute myeloid leukemia harboring a new NUP98-LEDGF rearrangement. Cancer Med. 2019, 8, 1765–1770. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Thiel, A.T.; Huang, J.; Lei, M.; Hua, X. Menin as a hub controlling mixed lineage leukemia. Bioessays 2012, 34, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Cermáková, K.; Tesina, P.; Demeulemeester, J.; El Ashkar, S.; Méreau, H.; Schwaller, J.; Rezáčová, P.; Veverka, V.; De Rijck, J. Validation and structural characterization of the LEDGF/p75-MLL interface as a new target for the treatment of MLL-dependent leukemia. Cancer Res. 2014, 74, 5139–5151. [Google Scholar] [CrossRef] [PubMed]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [CrossRef]
- Canella, A.; Van Belle, S.; Brouns, T.; Nigita, G.; Carlon, M.S.; Christ, F.; Debyser, Z. LEDGF/p75-mediated chemoresistance of mixed-lineage leukemia involves cell survival pathways and super enhancer activators. Cancer Gene Ther. 2021, 29, 133–140. [Google Scholar] [CrossRef]
- Demoen, L.; Matthijssens, F.; Reunes, L.; Palhais, B.; Lintermans, B.; T’Sas, S.; Fijalkowski, I.; Taminau, J.; Akele, M.Z.; Van Belle, S.; et al. A dual role for PSIP1/LEDGF in T cell acute lymphoblastic leukemia. Sci Adv. 2024, 10, eado6765. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, J.; Ortega, R.; Qian, W.; Casiano, C.A.; Zhang, J.Y. Preferential autoimmune response in PCa to cyclin B1 in a panel of tumor-associated antigens. J. Immunol. Res. 2014, 2014, 827827. [Google Scholar] [CrossRef]
- Xie, C.; Kim, H.J.; Haw, J.G.; Kalbasi, A.; Gardner, B.K.; Li, G.; Rao, J.; Chia, D.; Liong, M.; Punzalan, R.R.; et al. A novel multiplex assay combining autoantibodies plus PSA has potential implications for classification of PCa from non-malignant cases. J. Transl. Med. 2011, 9, 43. [Google Scholar] [CrossRef]
- O’Rourke, D.J.; DiJohnson, D.A.; Caiazzo, R.J., Jr.; Nelson, J.C.; Ure, D.; O’Leary, M.P.; Richie, J.P.; Liu, B.C. Autoantibody signatures as biomarkers to distinguish PCa from benign prostatic hyperplasia in patients with increased serum prostate specific antigen. Clin. Chim. Acta 2012, 413, 561–567. [Google Scholar] [CrossRef]
- Rios-Colon, L.; Cajigas-Du Ross, C.K.; Basu, A.; Elix, C.; Alicea-Polanco, I.; Sanchez, T.W.; Radhakrishnan, V.; Chen, C.S.; Casiano, C.A. Targeting the stress oncoprotein LEDGF/p75 to sensitize chemoresistant prostate cancer cells to taxanes. Oncotarget 2017, 8, 24915–24931. [Google Scholar] [CrossRef]
- Cajigas-Du Ross, C.K.; Martinez, S.R.; Woods-Burnham, L.; Duran, A.M.; Roy, S.; Basu, A.; Ramirez, J.A.; Ortiz-Hernandez, G.L.; Rios-Colon, L.; Chirshev, E.; et al. RNA sequencing reveals upregulation of a transcriptomic program associated with stemness in metastatic prostate cancer cells selected for taxane resistance. Oncotarget 2018, 9, 30363–30384. [Google Scholar] [CrossRef]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, D.M.; Marijt, K.A.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2016, 23, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Eigentler, A.; Handle, F.; Hackl, H.; Ploner, C.; Heidegger, I.; Schaefer, G.; Brandt, M.P.; Hoefer, J.; Van der Pluijm, G.; et al. Targeting the glucocorticoid receptor signature gene Mono Amine Oxidase-A enhances the efficacy of chemo- and anti-androgen therapy in advanced prostate cancer. Oncogene 2021, 40, 3087–3100. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.R.; Elix, C.C.; Ochoa, P.T.; Sanchez-Hernandez, E.S.; Alkashgari, H.R.; Ortiz-Hernandez, G.L.; Zhang, L.; Casiano, C.A. Glucocorticoid Receptor and beta-Catenin Interact in Prostate Cancer Cells and Their Co-Inhibition Attenuates Tumorsphere Formation, Stemness, and Docetaxel Resistance. Int. J. Mol. Sci. 2023, 24, 7130. [Google Scholar] [CrossRef]
- Woods-Burnham, L.; Cajigas-Du Ross, C.K.; Love, A.; Basu, A.; Sanchez-Hernandez, E.S.; Martinez, S.R.; Ortiz-Hernández, G.L.; Stiel, L.; Durán, A.M.; Wilson, C.; et al. Glucocorticoids Induce Stress Oncoproteins Associated with Therapy-Resistance in African American and European American Prostate Cancer Cells. Sci. Rep. 2018, 8, 15063. [Google Scholar] [CrossRef] [PubMed]
- Desfarges, S.; Abderrahmani, A.; Hernàndez-Novoa, B.; Munoz, M.; Ciuffi, A. LEDGF/p75 TATA-less promoter is driven by the transcription factor Sp1. J. Mol. Biol. 2011, 414, 177–193. [Google Scholar] [CrossRef]
- Singh, D.P.; Bhargavan, B.; Chhunchha, B.; Kubo, E.; Kumar, A.; Fatma, N. Transcriptional protein Sp1 regulates LEDGF transcription by directly interacting with its cis-elements in GC-rich region of TATA-less gene promoter. PLoS ONE 2012, 7, e37012. [Google Scholar] [CrossRef]
- Bhargavan, B.; Chhunchha, B.; Fatma, N.; Kubo, E.; Kumar, A.; Singh, D.P. Epigenetic repression of LEDGF during UVB exposure by recruitment of SUV39H1 and HDAC1 to the Sp1-responsive elements within LEDGF promoter CpG island. Epigenetics 2013, 8, 268–280. [Google Scholar] [CrossRef]
- Rammer, P.; Groth-Pedersen, L.; Kirkegaard, T.; Daugaard, M.; Rytter, A.; Szyniarowski, P.; Høyer-Hansen, M.; Povlsen, L.K.; Nylandsted, J.; Larsen, J.E.; et al. BAMLET activates a lysosomal cell death program in cancer cells. Mol. Cancer Ther. 2010, 9, 24–32. [Google Scholar] [CrossRef]
- Xu, X.; Powell, D.W.; Lambring, C.J.; Puckett, A.H.; Deschenes, L.; Prough, R.A.; Poeschla, E.M.; Samuelson, D.J. Human MCS5A1 candidate breast cancer susceptibility gene FBXO10 is induced by cellular stress and correlated with lens epithelium-derived growth factor (LEDGF). Mol. Carcinog. 2014, 53, 300–313. [Google Scholar] [CrossRef]
- Zammarchi, F.; de Stanchina, E.; Bournazou, E.; Supakorndej, T.; Martires, K.; Riedel, E.; Corben, A.D.; Bromberg, J.F.; Cartegni, L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 17779–17784. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.E.; Chen, S.U.; Lin, C.C.; Chang, C.H.; Lin, Y.C.; Tai, Y.L.; Shen, T.L.; Lee, H. Lysophosphatidic acid enhances vascular endothelial growth factor-C expression in human prostate cancer PC-3 cells. PLoS ONE 2012, 7, e41096. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, E.; Kuchenbaecker, K.B.; Beesley, J.; Adlard, J.; Agnarsson, B.A.; Andrulis, I.L.; Arun, B.K.; Barjhoux, L.; Belotti, M.; Benitez, J.; et al. Fine-Scale Mapping at 9p22.2 Identifies Candidate Causal Variants That Modify Ovarian Cancer Risk in BRCA1 and BRCA2 Mutation Carriers. PLoS ONE 2016, 11, e0158801. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Kartsonaki, C.; Gayther, S.A.; Pharoah, P.D.; Sinilnikova, O.M.; Beesley, J.; Chen, X.; McGuffog, L.; Healey, S.; Couch, F.J.; et al. Genetic variation at 9p22.2 and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2011, 103, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Garnett, T.O.; Duerksen-Hughes, P.J. Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Arch. Virol. 2006, 151, 2321–2335. [Google Scholar] [CrossRef]
- Yin, X.H.; Wang, Z.Q.; Guo, Q.H.; Wu, H.; Shi, M. Overexpressed LEDGF is a novel biomarker of poor prognosis in patients with cervical cancer. Eur. J. Gynaecol. Oncol. 2017, 38, 245–250. [Google Scholar]
- Huang, A.; Ho, C.S.; Ponzielli, R.; Barsyte-Lovejoy, D.; Bouffet, E.; Picard, D.; Hawkins, C.E.; Penn, L.Z. Identification of a novel c-Myc protein interactor, JPO2, with transforming activity in medulloblastoma cells. Cancer Res. 2005, 65, 5607–5619. [Google Scholar] [CrossRef]
- Guo, R.; Ma, Y.; Zhao, M.; Zhang, W.; An, G.; Chen, B.; Song, Y.; Xu, H.; Li, Y. Polymorphism rs2395655 affects LEDGF/p75 binding activity and p21WAF1/CIP1 gene expression in esophageal squamous cell carcinoma. Cancer Med. 2019, 8, 2313–2324. [Google Scholar] [CrossRef]
- Kanu, N.; Grönroos, E.; Martinez, P.; Burrell, R.A.; Yi Goh, X.; Bartkova, J.; Maya-Mendoza, A.; Mistrík, M.; Rowan, A.J.; Patel, H.; et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene 2015, 34, 5699–5708. [Google Scholar] [CrossRef]
- Debyser, Z.; Bruggemans, A.; Van Belle, S.; Janssens, J.; Christ, F. LEDGINs, Inhibitors of the Interaction Between HIV-1 Integrase and LEDGF/p75, Are Potent Antivirals with a Potential to Cure HIV Infection. Adv. Exp. Med. Biol. 2021, 1322, 97–114. [Google Scholar]
- Engelman, A.N. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J. Biol. Chem. 2019, 294, 15137–15157. [Google Scholar] [CrossRef]
- Zhao, Y.; Luo, Z. Recent Advances in the Development of Small-Molecular Inhibitors Target HIV Integrase-LEDGF/p75 Interaction. Mini. Rev. Med. Chem. 2015, 15, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.W.; Debnath, B.; Christ, F.; Otake, H.; Debyser, Z.; Neamati, N. Discovery of novel inhibitors of LEDGF/p75-IN protein-protein interactions. Bioorg. Med. Chem. 2013, 21, 957–963. [Google Scholar] [CrossRef]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef]
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Demeulemeester, J.; Chaltin, P.; Marchand, A.; De Maeyer, M.; Debyser, Z.; Christ, F. LEDGINs, non-catalytic site inhibitors of HIV-1 integrase: A patent review (2006–2014). Expert. Opin. Ther. Pat. 2014, 24, 609–632. [Google Scholar] [CrossRef]
- Sugiyama, S.; Iwaki, T.; Tamura, Y.; Tomita, K.; Matsuoka, E.; Arita, S.; Seki, T.; Yoshinaga, T.; Kawasuji, T. Discovery of novel integrase-LEDGF/p75 allosteric inhibitors based on a benzene scaffold. Bioorg. Med. Chem. 2020, 28, 115643. [Google Scholar] [CrossRef]
- George, A.; Gopi Krishna Reddy, A.; Satyanarayana, G.; Raghavendra, N.K. 1,2,3,4-Tetrahydroisoquinolines as inhibitors of HIV-1 integrase and human LEDGF/p75 interaction. Chem. Biol. Drug Des. 2018, 91, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Humbert, M.; Lescrinier, E.; Hendrix, J.; Vets, S.; Gijsbers, R.; Ruprecht, R.M.; Dietrich, U.; Debyser, Z.; Christ, F. Phage Display-Directed Discovery of LEDGF/p75 Binding Cyclic Peptide Inhibitors of HIV Replication. Mol. Ther. 2021, 29, 887. [Google Scholar] [CrossRef]
- Maehigashi, T.; Ahn, S.; Kim, U.I.; Lindenberger, J.; Oo, A.; Koneru, P.C.; Mahboubi, B.; Engelman, A.N.; Kvaratskhelia, M.; Kim, K.; et al. A highly potent and safe pyrrolopyridine-based allosteric HIV-1 integrase inhibitor targeting host LEDGF/p75-integrase interaction site. PLoS Pathog. 2021, 17, e1009671. [Google Scholar] [CrossRef]
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.; et al. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 2012, 287, 21189–21203. [Google Scholar] [CrossRef] [PubMed]
- Vantieghem, T.; Aslam, N.A.; Osipov, E.M.; Akele, M.; Van Belle, S.; Beelen, S.; Drexler, M.; Paulovcakova, T.; Lux, V.; Fearon, D.; et al. Rational fragment-based design of compounds targeting the PWWP domain of the HRP family. Eur. J. Med. Chem. 2024, 280, 116960. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Ochoa, P.T.; Casiano, C.A. The Emerging Roles of the Stress Epigenetic Reader LEDGF/p75 in Cancer Biology and Therapy Resistance: Mechanisms and Targeting Opportunities. Cancers 2024, 16, 3957. https://doi.org/10.3390/cancers16233957
Ortiz-Hernandez GL, Sanchez-Hernandez ES, Ochoa PT, Casiano CA. The Emerging Roles of the Stress Epigenetic Reader LEDGF/p75 in Cancer Biology and Therapy Resistance: Mechanisms and Targeting Opportunities. Cancers. 2024; 16(23):3957. https://doi.org/10.3390/cancers16233957
Chicago/Turabian StyleOrtiz-Hernandez, Greisha L., Evelyn S. Sanchez-Hernandez, Pedro T. Ochoa, and Carlos A. Casiano. 2024. "The Emerging Roles of the Stress Epigenetic Reader LEDGF/p75 in Cancer Biology and Therapy Resistance: Mechanisms and Targeting Opportunities" Cancers 16, no. 23: 3957. https://doi.org/10.3390/cancers16233957
APA StyleOrtiz-Hernandez, G. L., Sanchez-Hernandez, E. S., Ochoa, P. T., & Casiano, C. A. (2024). The Emerging Roles of the Stress Epigenetic Reader LEDGF/p75 in Cancer Biology and Therapy Resistance: Mechanisms and Targeting Opportunities. Cancers, 16(23), 3957. https://doi.org/10.3390/cancers16233957