
H1 Antihistamines—Promising Candidates for Repurposing in the Context of the Development of New Therapeutic Approaches to Cancer Treatment


Simple Summary
Abstract
1. Introduction
2. Drug Repurposing (DR)—A Strategy to Fight Cancer
3. Inflammation in Cancer
4. The Importance of the Histaminergic System in Carcinogenesis
4.1. Biological Role of Histamine
4.2. Histidine Decarboxylase Activity and Histamine Concentration in Tumor Tissues and Their Significance for Tumor Progression
4.3. Mechanisms of Action of Histamine on Cancer Cells
5. The Importance of the H1R Receptor in Modulating Processes Related to the Development and Progression of Cancer and the Mechanisms of the Anticancer Action of H1 Antihistamines
5.1. Histamine H1R Receptor
5.2. Generations of Antihistamines
5.3. H1R Receptor Function in Cancer
5.4. Mechanisms of the Potential Anticancer Action of H1 Antihistamines
5.4.1. Antihistamines in Cancer Immunotherapy
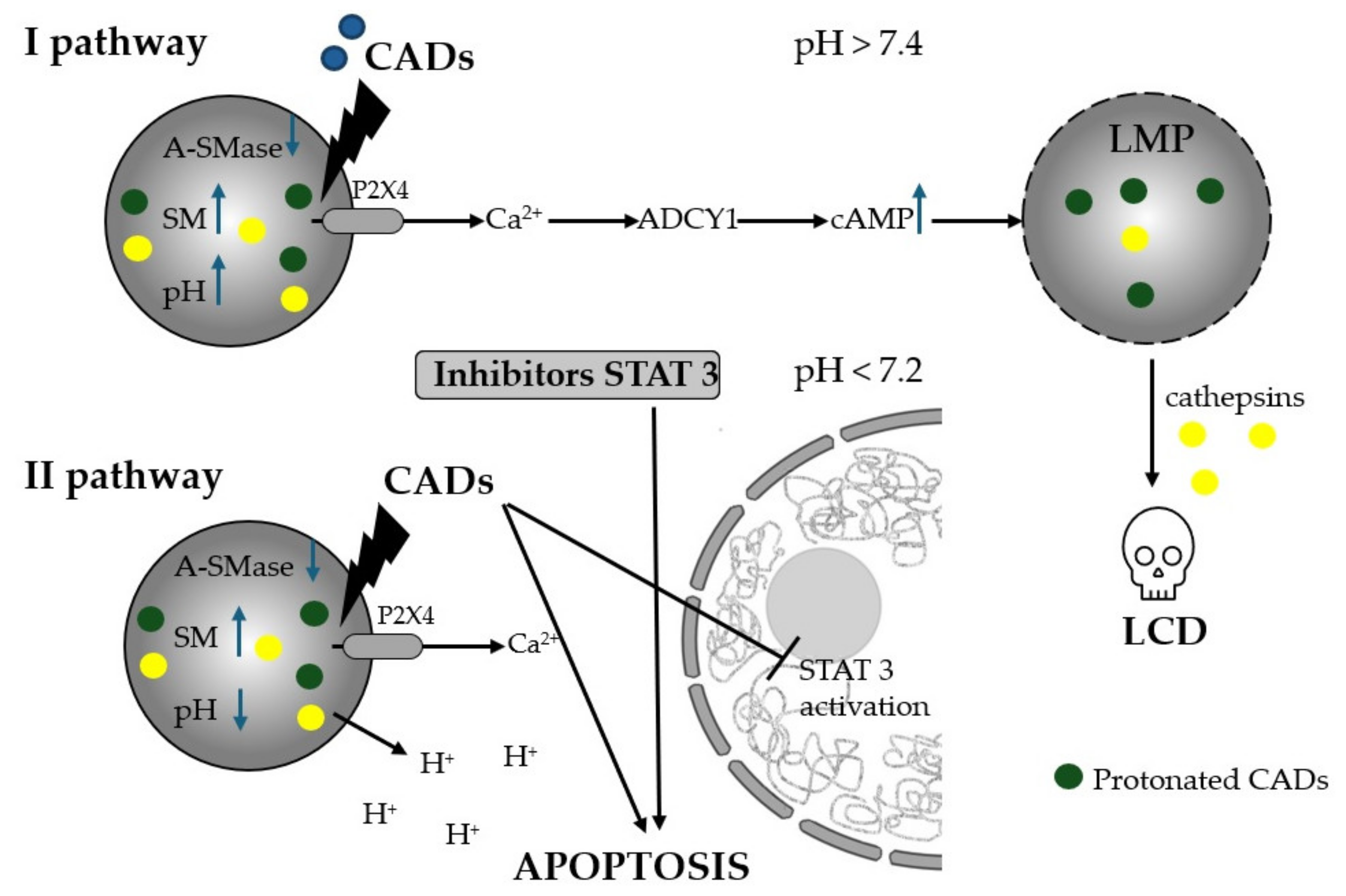
5.4.2. Cationic Amphiphilic Drug-Induction of Lysosomal Cell Death (LMP)
5.4.3. The Effect of Non-CAD on Apoptosis, Proliferation and Cell Cycle of Cancer Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saini, K.S.; Twelves, C. Determining lines of therapy in patients with solid cancers: A proposed new systematic and comprehensive framework. Br. J. Cancer 2021, 125, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Min, H.Y.; Lee, H.Y. Molecular targeted therapy for anticancer treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J. From ’Targeted Therapy’ to Targeted Therapy. Anticancer. Res. 2019, 39, 3341–3345. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Hamada, T.; Cao, Y.; Qian, Z.R.; Masugi, Y.; Nowak, J.A.; Yang, J.; Song, M.; Mima, K.; Kosumi, K.; Liu, L.; et al. Aspirin Use and Colorectal Cancer Survival According to Tumor CD274 (Programmed Cell Death 1 Ligand 1) Expression Status. J. Clin. Oncol. 2017, 35, 1836–1844. [Google Scholar] [CrossRef]
- Weth, F.R.; Hoggarth, G.B.; Weth, A.F.; Paterson, E.; White, M.P.J.; Tan, S.T.; Peng, L.; Gray, C. Unlocking hidden potential: Advancements, approaches, and obstacles in repurposing drugs for cancer therapy. Br. J. Cancer 2024, 130, 703–715. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, M.; Huang, H.; Jin, W.L. Drug repurposing for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 92. [Google Scholar] [CrossRef]
- Makhoba, X.H.; Viegas, C., Jr.; Mosa, R.A.; Viegas, F.P.D.; Pooe, O.J. Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Des. Devel Ther. 2020, 14, 3235–3249. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorg Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jonker, A.H.; O’Connor, D.; Cavaller-Bellaubi, M.; Fetro, C.; Gogou, M.; t Hoen, P.A.C.; de Kort, M.; Stone, H.; Valentine, N.; Pasmooij, A.M.G. Drug repurposing for rare: Progress and opportunities for the rare disease community. Front. Med. 2024, 11, 1352803. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Cho, W.C.S. Drug Repurposing for Cancer Therapy in the Era of Precision Medicine. Curr. Mol. Pharmacol. 2022, 15, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Badria, F.A. Drug Repurposing—Hypothesis, Molecular Aspects and Therapeutic Applications; IntechOpen: London, UK, 2020. [Google Scholar]
- Pantziarka, P. Scientific advice—Is drug repurposing missing a trick? Nat. Rev. Clin. Oncol. 2017, 14, 455–456. [Google Scholar] [CrossRef]
- Toumi, M.; Remuzat, C. Value added medicines: What value repurposed medicines might bring to society? J. Mark. Access Health Policy 2017, 5, 1264717. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Grimshaw, A.A.; Axson, S.A.; Choe, S.H.; Miller, J.E. Drug repurposing: A systematic review on root causes, barriers and facilitators. BMC Health Serv. Res. 2022, 22, 970. [Google Scholar] [CrossRef]
- Mohi-Ud-Din, R.; Chawla, A.; Sharma, P.; Mir, P.A.; Potoo, F.H.; Reiner, Z.; Reiner, I.; Atessahin, D.A.; Sharifi-Rad, J.; Mir, R.H.; et al. Repurposing approved non-oncology drugs for cancer therapy: A comprehensive review of mechanisms, efficacy, and clinical prospects. Eur. J. Med. Res. 2023, 28, 345. [Google Scholar] [CrossRef]
- van der Pol, K.H.; Aljofan, M.; Blin, O.; Cornel, J.H.; Rongen, G.A.; Woestelandt, A.G.; Spedding, M. Drug Repurposing of Generic Drugs: Challenges and the Potential Role for Government. Appl. Health Econ. Health Policy 2023, 21, 831–840. [Google Scholar] [CrossRef]
- Clohessy, J.G.; Pandolfi, P.P. Mouse hospital and co-clinical trial project--from bench to bedside. Nat. Rev. Clin. Oncol. 2015, 12, 491–498. [Google Scholar] [CrossRef]
- Mtewa, A.; Amanjot, A.; Yadesa, T.; Ngwira, K. Coronavirus Drug Discovery SARS-CoV-2 (COVID-19) Prevention, Diagnosis, and Treatment; Egbuna, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 205–226. [Google Scholar]
- Palve, V.; Liao, Y.; Remsing Rix, L.L.; Rix, U. Turning liabilities into opportunities: Off-target based drug repurposing in cancer. Semin. Cancer Biol. 2021, 68, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Mucke, H.A.M. Drug Repurposing: Then, Now, and in the Future. Drug Repurposing 2024, 1, 1–3. [Google Scholar] [CrossRef]
- Sleire, L.; Forde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Pfab, C.; Schnobrich, L.; Eldnasoury, S.; Gessner, A.; El-Najjar, N. Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know? Cancers 2021, 13, 3193. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Andricopulo, A.D. Drug repositioning approaches to parasitic diseases: A medicinal chemistry perspective. Drug Discov. Today 2016, 21, 1699–1710. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Abdelsayed, M.; Kort, E.J.; Jovinge, S.; Mercola, M. Repurposing drugs to treat cardiovascular disease in the era of precision medicine. Nat. Rev. Cardiol. 2022, 19, 751–764. [Google Scholar] [CrossRef]
- Liang, S.; Yu, H. Revealing new therapeutic opportunities through drug target prediction: A class imbalance-tolerant machine learning approach. Bioinformatics 2020, 36, 4490–4497. [Google Scholar] [CrossRef]
- Shih, H.P.; Zhang, X.; Aronov, A.M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications. Nat. Rev. Drug Discov. 2018, 17, 19–33. [Google Scholar] [CrossRef]
- Amiri Souri, E.; Chenoweth, A.; Karagiannis, S.N.; Tsoka, S. Drug repurposing and prediction of multiple interaction types via graph embedding. BMC Bioinform. 2023, 24, 202. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lonborg, A.; et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.; Pasquier, E.; Tomasini, P.; Ciccolini, J.; Greillier, L.; Andre, N.; Barlesi, F.; Mascaux, C. Drug repurposing in malignant pleural mesothelioma: A breath of fresh air? Eur. Respir. Rev. 2018, 27, 170098. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sintes, R.; Ledesma, M.D.; Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 2016, 32, 150–168. [Google Scholar] [CrossRef]
- Ellegaard, A.M.; Dehlendorff, C.; Vind, A.C.; Anand, A.; Cederkvist, L.; Petersen, N.H.T.; Nylandsted, J.; Stenvang, J.; Mellemgaard, A.; Osterlind, K.; et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. EBioMedicine 2016, 9, 130–139. [Google Scholar] [CrossRef]
- Hijazi, M.A.; Gessner, A.; El-Najjar, N. Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp. Cancers 2023, 15, 3199. [Google Scholar] [CrossRef]
- Irie, N.; Mizoguchi, K.; Warita, T.; Nakano, M.; Sasaki, K.; Tashiro, J.; Osaki, T.; Ishikawa, T.; Oltvai, Z.N.; Warita, K. Repurposing of the Cardiovascular Drug Statin for the Treatment of Cancers: Efficacy of Statin-Dipyridamole Combination Treatment in Melanoma Cell Lines. Biomedicines 2024, 12, 698. [Google Scholar] [CrossRef]
- Ishida, J.; Konishi, M.; Ebner, N.; Springer, J. Repurposing of approved cardiovascular drugs. J. Transl. Med. 2016, 14, 269. [Google Scholar] [CrossRef]
- Antoszczak, M.; Markowska, A.; Markowska, J.; Huczynski, A. Antidepressants and Antipsychotic Agents as Repurposable Oncological Drug Candidates. Curr. Med. Chem. 2021, 28, 2137–2174. [Google Scholar] [CrossRef]
- Racz, B.; Spengler, G. Repurposing Antidepressants and Phenothiazine Antipsychotics as Efflux Pump Inhibitors in Cancer and Infectious Diseases. Antibiotics 2023, 12, 137. [Google Scholar] [CrossRef]
- Dhas, Y.; Biswas, N.; Divyalakshmi, M.R.; Jones, L.D.; Ashili, S. Repurposing metabolic regulators: Antidiabetic drugs as anticancer agents. Mol. Biomed. 2024, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Bule, P.; Kolipaka, T.; Ranvare, S.; Chella, N. Drug Repurposing: Innovative Approaches to Drug Discovery and Development; Chella, N., Ranjan, O., Alexander, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2024; pp. 217–248. [Google Scholar]
- Pillai, U.J.; Ray, A.; Maan, M.; Dutta, M. Repurposing drugs targeting metabolic diseases for cancer therapeutics. Drug Discov. Today 2023, 28, 103684. [Google Scholar] [CrossRef] [PubMed]
- Bano, N.; Parveen, S.; Saeed, M.; Siddiqui, S.; Abohassan, M.; Mir, S.S. Drug Repurposing of Selected Antibiotics: An Emerging Approach in Cancer Drug Discovery. ACS Omega 2024, 9, 26762–26779. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Jin, W.; Zhang, J.; Zhu, L.; Lu, J.; Zhen, Y.; Zhang, L.; Ouyang, L.; Liu, B.; Yu, H. Repurposing non-oncology small-molecule drugs to improve cancer therapy: Current situation and future directions. Acta Pharm. Sin. B 2022, 12, 532–557. [Google Scholar] [CrossRef] [PubMed]
- Ozleyen, A.; Yilmaz, Y.B.; Donmez, S.; Atalay, H.N.; Antika, G.; Tumer, T.B. Looking at NSAIDs from a historical perspective and their current status in drug repurposing for cancer treatment and prevention. J. Cancer Res. Clin. Oncol. 2023, 149, 2095–2113. [Google Scholar] [CrossRef]
- Sousa, S.M.; Xavier, C.P.R.; Vasconcelos, M.H.; Palmeira, A. Repurposing some of the Well-known Non-steroid Anti-inflammatory Drugs (NSAIDs) for Cancer Treatment. Curr. Top. Med. Chem. 2023, 23, 1171–1195. [Google Scholar] [CrossRef]
- Rosenzweig, M.; Palmer, J.; Tsai, N.C.; Synold, T.; Wu, X.; Tao, S.; Hammond, S.N.; Buettner, R.; Duarte, L.; Htut, M.; et al. Repurposing leflunomide for relapsed/refractory multiple myeloma: A phase 1 study. Leuk. Lymphoma 2020, 61, 1669–1677. [Google Scholar] [CrossRef]
- Zhang, C.; Chu, M. Leflunomide: A promising drug with good antitumor potential. Biochem. Biophys. Res. Commun. 2018, 496, 726–730. [Google Scholar] [CrossRef]
- Kelly-Irving, M.; Delpierre, C.; Vineis, P. Beyond bad luck: Induced mutations and hallmarks of cancer. Lancet Oncol. 2017, 18, 999–1000. [Google Scholar] [CrossRef]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef]
- Sonaye, H.V.; Sheikh, R.Y.; Doifode, C.A. Drug repurposing: Iron in the fire for older drugs. Biomed. Pharmacother. 2021, 141, 111638. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Mi, W.; Li, F.; Zhu, L.; Ou, Q.; Li, M.; Li, T.; Ma, Y.; Zhang, Y.; Xu, Y. Optimizing drug combination and mechanism analysis based on risk pathway crosstalk in pan cancer. Sci. Data 2024, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; de Wolf, E.; Jawad, M.J.; Richardson, A. The poor design of clinical trials of statins in oncology may explain their failure—Lessons for drug repurposing. Cancer Treat. Rev. 2018, 69, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B. Statins in the Cause and Prevention of Cancer: Confounding by Indication and Mediation by Rhabdomyolysis and Phosphate Toxicity. J. Cardiovasc. Dev. Dis. 2024, 11, 296. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P.; Vikas, P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience 2014, 8, 442. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Malla, R.R.; Kiran, P. Tumor microenvironment pathways: Cross regulation in breast cancer metastasis. Genes. Dis. 2022, 9, 310–324. [Google Scholar] [CrossRef]
- Archilla-Ortega, A.; Domuro, C.; Martin-Liberal, J.; Munoz, P. Blockade of novel immune checkpoints and new therapeutic combinations to boost antitumor immunity. J. Exp. Clin. Cancer Res. 2022, 41, 62. [Google Scholar] [CrossRef]
- Davoudi, F.; Moradi, A.; Sadeghirad, H.; Kulasinghe, A. Tissue biomarkers of immune checkpoint inhibitor therapy. Immunol. Cell Biol. 2024, 102, 179–193. [Google Scholar] [CrossRef]
- Sadeghi Rad, H.; Bazaz, S.R.; Monkman, J.; Ebrahimi Warkiani, M.; Rezaei, N.; O’Byrne, K.; Kulasinghe, A. The evolving landscape of predictive biomarkers in immuno-oncology with a focus on spatial technologies. Clin. Transl. Immunol. 2020, 9, e1215. [Google Scholar] [CrossRef]
- Walsh, L.A.; Quail, D.F. Decoding the tumor microenvironment with spatial technologies. Nat. Immunol. 2023, 24, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Nigam, M.; Mishra, A.P.; Deb, V.K.; Dimri, D.B.; Tiwari, V.; Bungau, S.G.; Bungau, A.F.; Radu, A.F. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomed. Pharmacother. 2023, 164, 115015. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Y.; Li, Q.; Yao, J.; Yuan, X.; Zhang, Y.; Yin, X.; Saito, Y.; Fan, H.; Li, P.; et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients via activation of the macrophage histamine receptor H1. Cancer Cell 2022, 40, 36–52.e9. [Google Scholar] [CrossRef]
- Qu, X.; Tang, Y.; Hua, S. Immunological Approaches Towards Cancer and Inflammation: A Cross Talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef]
- Sadreddini, S.; Baradaran, B.; Aghebati-Maleki, A.; Sadreddini, S.; Shanehbandi, D.; Fotouhi, A.; Aghebati-Maleki, L. Immune checkpoint blockade opens a new way to cancer immunotherapy. J. Cell Physiol. 2019, 234, 8541–8549. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Poliezhaieva, T.; Ermolaeva, M.A. DNA damage in protective and adverse inflammatory responses: Friend of foe? Mech. Ageing Dev. 2017, 165, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Omidvar, S.; Vahedian, V.; Sourani, Z.; Yari, D.; Asadi, M.; Jafari, N.; Khodavirdilou, L.; Bagherieh, M.; Shirzad, M.; Hosseini, V. The molecular crosstalk between innate immunity and DNA damage repair/response: Interactions and effects in cancers. Pathol. Res. Pract. 2024, 260, 155405. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Greten, T.F.; Xia, Q. Immunosuppressive cell death in cancer. Nat. Rev. Immunol. 2017, 17, 401. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Srivatsa, S.; Paul, M.C.; Cardone, C.; Holcmann, M.; Amberg, N.; Pathria, P.; Diamanti, M.A.; Linder, M.; Timelthaler, G.; Dienes, H.P.; et al. EGFR in Tumor-Associated Myeloid Cells Promotes Development of Colorectal Cancer in Mice and Associates With Outcomes of Patients. Gastroenterology 2017, 153, 178–190e110. [Google Scholar] [CrossRef]
- Kesh, K.; Gupta, V.K.; Durden, B.; Garrido, V.; Mateo-Victoriano, B.; Lavania, S.P.; Banerjee, S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers 2020, 12, 3067. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e1396. [Google Scholar] [CrossRef]
- Solimando, A.G.; Desantis, V.; Ribatti, D. Mast Cells and Interleukins. Int. J. Mol. Sci. 2022, 23, 14004. [Google Scholar] [CrossRef] [PubMed]
- Faustino-Rocha, A.I.; Ferreira, R.; Gama, A.; Oliveira, P.A.; Ginja, M. Antihistamines as promising drugs in cancer therapy. Life Sci. 2017, 172, 27–41. [Google Scholar] [CrossRef]
- Baran, J.; Sobiepanek, A.; Mazurkiewicz-Pisarek, A.; Rogalska, M.; Gryciuk, A.; Kuryk, L.; Abraham, S.N.; Staniszewska, M. Mast Cells as a Target-A Comprehensive Review of Recent Therapeutic Approaches. Cells 2023, 12, 1187. [Google Scholar] [CrossRef] [PubMed]
- Lichterman, J.N.; Reddy, S.M. Mast Cells: A New Frontier for Cancer Immunotherapy. Cells 2021, 10, 1270. [Google Scholar] [CrossRef] [PubMed]
- Derakhshani, A.; Vahidian, F.; Alihasanzadeh, M.; Mokhtarzadeh, A.; Lotfi Nezhad, P.; Baradaran, B. Mast cells: A double-edged sword in cancer. Immunol. Lett. 2019, 209, 28–35. [Google Scholar] [CrossRef]
- Ribatti, D. New insights into the role of mast cells as a therapeutic target in cancer through the blockade of immune checkpoint inhibitors. Front. Med. 2024, 11, 1373230. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef]
- Sarasola, M.P.; Taquez Delgado, M.A.; Nicoud, M.B.; Medina, V.A. Histamine in cancer immunology and immunotherapy. Current status and new perspectives. Pharmacol. Res. Perspect. 2021, 9, e00778. [Google Scholar] [CrossRef]
- Linton, S.; Hossenbaccus, L.; Ellis, A.K. Evidence-based use of antihistamines for treatment of allergic conditions. Ann. Allergy Asthma Immunol. 2023, 131, 412–420. [Google Scholar] [CrossRef]
- Blaya, B.; Nicolau-Galmes, F.; Jangi, S.M.; Ortega-Martinez, I.; Alonso-Tejerina, E.; Burgos-Bretones, J.; Perez-Yarza, G.; Asumendi, A.; Boyano, M.D. Histamine and histamine receptor antagonists in cancer biology. Inflamm. Allergy Drug Targets 2010, 9, 146–157. [Google Scholar] [CrossRef]
- Shahid, M.; Tripathi, T.; Sobia, F.; Moin, S.; Siddiqui, M.; Khan, R.A. Histamine, Histamine Receptors, and their Role in Immunomodulation: An Updated Systematic Review. Open Immunol. J. 2009, 2, 9–41. [Google Scholar] [CrossRef]
- Dileepan, K.N.; Raveendran, V.V.; Sharma, R.; Abraham, H.; Barua, R.; Singh, V.; Sharma, R.; Sharma, M. Mast cell-mediated immune regulation in health and disease. Front. Med. 2023, 10, 1213320. [Google Scholar] [CrossRef] [PubMed]
- Falcone, F.H.; Zillikens, D.; Gibbs, B.F. The 21st century renaissance of the basophil? Current insights into its role in allergic responses and innate immunity. Exp. Dermatol. 2006, 15, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Ziętkowski, Z.; Łukaszyk, M.; Skiepko, U.; Bodzenta-Łukaszyk, A. Antihistaminic drugs in treatment of pollinosis. Alerg. Astma Immunol. 2016, 21, 28–32. [Google Scholar]
- Huang, H.; Li, Y.; Liang, J.; Finkelman, F.D. Molecular Regulation of Histamine Synthesis. Front. Immunol. 2018, 9, 1392. [Google Scholar] [CrossRef]
- Dy, M.; Schneider, E. Histamine-cytokine connection in immunity and hematopoiesis. Cytokine Growth Factor. Rev. 2004, 15, 393–410. [Google Scholar] [CrossRef]
- Kazumori, H.; Ishihara, S.; Rumi, M.A.; Ortega-Cava, C.F.; Kadowaki, Y.; Kinoshita, Y. Transforming growth factor-alpha directly augments histidine decarboxylase and vesicular monoamine transporter 2 production in rat enterochromaffin-like cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G508–G514. [Google Scholar] [CrossRef]
- Stojković, S.; Cekić, S.; Ristov, M.; Ristić, M.; Đukić, D.; Binić, M.; Virijević, D. Histamine and Antihistamines. Acta Fac. Med. Naissensis 2015, 32, 7–22. [Google Scholar] [CrossRef]
- Tatarkiewicz, J.; Rzodkiewicz, P.; Zochowska, M.; Staniszewska, A.; Bujalska-Zadrozny, M. New antihistamines—Perspectives in the treatment of some allergic and inflammatory disorders. Arch. Med. Sci. 2019, 15, 537–553. [Google Scholar] [CrossRef]
- Leurs, R.; Church, M.K.; Taglialatela, M. H1-antihistamines: Inverse agonism, anti-inflammatory actions and cardiac effects. Clin. Exp. Allergy 2002, 32, 489–498. [Google Scholar] [CrossRef]
- Neumann, J.; Hofmann, B.; Kirchhefer, U.; Dhein, S.; Gergs, U. Function and Role of Histamine H1 Receptor in the Mammalian Heart. Pharmaceuticals 2023, 16, 734. [Google Scholar] [CrossRef] [PubMed]
- Simons, F.E.; Simons, K.J. H1 antihistamines: Current status and future directions. World Allergy Organ. J. 2008, 1, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.L.; Cho, J. Pathophysiological Roles of Histamine Receptors in Cancer Progression: Implications and Perspectives as Potential Molecular Targets. Biomolecules 2021, 11, 1232. [Google Scholar] [CrossRef] [PubMed]
- Jemima, E.A.; Prema, A.; Thangam, E.B. Functional characterization of histamine H4 receptor on human mast cells. Mol. Immunol. 2014, 62, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zampeli, E.; Tiligada, E. The role of histamine H4 receptor in immune and inflammatory disorders. Br. J. Pharmacol. 2009, 157, 24–33. [Google Scholar] [CrossRef]
- Falus, A.; Hegyesi, H.; Lazar-Molnar, E.; Pos, Z.; Laszlo, V.; Darvas, Z. Paracrine and autocrine interactions in melanoma: Histamine is a relevant player in local regulation. Trends Immunol. 2001, 22, 648–652. [Google Scholar] [CrossRef]
- Rivera, E.S.; Cricco, G.P.; Engel, N.I.; Fitzsimons, C.P.; Martin, G.A.; Bergoc, R.M. Histamine as an autocrine growth factor: An unusual role for a widespread mediator. Semin. Cancer Biol. 2000, 10, 15–23. [Google Scholar] [CrossRef]
- Haak-Frendscho, M.; Darvas, Z.; Hegyesi, H.; Karpati, S.; Hoffman, R.L.; Laszlo, V.; Bencsath, M.; Szalai, C.; Furesz, J.; Timar, J.; et al. Histidine decarboxylase expression in human melanoma. J. Invest. Dermatol. 2000, 115, 345–352. [Google Scholar] [CrossRef]
- Massari, N.A.; Nicoud, M.B.; Medina, V.A. Histamine receptors and cancer pharmacology: An update. Br. J. Pharmacol. 2020, 177, 516–538. [Google Scholar] [CrossRef]
- August, E.M.; Patnaude, L.; Hopkins, J.; Studts, J.; Gautschi, E.; Shrutkowski, A.; Kronkaitis, A.; Brown, M.; Kabcenell, A.; Rajotte, D. Development of a high-throughput assay to measure histidine decarboxylase activity. J. Biomol. Screen. 2006, 11, 816–821. [Google Scholar] [CrossRef]
- Cricco, G.; Martin, G.; Medina, V.; Nunez, M.; Gutierrez, A.; Cocca, C.; Bergoc, R.; Rivera, E. Histamine regulates the MAPK pathway via the H(2) receptor in PANC-1 human cells. Inflamm. Res. 2004, 53 (Suppl. S1), S65–S66. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Hodges, K.; Meng, F.; Alpini, G.; Francis, H. Histamine and histamine receptor regulation of gastrointestinal cancers. Transl. Gastrointest. Cancer 2012, 1, 215–227. [Google Scholar] [PubMed]
- Krauth, M.T.; Agis, H.; Aichberger, K.J.; Simonitsch-Klupp, I.; Mullauer, L.; Mayerhofer, M.; Bohm, A.; Horny, H.P.; Valent, P. Immunohistochemical detection of histidine decarboxylase in neoplastic mast cells in patients with systemic mastocytosis. Hum. Pathol. 2006, 37, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Darvas, Z.; Sakurai, E.; Schwelberger, H.G.; Hegyesi, H.; Rivera, E.; Othsu, H.; Watanabe, T.; Pallinger, E.; Falus, A. Autonomous histamine metabolism in human melanoma cells. Melanoma Res. 2003, 13, 239–246. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Akhter, J.; Morris, D.L. In vitro effect of histamine and histamine H1 and H2 receptor antagonists on cellular proliferation of human malignant melanoma cell lines. Melanoma Res. 1996, 6, 95–99. [Google Scholar] [CrossRef]
- Stanosz, S.; Stanosz, M.; von Mach-Szczypiński, J. Metabolizm histaminy w tkance pierwotnych raków przewodowych gruczołu piersiowego. Contemp. Oncol. 2007, 11, 6–11. [Google Scholar]
- Garcia-Caballero, M.; Neugebauer, E.; Rodriguez, F.; Nunez de Castro, I.; Vara-Thorbeck, C. Histamine synthesis and content in benign and malignant breast tumours. Its effects on other host tissues. Surg. Oncol. 1994, 3, 167–173. [Google Scholar] [CrossRef]
- Moriarty, C.M.; Stucky, J.L.; Hamburger, K.W.; Patil, K.D.; Foley, J.F.; Koefoot, R.R. Blood histamine and solid malignant tumors. J. Cancer Res. Clin. Oncol. 1988, 114, 588–592. [Google Scholar] [CrossRef]
- Graff, L.; Frungieri, M.; Zanner, R.; Pohlinger, A.; Prinz, C.; Gratzl, M. Expression of histidine decarboxylase and synthesis of histamine by human small cell lung carcinoma. Am. J. Pathol. 2002, 160, 1561–1565. [Google Scholar] [CrossRef]
- Stoyanov, E.; Uddin, M.; Mankuta, D.; Dubinett, S.M.; Levi-Schaffer, F. Mast cells and histamine enhance the proliferation of non-small cell lung cancer cells. Lung Cancer 2012, 75, 38–44. [Google Scholar] [CrossRef]
- Matsumoto, N.; Ebihara, M.; Oishi, S.; Fujimoto, Y.; Okada, T.; Imamura, T. Histamine H1 receptor antagonists selectively kill cisplatin-resistant human cancer cells. Sci. Rep. 2021, 11, 1492. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.; Hawkins, R.E. Cimetidine in colorectal cancer--are the effects immunological or adhesion-mediated? Br. J. Cancer 2002, 86, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Previati, M.; Raspadori, A.; Bertolaso, L.; Parmeggiani, A.; Bindini, D.; Vitali, C.; Lanzoni, I.; Corbacella, E.; Saviano, M.; Fagioli, F.; et al. Determination of histamine in the whole blood of colon cancer patients. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2002, 780, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Klapan, I.; Katic, V.; Culo, F.; Sabolovic, D.; Cuk, V.; Fumic, K.; Simovic, S. Lipid-bound sialic acid, prostaglandin E and histamine in head and neck cancer. Eur. J. Cancer 1993, 29A, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Cianchi, F.; Cortesini, C.; Schiavone, N.; Perna, F.; Magnelli, L.; Fanti, E.; Bani, D.; Messerini, L.; Fabbroni, V.; Perigli, G.; et al. The role of cyclooxygenase-2 in mediating the effects of histamine on cell proliferation and vascular endothelial growth factor production in colorectal cancer. Clin. Cancer Res. 2005, 11, 6807–6815. [Google Scholar] [CrossRef] [PubMed]
- Moya-Garcia, A.A.; Pino-Angeles, A.; Sanchez-Jimenez, F.; Urdiales, J.L.; Medina, M.A. Histamine, Metabolic Remodelling and Angiogenesis: A Systems Level Approach. Biomolecules 2021, 11, 415. [Google Scholar] [CrossRef]
- Medina, V.A.; Rivera, E.S. Histamine receptors and cancer pharmacology. Br. J. Pharmacol. 2010, 161, 755–767. [Google Scholar] [CrossRef]
- Garcia-Quiroz, J.; Camacho, J. Astemizole: An old anti-histamine as a new promising anti-cancer drug. Anticancer. Agents Med. Chem. 2011, 11, 307–314. [Google Scholar] [CrossRef]
- Medina, V.; Cricco, G.; Nunez, M.; Martin, G.; Mohamad, N.; Correa-Fiz, F.; Sanchez-Jimenez, F.; Bergoc, R.; Rivera, E.S. Histamine-mediated signaling processes in human malignant mammary cells. Cancer Biol. Ther. 2006, 5, 1462–1471. [Google Scholar] [CrossRef]
- Cricco, G.; Martin, G.; Labombarda, F.; Cocca, C.; Bergoc, R.; Rivera, E. Human pancreatic carcinoma cell line Panc-I and the role of histamine in growth regulation. Inflamm. Res. 2000, 49 (Suppl. S1), S68–S69. [Google Scholar] [CrossRef]
- Chen, J.; Hu, X.Y. Inhibition of histamine receptor H3R suppresses prostate cancer growth, invasion and increases apoptosis via the AR pathway. Oncol. Lett. 2018, 16, 4921–4928. [Google Scholar] [CrossRef] [PubMed]
- Bowrey, P.F.; King, J.; Magarey, C.; Schwartz, P.; Marr, P.; Bolton, E.; Morris, D.L. Histamine, mast cells and tumour cell proliferation in breast cancer: Does preoperative cimetidine administration have an effect? Br. J. Cancer 2000, 82, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Falus, A.; Gilicze, A. Tumor formation and antitumor immunity; the overlooked significance of histamine. J. Leukoc. Biol. 2014, 96, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, L.T.; Van Buul-Offers, S.C.; Gloudemans, T.; Roholl, P.J.; Sussenbach, J.S.; Den Otter, W. Histamine-stimulated expression of insulin-like growth factors in human glioma cells. Br. J. Cancer 1997, 75, 1091–1097. [Google Scholar] [CrossRef]
- Watson, S.A.; Wilkinson, L.J.; Robertson, J.F.; Hardcastle, J.D. Effect of histamine on the growth of human gastrointestinal tumours: Reversal by cimetidine. Gut 1993, 34, 1091–1096. [Google Scholar] [CrossRef]
- Park, C.; Lee, J.W.; Kim, K.; Seen, D.S.; Jeong, J.Y.; Huh, W.K. Simultaneous activation of CXC chemokine receptor 4 and histamine receptor H1 enhances calcium signaling and cancer cell migration. Sci. Rep. 2023, 13, 1894. [Google Scholar] [CrossRef]
- Davio, C.A.; Cricco, G.; Andrade, N.; Bergoc, R.; Rivera, E. H1 and H2 histamine receptors in human mammary carcinomas. Agents Actions 1993, 38, C172–C174. [Google Scholar] [CrossRef]
- LaBella, F.S.; Brandes, L.J. Interaction of histamine and other bioamines with cytochromes P450: Implications for cell growth modulation and chemopotentiation by drugs. Semin. Cancer Biol. 2000, 10, 47–53. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Xu, Q.; Liu, G.; Sun, N.; Che, H.; He, T. Apigenin Inhibits the Histamine-Induced Proliferation of Ovarian Cancer Cells by Downregulating ERalpha/ERbeta Expression. Front. Oncol. 2021, 11, 682917. [Google Scholar] [CrossRef]
- Garbuzenko, E.; Nagler, A.; Pickholtz, D.; Gillery, P.; Reich, R.; Maquart, F.X.; Levi-Schaffer, F. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: A direct role for mast cells in skin fibrosis. Clin. Exp. Allergy 2002, 32, 237–246. [Google Scholar] [CrossRef]
- Lazar-Molnar, E.; Hegyesi, H.; Pallinger, E.; Kovacs, P.; Toth, S.; Fitzsimons, C.; Cricco, G.; Martin, G.; Bergoc, R.; Darvas, Z.; et al. Inhibition of human primary melanoma cell proliferation by histamine is enhanced by interleukin-6. Eur. J. Clin. Invest. 2002, 32, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Fritz, I.; Wagner, P.; Olsson, H. Improved survival in several cancers with use of H1-antihistamines desloratadine and loratadine. Transl. Oncol. 2021, 14, 101029. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.F.; Lin, Y.W.; Chiu, W.K.; Lin, C.W.; Yang, Y.C.; Chang, L.C.; Chang, J.; Yang, S.F.; Chien, M.H. Combined impacts of histamine receptor H1 gene polymorphisms and an environmental carcinogen on the susceptibility to and progression of oral squamous cell carcinoma. Aging 2022, 14, 4500–4512. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, H.; Miyamoto, Y.; Terao, T.; Yoshida, H.; Kuroda, W.; Kitamura, Y.; Takeda, N.; Fukui, H. Signaling Pathway of Histamine H1 Receptor-Mediated Histamine H1 Receptor Gene Upregulation Induced by Histamine in U-373 MG Cells. Curr. Issues Mol. Biol. 2021, 43, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Mandola, A.; Nozawa, A.; Eiwegger, T. Histamine, histamine receptors, and anti-histamines in the context of allergic responses. LymphoSign J. 2019, 6, 35–51. [Google Scholar] [CrossRef]
- Travi, B.L. Current status of antihistamine drugs repurposing for infectious diseases. Med. Drug Discov. 2022, 15, 100140. [Google Scholar] [CrossRef]
- Kou, E.; Zhang, X.; Dong, B.; Wang, B.; Zhu, Y. Combination of H1 and H2 Histamine Receptor Antagonists: Current Knowledge and Perspectives of a Classic Treatment Strategy. Life 2024, 14, 164. [Google Scholar] [CrossRef]
- Criado, P.R.; Criado, R.F.; Maruta, C.W.; Machado Filho, C. Histamine, histamine receptors and antihistamines: New concepts. An. Bras. Dermatol. 2010, 85, 195–210. [Google Scholar] [CrossRef]
- Devillier, P.; Roche, N.; Faisy, C. Clinical pharmacokinetics and pharmacodynamics of desloratadine, fexofenadine and levocetirizine: A comparative review. Clin. Pharmacokinet. 2008, 47, 217–230. [Google Scholar] [CrossRef]
- Wu, R.L.; Anthes, J.C.; Kreutner, W.; Harris, A.G.; West, R.E., Jr. Desloratadine inhibits constitutive and histamine-stimulated nuclear factor-kappaB activity consistent with inverse agonism at the histamine H1 Receptor. Int. Arch. Allergy Immunol. 2004, 135, 313–318. [Google Scholar] [CrossRef]
- Wang, M.; Wei, X.; Shi, L.; Chen, B.; Zhao, G.; Yang, H. Integrative genomic analyses of the histamine H1 receptor and its role in cancer prediction. Int. J. Mol. Med. 2014, 33, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Yanai, K.; Yoshikawa, T.; Yanai, A.; Nakamura, T.; Iida, T.; Leurs, R.; Tashiro, M. The clinical pharmacology of non-sedating antihistamines. Pharmacol. Ther. 2017, 178, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, Q.; Wu, Z.; Li, M.; He, B.; Du, Y.; Zhang, K.; Tao, Y. Molecular mechanism of antihistamines recognition and regulation of the histamine H1 receptor. Nat. Commun. 2024, 15, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, R.; Peng, C.; Chen, X.; Li, J. Pharmacogenomics for the efficacy and side effects of antihistamines. Exp. Dermatol. 2022, 31, 993–1004. [Google Scholar] [CrossRef]
- Parisi, G.F.; Leonardi, S.; Ciprandi, G.; Corsico, A.; Licari, A.; Miraglia Del Giudice, M.; Peroni, D.; Salpietro, C.; Marseglia, G.L. Antihistamines in children and adolescents: A practical update. Allergol. Immunopathol. 2020, 48, 753–762. [Google Scholar] [CrossRef]
- Fein, M.N.; Fischer, D.A.; O’Keefe, A.W.; Sussman, G.L. CSACI position statement: Newer generation H1-antihistamines are safer than first-generation H1-antihistamines and should be the first-line antihistamines for the treatment of allergic rhinitis and urticaria. Allergy Asthma Clin. Immunol. 2019, 15, 61. [Google Scholar] [CrossRef]
- Farzam, K.; Sabir, S.; O’Rourke, M.C. Antihistamines; StatPearls Publishing LLC.: Tampa, FL, USA, 2024. [Google Scholar]
- Meltzer, E.O.; Rosario, N.A.; Van Bever, H.; Lucio, L. Correction: Fexofenadine: Review of safety, efficacy and unmet needs in children with allergic rhinitis. Allergy Asthma Clin. Immunol. 2022, 18, 112. [Google Scholar] [CrossRef]
- Ferrer, M.; Morais-Almeida, M.; Guizova, M.; Khanferyan, R. Evaluation of treatment satisfaction in children with allergic disease treated with an antihistamine: An international, non-interventional, retrospective study. Clin. Drug Investig. 2010, 30, 15–34. [Google Scholar] [CrossRef]
- Baniya, M.K.; Kim, E.H.; Chun, K.S. Terfenadine, a histamine H1 receptor antagonist, induces apoptosis by suppressing STAT3 signaling in human colorectal cancer HCT116 cells. Front. Pharmacol. 2024, 15, 1418266. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Akhter, J.; Adams, W.J.; Morris, D.L. Histamine content in colorectal cancer. Are there sufficient levels of histamine to affect lymphocyte function? Eur. J. Surg. Oncol. 1997, 23, 224–227. [Google Scholar] [CrossRef]
- Shi, Z.; Fultz, R.S.; Engevik, M.A.; Gao, C.; Hall, A.; Major, A.; Mori-Akiyama, Y.; Versalovic, J. Distinct roles of histamine H1- and H2-receptor signaling pathways in inflammation-associated colonic tumorigenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 16, G205–G216. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Qi, J.; Li, S.; Zhang, C.; Wang, H.; Shao, L.; Yuan, X.; Sha, Q. Desloratadine, a Novel Antigrowth Reagent for Bladder Cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820926591. [Google Scholar] [CrossRef] [PubMed]
- Verdoodt, F.; Pottegard, A.; Dehlendorff, C.; Jaattela, M.; Hallas, J.; Friis, S.; Kjaer, S.K. Antihistamine use and risk of ovarian cancer: A population-based case-control study. Maturitas 2019, 120, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.B.T.; Topalov, N.E.; Deuster, E.; Hysenaj, I.; Mayr, D.; Chelariu-Raicu, A.; Beyer, S.; Kolben, T.; Burges, A.; Mahner, S.; et al. Expression pattern and prognostic potential of histamine receptors in epithelial ovarian cancer. J. Cancer Res. Clin. Oncol. 2023, 149, 2501–2511. [Google Scholar] [CrossRef]
- Fernandez-Nogueira, P.; Noguera-Castells, A.; Fuster, G.; Recalde-Percaz, L.; Moragas, N.; Lopez-Plana, A.; Enreig, E.; Jauregui, P.; Carbo, N.; Almendro, V.; et al. Histamine receptor 1 inhibition enhances antitumor therapeutic responses through extracellular signal-regulated kinase (ERK) activation in breast cancer. Cancer Lett. 2018, 424, 70–83. [Google Scholar] [CrossRef]
- Sieja, K.; Stanosz, S.; von Mach-Szczypinski, J.; Olewniczak, S.; Stanosz, M. Concentration of histamine in serum and tissues of the primary ductal breast cancers in women. Breast 2005, 14, 236–241. [Google Scholar] [CrossRef]
- von Mach-Szczypinski, J.; Stanosz, S.; Sieja, K.; Stanosz, M. Metabolism of histamine in tissues of primary ductal breast cancer. Metabolism 2009, 58, 867–870. [Google Scholar] [CrossRef]
- Grimm, M.; Krimmel, M.; Alexander, D.; Munz, A.; Kluba, S.; Keutel, C.; Hoffmann, J.; Polligkeit, J.; Reinert, S.; Hoefert, S. Prognostic value of histamine H1 receptor expression in oral squamous cell carcinoma. Clin. Oral. Investig. 2013, 17, 949–955. [Google Scholar] [CrossRef]
- Francis, T.; Graf, A.; Hodges, K.; Kennedy, L.; Hargrove, L.; Price, M.; Kearney, K.; Francis, H. Histamine regulation of pancreatitis and pancreatic cancer: A review of recent findings. Hepatobiliary Surg. Nutr. 2013, 2, 216–226. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef]
- Chen, J.; Liu, G.; Wang, X.; Hong, H.; Li, T.; Li, L.; Wang, H.; Xie, J.; Li, B.; Li, T.; et al. Glioblastoma stem cell-specific histamine secretion drives pro-angiogenic tumor microenvironment remodeling. Cell Stem Cell 2022, 29, 1531–1546.E7. [Google Scholar] [CrossRef] [PubMed]
- Cornet-Masana, J.M.; Banus-Mulet, A.; Cuesta-Casanovas, L.; Carbo, J.M.; Guijarro, F.; Torrente, M.A.; Esteve, J.; Risueno, R.M. Histamine receptor 1 is expressed in leukaemic cells and affects differentiation sensitivity. J. Cell Mol. Med. 2020, 24, 13536–13541. [Google Scholar] [CrossRef] [PubMed]
- Davio, C.; Baldi, A.; Mladovan, A.; Cricco, G.; Fitzsimons, C.; Bergoc, R.; Rivera, E. Expression of histamine receptors in different cell lines derived from mammary gland and human breast carcinomas. Inflamm. Res. 1995, 44 (Suppl. S1), S70–S71. [Google Scholar] [CrossRef] [PubMed]
- Specht, T.; Seifert, R. Repurposing of H1-receptor antagonists (levo)cetirizine, (des)loratadine, and fexofenadine as a case study for systematic analysis of trials on clinicaltrials.gov using semi-automated processes with custom-coded software. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 2995–3018. [Google Scholar] [CrossRef]
- Laverdiere, I.; Boileau, M.; Neumann, A.L.; Frison, H.; Mitchell, A.; Ng, S.W.K.; Wang, J.C.Y.; Minden, M.D.; Eppert, K. Leukemic stem cell signatures identify novel therapeutics targeting acute myeloid leukemia. Blood Cancer J. 2018, 8, 52. [Google Scholar] [CrossRef]
- Jangi, S.M.; Ruiz-Larrea, M.B.; Nicolau-Galmes, F.; Andollo, N.; Arroyo-Berdugo, Y.; Ortega-Martinez, I.; Diaz-Perez, J.L.; Boyano, M.D. Terfenadine-induced apoptosis in human melanoma cells is mediated through Ca2+ homeostasis modulation and tyrosine kinase activity, independently of H1 histamine receptors. Carcinogenesis 2008, 29, 500–509. [Google Scholar] [CrossRef]
- Anand, A.; Liu, B.; Dicroce Giacobini, J.; Maeda, K.; Rohde, M.; Jaattela, M. Cell Death Induced by Cationic Amphiphilic Drugs Depends on Lysosomal Ca2+ Release and Cyclic AMP. Mol. Cancer Ther. 2019, 18, 1602–1614. [Google Scholar] [CrossRef]
- Liu, B.; Chen, R.; Zhang, Y.; Huang, J.; Luo, Y.; Rosthoj, S.; Zhao, C.; Jaattela, M. Cationic amphiphilic antihistamines inhibit STAT3 via Ca2+-dependent lysosomal H+ efflux. Cell Rep. 2023, 42, 112137. [Google Scholar] [CrossRef]
- Chen, J.S.; Lin, S.Y.; Tso, W.L.; Yeh, G.C.; Lee, W.S.; Tseng, H.; Chen, L.C.; Ho, Y.S. Checkpoint kinase 1-mediated phosphorylation of Cdc25C and bad proteins are involved in antitumor effects of loratadine-induced G2/M phase cell-cycle arrest and apoptosis. Mol. Carcinog. 2006, 45, 461–478. [Google Scholar] [CrossRef]
- Trybus, E.; Krol, T.; Trybus, W. The Multidirectional Effect of Azelastine Hydrochloride on Cervical Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5890. [Google Scholar] [CrossRef]
- Lin, J.C.; Ho, Y.S.; Lee, J.J.; Liu, C.L.; Yang, T.L.; Wu, C.H. Induction of apoptosis and cell-cycle arrest in human colon cancer cells by meclizine. Food Chem. Toxicol. 2007, 45, 935–944. [Google Scholar] [CrossRef] [PubMed]
- de Guadalupe Chavez-Lopez, M.; Hernandez-Gallegos, E.; Vazquez-Sanchez, A.Y.; Gariglio, P.; Camacho, J. Antiproliferative and proapoptotic effects of astemizole on cervical cancer cells. Int. J. Gynecol. Cancer 2014, 24, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Ramos, G.; Hernandez-Gallegos, E.; Vera, E.; Chavez-Lopez, M.G.; Zuniga-Garcia, V.; Sanchez-Perez, Y.; Garrido, E.; Camacho, J. Astemizole inhibits cell proliferation in human prostate tumorigenic cells expressing ether a-go-go-1 potassium channels. Cell. Mol. Biol. 2017, 63, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Yao, W.; Xiong, Y.; Li, J.; Liu, L.; Shi, L.; Zhang, W.; Zhang, C.; Nie, L.; Wan, J. Attenuated expression of HRH4 in colorectal carcinomas: A potential influence on tumor growth and progression. BMC Cancer 2011, 11, 195. [Google Scholar] [CrossRef]
- Chiang, C.H.; Chiang, C.H.; Peng, C.Y.; Hsia, Y.P.; See, X.Y.; Horng, C.S.; Chang, Y.C.; Shen, X.E.; Wang, S.S.; Tsai, T.C.; et al. Efficacy of cationic amphiphilic antihistamines on outcomes of patients treated with immune checkpoint inhibitors. Eur. J. Cancer 2022, 174, 1–9. [Google Scholar] [CrossRef]
- Fritz, I.; Wagner, P.; Broberg, P.; Einefors, R.; Olsson, H. Desloratadine and loratadine stand out among common H1-antihistamines for association with improved breast cancer survival. Acta Oncol. 2020, 59, 1103–1109. [Google Scholar] [CrossRef]
- Chen, S.; Luster, A.D. Antihistamines for cancer immunotherapy: More than just treating allergies. Cancer Cell 2022, 40, 9–11. [Google Scholar] [CrossRef]
- Hu, H.F.; Xu, W.W.; Li, Y.J.; He, Y.; Zhang, W.X.; Liao, L.; Zhang, Q.H.; Han, L.; Yin, X.F.; Zhao, X.X.; et al. Anti-allergic drug azelastine suppresses colon tumorigenesis by directly targeting ARF1 to inhibit IQGAP1-ERK-Drp1-mediated mitochondrial fission. Theranostics 2021, 11, 1828–1844. [Google Scholar] [CrossRef]
- Jangi, S.M.; Diaz-Perez, J.L.; Ochoa-Lizarralde, B.; Martin-Ruiz, I.; Asumendi, A.; Perez-Yarza, G.; Gardeazabal, J.; Diaz-Ramon, J.L.; Boyano, M.D. H1 histamine receptor antagonists induce genotoxic and caspase-2-dependent apoptosis in human melanoma cells. Carcinogenesis 2006, 27, 1787–1796. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, X.; Li, E.; Zhang, G.; Wang, X.; Tang, T.; Bai, X.; Liang, T. VISTA: An immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 83. [Google Scholar] [CrossRef]
- Hamid, O.; Hamidi, N. Enhancing immuno-oncology efficacy with H1-antihistamine in cancer therapy: A review of current research and findings. Curr. Med. Res. Opin. 2024, 40, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, P.; Geca, K.; Michalski, A.; Klosinska, M.; Kaczynska, A.; Polkowski, W.P.; Pelc, Z.; Skorzewska, M. Vista of the Future: Novel Immunotherapy Based on the Human V-Set Immunoregulatory Receptor for Digestive System Tumors. Int. J. Mol. Sci. 2023, 24, 9945. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; He, Y.; Tang, Y.; Chen, Z.S.; Qu, M. VISTA: A Novel Checkpoint for Cancer Immunotherapy. Drug Discov. Today 2024, 29, 104045. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lin, J.R.; Robitschek, E.J.; Kasumova, G.G.; Heyde, A.; Shi, A.; Kraya, A.; Zhang, G.; Moll, T.; Frederick, D.T.; et al. Evolution of delayed resistance to immunotherapy in a melanoma responder. Nat. Med. 2021, 27, 985–992. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Kugelmann, D.; Rotkopf, L.T.; Radeva, M.Y.; Garcia-Ponce, A.; Walter, E.; Waschke, J. Histamine causes endothelial barrier disruption via Ca2+-mediated RhoA activation and tension at adherens junctions. Sci. Rep. 2018, 8, 13229. [Google Scholar] [CrossRef]
- Fritz, I.; Wagner, P.; Bottai, M.; Eriksson, H.; Ingvar, C.; Krakowski, I.; Nielsen, K.; Olsson, H. Desloratadine and loratadine use associated with improved melanoma survival. Allergy 2020, 75, 2096–2099. [Google Scholar] [CrossRef]
- Liu, X.; Zhong, R.; Huang, J.; Chen, Z.; Xu, H.; Lin, L.; Cai, Q.; He, M.; Lao, S.; Deng, H.; et al. Loratidine is associated with improved prognosis and exerts antineoplastic effects via apoptotic and pyroptotic crosstalk in lung cancer. J. Exp. Clin. Cancer Res. 2024, 43, 5. [Google Scholar] [CrossRef]
- Kalpaklioglu, F.; Baccioglu, A. Efficacy and safety of H1-antihistamines: An update. Antiinflamm Antiallergy Agents Med. Chem. 2012, 11, 230–237. [Google Scholar] [CrossRef]
- Dobbeling, U.; Waeckerle-Men, Y.; Zabel, F.; Graf, N.; Kundig, T.M.; Johansen, P. The antihistamines clemastine and desloratadine inhibit STAT3 and c-Myc activities and induce apoptosis in cutaneous T-cell lymphoma cell lines. Exp. Dermatol. 2013, 22, 119–124. [Google Scholar] [CrossRef]
- Hadzijusufovic, E.; Peter, B.; Gleixner, K.V.; Schuch, K.; Pickl, W.F.; Thaiwong, T.; Yuzbasiyan-Gurkan, V.; Mirkina, I.; Willmann, M.; Valent, P. H1-receptor antagonists terfenadine and loratadine inhibit spontaneous growth of neoplastic mast cells. Exp. Hematol. 2010, 38, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Olasinska-Wisniewska, A.; Olasinski, J.; Grajek, S. Cardiovascular safety of antihistamines. Postepy Dermatol. Alergol. 2014, 31, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Rosania, G.R.; Horobin, R.W.; Kornhuber, J. Quantitative modeling of selective lysosomal targeting for drug design. Eur. Biophys. J. 2008, 37, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef]
- Joris, F.; De Backer, L.; Van de Vyver, T.; Bastiancich, C.; De Smedt, S.C.; Raemdonck, K. Repurposing cationic amphiphilic drugs as adjuvants to induce lysosomal siRNA escape in nanogel transfected cells. J. Control Release 2018, 269, 266–276. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Toprak, M.; Noory, M.A.; Robertson, G.P. Effect of lysosomotropic molecules on cellular homeostasis. Pharmacol. Res. 2017, 117, 177–184. [Google Scholar] [CrossRef]
- Kolzer, M.; Werth, N.; Sandhoff, K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004, 559, 96–98. [Google Scholar] [CrossRef]
- Repnik, U.; Hafner Cesen, M.; Turk, B. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion 2014, 19 Pt A, 49–57. [Google Scholar] [CrossRef]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem. Soc. Trans. 2018, 46, 207–215. [Google Scholar] [CrossRef]
- Chanas-Larue, A.; Villalpando-Rodriguez, G.E.; Henson, E.S.; Johnston, J.B.; Gibson, S.B. Antihistamines are synergistic with Bruton’s tyrosine kinase inhibiter ibrutinib mediated by lysosome disruption in chronic lymphocytic leukemia (CLL) cells. Leuk. Res. 2020, 96, 106423. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Barri, M.; Atakpa-Adaji, P.; Taylor, C.W.; St John Smith, E.; Murrell-Lagnado, R.D. P2X4 Receptors Mediate Ca2+ Release from Lysosomes in Response to Stimulation of P2X7 and H1 Histamine Receptors. Int. J. Mol. Sci. 2021, 22, 10492. [Google Scholar] [CrossRef] [PubMed]
- Kanellopoulos, J.M.; Almeida-da-Silva, C.L.C.; Ruutel Boudinot, S.; Ojcius, D.M. Structural and Functional Features of the P2X4 Receptor: An Immunological Perspective. Front. Immunol. 2021, 12, 645834. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, I.O.; Groth-Pedersen, L.; Dicroce-Giacobini, J.; Jonassen, A.S.H.; Mortensen, M.; Bilgin, M.; Schmiegelow, K.; Jaattela, M.; Maeda, K. Cationic amphiphilic drugs induce elevation in lysoglycerophospholipid levels and cell death in leukemia cells. Metabolomics 2020, 16, 91. [Google Scholar] [CrossRef]
- Pegram, M.; Jackisch, C.; Johnston, S.R.D. Estrogen/HER2 receptor crosstalk in breast cancer: Combination therapies to improve outcomes for patients with hormone receptor-positive/HER2-positive breast cancer. NPJ Breast Cancer 2023, 9, 45. [Google Scholar] [CrossRef]
- Jangi, S.M.; Asumendi, A.; Arlucea, J.; Nieto, N.; Perez-Yarza, G.; Morales, M.C.; de la Fuente-Pinedo, M.; Boyano, M.D. Apoptosis of human T-cell acute lymphoblastic leukemia cells by diphenhydramine, an H1 histamine receptor antagonist. Oncol. Res. 2004, 14, 363–372. [Google Scholar] [CrossRef]
- Bhat, M.A.; Roy, S.; Dhaneshwar, S.; Kumar, S.; Saxena, S.K. Desloratadine via its anti-inflammatory and antioxidative properties ameliorates TNBS-induced experimental colitis in rats. Immunopharmacol. Immunotoxicol. 2024, 46, 436–449. [Google Scholar] [CrossRef]
- Cassano, N.; Raho, G.; Filieri, M.; D’Argento, V.; Amoruso, A.; Filotico, R.; Vena, G.A. Influence of desloratadine on oxidative stress markers in patients with chronic idiopathic urticaria. Int. J. Dermatol. 2006, 45, 394–396. [Google Scholar] [CrossRef]
- Caron, G.; Delneste, Y.; Roelandts, E.; Duez, C.; Bonnefoy, J.Y.; Pestel, J.; Jeannin, P. Histamine polarizes human dendritic cells into Th2 cell-promoting effector dendritic cells. J. Immunol. 2001, 167, 3682–3686. [Google Scholar] [CrossRef]
- McIlroy, A.; Caron, G.; Blanchard, S.; Fremaux, I.; Duluc, D.; Delneste, Y.; Chevailler, A.; Jeannin, P. Histamine and prostaglandin E up-regulate the production of Th2-attracting chemokines (CCL17 and CCL22) and down-regulate IFN-gamma-induced CXCL10 production by immature human dendritic cells. Immunology 2006, 117, 507–516. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| ACTION OF H1 ANTIHISTAMINES IN CANCER | ||||
|---|---|---|---|---|
| Induction of LMP and Lysosomal Cell Death | Antiproliferative Effect | Increasing Anticancer Immunity. Sensitization to Immunotherapy | Stimulation of Apoptosis | Induction of DNA Damage |
| Astemizole [37,181,182] Clemastine [37] Ebastine [37,181,182] Loratadine [37] Desloratadine [37] Terfenadine [34,37,182] | Loratadine [183] Desloratadine [166] Azelastine [184] Meclizine [185] Terfenadine [169] Astemizole [186,187] Cyproheptadine [188] | Ebastine [189,190] Loratadine [145,190] Fexofenadine [71,191] Desloratadine [145,189,190] Cyproheptadine [189] | Cloperastine [124] Azelastine [184,192] Desloratadine [166] Terfenadine [169,180,193] Loratadine [183] Meclizine [185] Astemizole [180,186] Triploidine [193] Diphenhydramine [193] Astemizole [193] | Terfenadine [193] Triploidine [193] Astemizole [193] Diphenhydramine [193] Loratadine [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trybus, E.; Trybus, W. H1 Antihistamines—Promising Candidates for Repurposing in the Context of the Development of New Therapeutic Approaches to Cancer Treatment. Cancers 2024, 16, 4253. https://doi.org/10.3390/cancers16244253
Trybus E, Trybus W. H1 Antihistamines—Promising Candidates for Repurposing in the Context of the Development of New Therapeutic Approaches to Cancer Treatment. Cancers. 2024; 16(24):4253. https://doi.org/10.3390/cancers16244253
Chicago/Turabian StyleTrybus, Ewa, and Wojciech Trybus. 2024. "H1 Antihistamines—Promising Candidates for Repurposing in the Context of the Development of New Therapeutic Approaches to Cancer Treatment" Cancers 16, no. 24: 4253. https://doi.org/10.3390/cancers16244253
APA StyleTrybus, E., & Trybus, W. (2024). H1 Antihistamines—Promising Candidates for Repurposing in the Context of the Development of New Therapeutic Approaches to Cancer Treatment. Cancers, 16(24), 4253. https://doi.org/10.3390/cancers16244253