
Stem Cell Origin of Cancer: Clinical Implications beyond Immunotherapy for Drug versus Therapy Development in Cancer Care

 , and
, and
Abstract
:Simple Summary
Abstract
Science is the belief in the ignorance of experts.Richard Feynman.
1. Introduction
2. Brief History
3. Immune Privilege
3.1. Does Anti-PD1/L1 Provide Immediate Clinical Efficacy (e.g., within 7 Days)?
3.2. Does a Brief or Bolus Anti-PD1/L1 Treatment Provide Durable If Not Permanent Clinical Benefits?
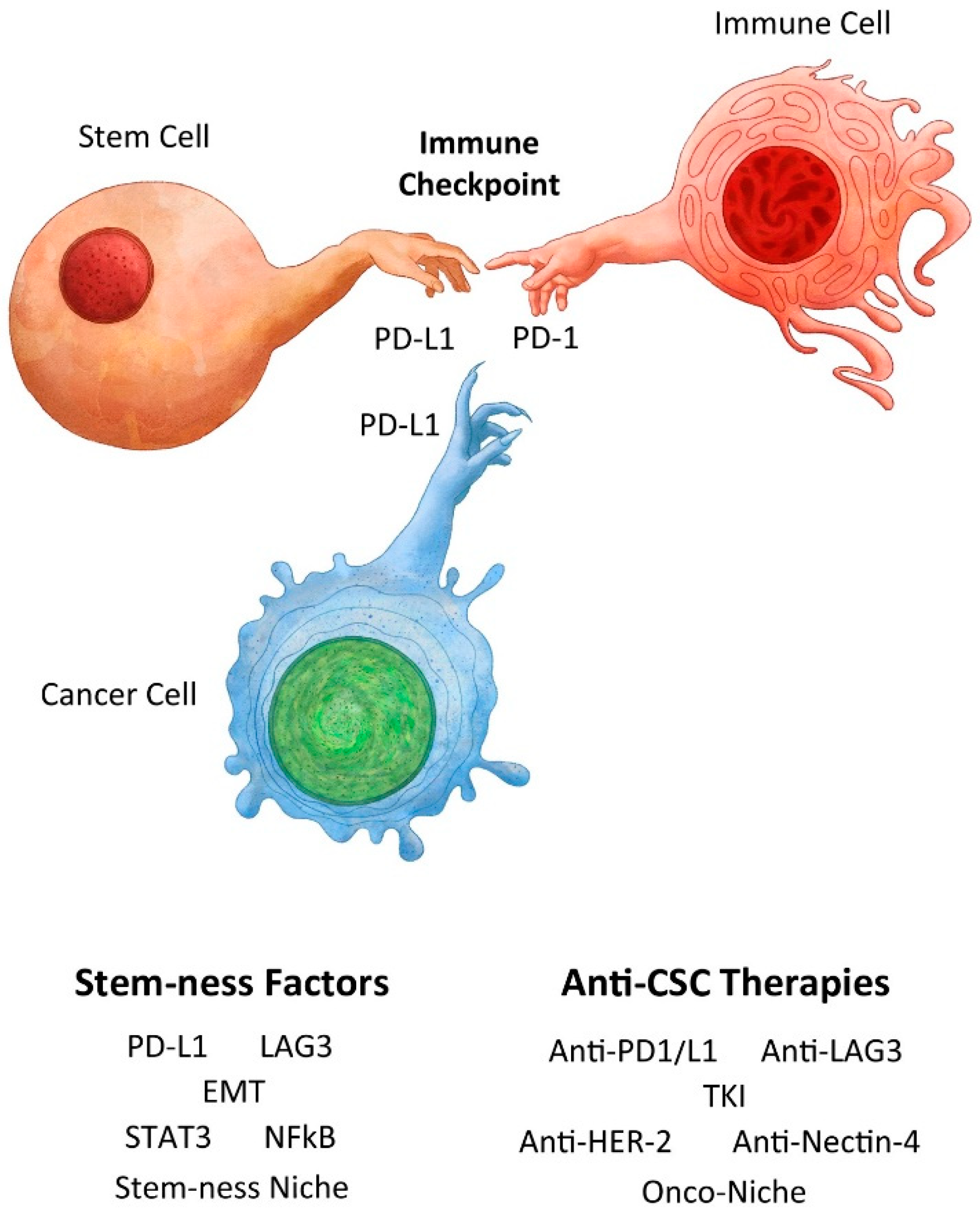
4. A Tale of Two Checkpoint Inhibitors
4.1. PD1/L1
4.2. LAG3
5. Anti-Cancer and/or Immune Modulation
6. Immune Activation/Modulation
6.1. Animal Models
6.2. Pregnancy
6.3. Clinical Trials
6.3.1. IDO1
6.3.2. TIGIT
6.3.3. IL-2
7. Checkpoint Inhibitors Are Not Equal
7.1. CTLA-4
7.2. Melanoma-Advanced
7.3. Melanoma-Adjuvant
7.4. SCLC-ED
7.5. NSCLC-Metastatic
7.6. NSCLC-Locally Advanced
7.7. Additional Malignancies
8. Tumor Subtypes and Phenotypes
8.1. Melanoma
8.2. Renal Cell Carcinoma (RCC)
8.3. Bladder Cancer
8.4. Multiple Myeloma
8.5. Prostate Cancer
9. Beyond Immune Modulation
10. Drug vs. Therapy Development
11. Combination Strategies
11.1. Anti-PD1/PDL-1 Combined with Immunomodulatory Therapy
11.1.1. Advanced
11.1.2. Neoadjuvant
11.2. Anti-PD1/PDL-1 Combined with Anti-Cancer Therapy
11.2.1. Extensive Disease
11.2.2. Unresectable, Stage III
11.2.3. Metastatic
11.2.4. Neoadjuvant, Stage II/III
11.2.5. Metastatic/Unresectable
11.3. Anti-PD1/PDL-1 Combined with Anti-CSC Agents
11.3.1. BRAF+
11.3.2. HER2+
11.3.3. Nectin4+
11.4. Anti-PD1/PDL-1 Combined with TKI (Including Anti-VEGFR)
11.4.1. Renal Cell Carcinoma
11.4.2. Hepatocellular Carcinoma
11.5. Anti-PD1/PDL-1 Combined with Anti-Metabolic Agents
11.6. Anti-PD1/PDL-1 Combined with Anti-Inflammatory Agents
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ebbel, B. The Papyrus Ebers: The Greatest Egyptian Medical Documents; Oxford University Press: London, UK, 1937. [Google Scholar]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas, with a report of ten original cases. Am. J. Med. Sci. 1893, 105, 487–511. [Google Scholar] [CrossRef]
- Richardson, M.A.; Ramirez, T.; Russell, N.C.; Moye, L.A. Coley toxins immunotherapy: A retrospective review. Altern. Ther. Health Med. 1999, 5, 42–47. [Google Scholar]
- Morales, A.; Eidinger, D.; Bruce, A.W. Intracavitary bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J. Urol. 1976, 116, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with Sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Aydin, A.M.; Maraboyina, S.; Chen, Z.; Singh, S.; Gokden, N.; Langford, T. Stem cell origin of cancer: Clinical implications for cancer immunity and immunotherapy. Cancers 2023, 15, 5385. [Google Scholar] [CrossRef]
- Hodi, F.S.; Hwu, W.J.; Kefford, R.; Weber, J.S.; Daud, A.; Hamid, O.; Patnaik, A.; Ribas, A.; Robert, C.; Gandadhar, T.C.; et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J. Clin. Oncol. 2016, 34, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.J.; Mesquita, A. Complete and long-lasting response to immunotherapy: A case report of urothelial cancer. Medicine 2022, 101, e28940. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumor DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Waterhouse, D.M.; Garon, E.B.; Chandler, J.; McLeod, M.; Hussein, M.A.; Jotte, R.; Horn, L.; Daniel, D.B.; Keogh, G.; Creelan, B.; et al. Continuous versus 1-year fixed duration nivolumab in previously treated advanced non-small cell lung cancer: CheckMate 153. J. Clin. Oncol. 2020, 38, 3863–3873. [Google Scholar] [CrossRef]
- Jansen, Y.J.L.; Rozeman, E.A.; Mason, R.; Goldinger, S.M.; Foppen, M.H.G.; Hoejberg, L.; Schmidt, H.; van Thienen, J.V.; Haanen, J.B.A.G.; Tiainen, L.; et al. Discontinuation of anti-PD-1 antibody therapy in the absence of disease progression or treatment limiting toxicity; clinical outcomes in advanced melanoma. Ann. Oncol. 2019, 30, 1154–1161. [Google Scholar] [CrossRef]
- Yadollahi, P.; Jeon, Y.K.; Ng, W.L.; Choi, I. Current understanding of cancer-intrinsic PD-L1: Regulation of expression and its protumoral activity. BMB Rep. 2021, 54, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.; Maijenburg, M.M.W.; Li, Y.; Gao, L.; Hadland, B.K.; Gao, P.; Minoura, K.; Bernstein, I.D.; Tan, K.; Speck, N.A. Maturation of hematopoietic stem cells from prehematopoietic stem cells is accompanied by upregulation of PD-L1. J. Exp. Med. 2018, 215, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Eljaafari, A.; Pestel, J.; Magueresse-Battistoni, B.L.; Chanon, S.; Watson, J.; Robert, M.; Disse, E.; Vidal, H. Adipose-tissue-derived mesenchymal stem cells mediate PD-L1 overexpression in the white adipose tissue of obese individuals, resulting in T cell dysfunction. Cells 2021, 10, 2645. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Chen, P.Y.; Yang, Y.P.; Zheng, M.Y.; Miao, C.C.; Wen, K.C.; Chang, K.M.; Chou, S.J.; Wang, M.L.; Chiou, S.H.; et al. Cytokine and epigenetic regulation of programmed death-ligand 1 in stem cell differentiation and cancer cell plasticity. Stem Cells 2021, 39, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Huang, F.; Wei, W.; Zhou, Y.; Ye, Z.; Yu, L.; Hu, J.; Cai, C. Programmed cell death ligand 1 is enriched in mammary stem cells and promotes mammary development and regeneration. Front. Cell Dev. Biol. 2021, 9, 772669. [Google Scholar] [CrossRef]
- Palmer, J.W.; Villavicencio, K.M.; Idris, M.; Weddle, D.; Filipp, F.V.; NISC Comparative Sequencing Program; Pavan, W.J.; Harris, M.L. The relationship between PD-L1 and quiescence in melanocyte stem cell aging. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef]
- Rong, Z.; Wang, M.; Hu, Z.; Stradner, M.; Zhu, S.; Kong, H.; Yi, H.; Goldrath, A.; Yang, Y.G.; Xu, Y.; et al. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell 2014, 14, 121–130. [Google Scholar] [CrossRef]
- Liu, D.; Lu, Q.; Wang, X.; Wang, J.; Lu, N.; Jiang, Z.; Hao, X.; Li, J.; Liu, J.; Cao, P.; et al. LSECtin on tumor-associated macrophages enhances breast cancer stemness via interaction with its receptor BTN3A3. Cell Res. 2019, 29, 365–378. [Google Scholar] [CrossRef]
- Ilmer, M.; Mazurek, N.; Gilcrease, M.Z.; Byrd, J.C.; Woodward, W.A.; Buchholz, T.A.; Acklin, K.; Ramirez, K.; Hafley, M.; Alt, E.; et al. Low pression of galectin-3 is associated with poor survival in node-positive breast cancers and mesenchymal phenotype in breast cancer stem cells. Breast Cancer Res. 2016, 18, 97. [Google Scholar] [CrossRef]
- Bie, F.; Wang, G.; Qu, X.; Wang, Y.; Huang, C.; Wang, Y.; Du, J. Loss of FGL1 induces epithelial-mesenchymal transition and angiogenesis in LKB1 mutant lung adenocarcinoma. Int. J. Oncol. 2019, 55, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Lin, H.M.; Zhu, K.; Cao, Y.; Xu, X.L.; Zhou, Z.Y.; Xu, L.B.; Liu, C.; Zhang, R. Immune checkpoint FGL1 expression of circulating tumor cells is associated with poor survival in curatively resected hepatocellular carcinoma. Front. Oncol. 2022, 12, 810269. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutierrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. NEJM 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Ahn, J.H.; Lee, B.H.; Kim, S.E.; Kwon, B.E.; Jeong, H.; Choi, J.P.; Kim, M.J.; Park, Y.; Kim, B.S.; Kim, D.H.; et al. A novel anti-PDL1 antibody exhibits antitumor effects on myeloma in murine models via antibody-dependent cellular cytotoxicity. Biomol. Ther. 2021, 29, 166–174. [Google Scholar] [CrossRef]
- Miyazaki, T.; Dierich, A.; Benoist, C.; Mathis, D. Independent modes of natural killing distinguished in mice lacking Lag3. Science 1996, 272, 405–408. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Horak, I. Immunodeficiency in IL-2-knock-out mice. Clin. Immunol. Immunopathol. 1995, 76, S172–S173. [Google Scholar] [CrossRef]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 253–261. [Google Scholar] [CrossRef]
- Andrikopoulou, A.; Korakiti, A.M.; Apostolidou, K.; Dimopoulos, M.A.; Zagouri, F. Immune checkpoint inhibitor administration during pregnancy: A case series. ESMO Open 2021, 6, 100262. [Google Scholar] [CrossRef]
- Mittra, A.; Naqash, A.R.; Murray, J.H.; Finnigan, S.; Kwak-Kim, J.; Ivy, S.P.; Chen, A.P.; Sharon, E. Outcomes of pregnancy during immunotherapy treatment for cancer: Analysis of clinical trials sponsored by the National Cancer Institute. Oncologist 2021, 26, e1883–e1886. [Google Scholar] [CrossRef] [PubMed]
- Salehi, I.; Porto, L.; Elser, C.; Singh, J.; Saibil, S.; Maxwell, C. Immune checkpoint inhibitor exposure in pregnancy: A scoping review. J. Immunother. 2002, 45, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Abruzzese, E. Chronic myeloid leukemia and pregnancy. In Proceedings of the 11th Annual Meeting of the Society of Hematologic Oncology (SOHO 2023), Houston, TX, USA, 8 September 2023. [Google Scholar]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-term outcomes with nivolumab plus ipilimuamb or nivollumab alone versus ipilimumab in patients with advanced melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczesna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Ozguroglu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomized, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive stage small-cell lung cancer (CASPIAN): Updated results from a randomized, controlled, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 51–65. [Google Scholar] [CrossRef]
- Rudin, C.M.; Liu, S.V.; Soo, R.A.; Lu, S.; Hong, M.H.; Lee, J.S.; Bryl, M.; Dumoulin, D.W.; Rittmeyer, A.; Chiu, C.H.; et al. SKYSCRAPER-02: Tiragolumab in combination with atezolumab plus chemotherapy in untreatment extensive-stage small-cell lung cancer. J. Clin. Oncol. 2024, 42, 324–335. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Kim, S.W.; Ursol, G.; Hussein, M.; Lim, F.L.; et al. Durvalumab with or without tremelimumab in combination with chemotherapy as first-line therapy for metastatic non-small cell lung cancer: The phase III POSEIDON study. J. Clin. Oncol. 2023, 41, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Senler, F.C.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.J.; van den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: The MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Boyer, M.; Sendur, M.A.N.; Rodriguez-Abreu, D.; Park, K.; Lee, D.H.; Cicin, I.; Yumuk, P.F.; Orlandi, F.J.; Leal, T.A.; Molinier, O.; et al. Pembrolizumab plus ipilimumab or placebo for metastatic non-small cell lung cancer with PDL1 tumor proportion score >50: Randomized, double-blind phase III KEYNOTE-598 study. J. Clin. Oncol. 2021, 39, 2327–2338. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Wright, G.H.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.B.; Im, S.A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): A phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Huober, J.; Barrios, C.H.; Niikura, N.; Jarzab, M.; Chang, Y.C.; Huggins-Puhalla, S.L.; Graupner, V.; Eiger, D.; Henschel, V.; Gochitashvili, N.; et al. VP6-2021: IMpassion050: A phase III study of neoadjuvant atezolizumab + pertuzumab + trastuzumab + chemotherapy in high-risk, HER2-positive early breast Cancer. Ann. Oncol. 2021, 32, 1061–1062. [Google Scholar] [CrossRef]
- Plimack, E.R.; Powles, T.; Stus, V.; Gafanov, R.; Nosov, D.; Waddell, T.; Alekseev, B.; Pouliot, F.; Melichar, B.; Soulieres, D.; et al. Pembrolizumab plus axitinib versus sunitinib as first-line treatment of advanced renal cell carcinoma: 43-month follow-up of the phase 3 KEYNOTE-426 study. Eur. Urol. 2023, 84, 449–454. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Eto, M.; Motzer, R.; De Giorgi, U.; Buchler, T.; Basappa, N.S.; Mendez-Vidal, M.J.; Tjulandin, S.; Park, S.H.; Melichar, B.; et al. Lenvatinib plus pembrolizumab versus sunitinib as first-line treatment of patients with advanced renal cell carcinoma (CLEAR): Extended follow-up from the phase 3, randomized, open-label study. Lancet Oncol. 2023, 24, 228–238. [Google Scholar] [CrossRef]
- Burotto, M.; Powles, T.; Escudier, B.; Apolo, A.B.; Bourlon, M.T.; Shah, A.Y.; Suarez, C.; Porta, C.; Barrios, C.H.; Richardet, M.; et al. Nivolumab plus cabozantinib vs sunitinib for first-line treatment of advanced renal cell carcinoma: 3-year follow-up from the phase 3 CheckMate 9ER trial. J. Clin. Oncol. 2023, 41 (Suppl. S6), abstr 603. [Google Scholar] [CrossRef]
- Motzer, R.J.; McDermott, D.F.; Escudier, B.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; Barthelemy, P.; Plimack, E.R.; Porta, C.; George, S.; et al. Conditional survival and long-term efficacy with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma. Cancer 2022, 128, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Powles, T.; Albiges, L.; Burotto, M.; Szczylik, C.; Zurawski, B.; Ruiz, E.Y.; Maruzzo, M.; Zaizar, A.S.; Fein, L.E.; et al. Cabozantinib plus nivolumab and ipilimumab in renal cell carcinoma. N. Engl. J. Med. 2023, 388, 1767–1778. [Google Scholar] [CrossRef]
- Tannir, N.; Formiga, M.N.; Agarwal, N.; Pal, S.K.; Cho, D.; George, D.J.; Hong, W.; Tang, L.; Qureshi, A.; Tagliaferri, M.A.; et al. LBA68—Bempegaldesleukin plus nivolumab compared to investigator’s choice of sunitinib or cabozantinib in previously untreated advanced renal cell carcinoma: Results from a phase III randomized study (PIVOT-09). Ann. Oncol. 2022, 33 (Suppl. S7), S808–S869. [Google Scholar] [CrossRef]
- Santoni, M.; Buti, S.; Myint, Z.W.; Maruzzo, M.; Iacovelli, R.; Pichler, M.; Kopecky, J.; Kucharz, J.; Rizzo, M.; Galli, L.; et al. Real-world outcome of patients with advanced renal cell carcinoma and intermediate- or poor-risk international metastatic renal cell carcinoma database consortium criteria treated by immune-oncology combinations: Differential effectiveness by risk group? Eur. Urol. Oncol. 2024, 7, 102–111. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Dao, T.V.; De Toni, E.N.; et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022, 1, 8. [Google Scholar] [CrossRef]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lievre, A.; et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: Interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet 2023, 402, 2197–2208. [Google Scholar] [CrossRef]
- Sun, J.M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.P.; Li, Z.; Kim, S.B.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): A randomised, placebo-controlled, phase 3 study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef]
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.H.; Adenis, A.; et al. Nivolumab combination therapy in advanced esophageal squamous-cell carcinoma. N. Engl. J. Med. 2022, 386, 449–462. [Google Scholar] [CrossRef]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Lee, M.A.; Kitano, M.; et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Haddad, R.; Even, C.; Tahara, M.; Dvorkin, M.; Ciuleanu, T.E.; Clement, P.M.; Mesia, R.; Kutukova, S.; Zholudeva, L.; et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann. Oncol. 2020, 31, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Even, C.; Mesia, R.; Remenar, E.; Daste, A.; Delord, J.P.; Krauss, J.; Sab, N.F.; Nabell, L.; Ready, N.E.; et al. Safety and efficacy of durvalumab with or without tremelimumab in patients with PD-L1-low/negative recurrent or metastatic HNSCC: The phase 2 CONDOR randomized clinical trial. JAMA Oncol. 2019, 5, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Ferris, R.L.; Gillison, M.; Tahara, M.; Argiris, A.; Fayette, J.; Schenker, M.; Bratland, A.; Walker, J.W.T.; Grell, P.; et al. Efficacy and safety of nivolumab plus ipilimumab vs nivolumab alone for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck: The phase 2 CheckMate 714 randomized Clinical trial. JAMA Oncol. 2023, 9, 779–789. [Google Scholar] [CrossRef]
- Powles, T.; van der Heijden, M.S.; Castellano, D.; Galsky, M.D.; Loriot, Y.; Petrylak, D.P.; Ogawa, O.; Park, S.H.; Lee, J.L.; De Giorgi, U.; et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1574–1588. [Google Scholar] [CrossRef]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J., Jr.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized phase II trial of nivolumab and ipilimumab for recurrent or persistent ovarian cancer: An NRG Oncology study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef]
- Hayakawa, T.; Yaguchi, T.; Kawakami, Y. Enhanced anti-tumor effects of anti-PD1 blockade combined with a highly absorptive form of curcumin targeting STAT3. Cancer Sci. 2020, 111, 4326–4335. [Google Scholar] [CrossRef]
- Kooshkaki, O.; Derakhshani, A.; Hosseinkhani, N.; Torabi, M.; Safaei, S.; Brunetti, O.; Racanelli, V.; Silvestris, N.; Baradaran, B. Combination of ipilimumab and nivolumab in cancers: From clinical practice to ongoing clinical trials. Int. J. Mol. Sci. 2020, 21, 4427. [Google Scholar] [CrossRef]
- Serritella, A.V.; Shenoy, N.K. Nivolumab plus ipilimumab vs nivolumab alone in advanced cancers other than melanoma: A meta-analysis. JAMA Oncol. 2023, 9, 1441–1446. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Knight, A.; Karapetyan, L.; Kirkwood, J.M. Immunotherapy in melanoma: Recent advances and future directions. Cancers 2023, 15, 1106. [Google Scholar] [CrossRef]
- Boutros, A.; Tanda, E.T.; Croce, E.; Catalano, F.; Ceppi, M.; Bruzzone, M.; Cecchi, F.; Arecco, L.; Fraguglia, M.; Pronzato, P.; et al. Activity and safety of first-line treatments for advanced melanoma: A network meta-analysis. Eur. J. Cancer 2023, 188, 64–79. [Google Scholar] [CrossRef]
- Atkins, M.B.; Abu-Sbeih, H.; Ascierto, P.A.; Bishop, M.R.; Chen, D.S.; Dhodapkar, M.; Emens, L.A.; Ernstoff, M.S.; Ferris, R.L.; Greten, T.F.; et al. Maximizing the value of phase III trials in immune-oncology: A checklist from the society for immunotherapy of cancer (SITC). J. Immunother. Cancer 2022, 10, e005413. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616. [Google Scholar] [CrossRef]
- Zimmer, L.; Livingstone, E.; Hassel, J.C.; Fluck, M.; Eigentler, T.; Loquai, C.; Haferkamp, S.; Gutzmer, R.; Meier, F.; Mohr, P. Adjuvant nivolumab plus ipilimumab or nivolumab monotherapy versus placebo in patients with resected stage IV melanoma with no evidence of disease (IMMUNED): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 395, 1558–1568. [Google Scholar] [CrossRef]
- Long, G.V.; Schadendorf, D.; Vecchio, M.D.; Larkin, J.; Atkinson, V.; Schenker, M.; Pigozzo, J.; Gogas, H.J.; Dalle, S.; Meyer, N. Abstract CT004: Adjuvant therapy with nivolumab combined with ipilimumab vs nivolumab alone in patients with resected stage IIIB-D/IV melanoma (CheckMate 915). Cancer Res. 2021, 81, CT004. [Google Scholar] [CrossRef]
- Owonikoko, T.; Park, K.; Govindan, R.; Ready, N.; Reck, M.; Peters, S.; Dakhil, S.R.; Navarro, A.; Rodriguez-Cid, J.; Schenker, M.; et al. Nivolumab and ipilimumab as maintenance therapy in extensive-disease small-cell lung cancer: CheckMate 451. J. Clin. Oncol. 2021, 39, 1349–1359. [Google Scholar] [CrossRef]
- Remon, J.; Collazo, A.; Jimenez, B. CheckMate 227 trial has not checked the immune-strategy in first-line setting in advanced non-small cell lung cancer. Transl. Cancer Res. 2020, 9, 2168–2170. [Google Scholar] [CrossRef]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef]
- Cascone, T.; Leung, C.H.; Weissferdt, A.; Pataer, A.; Carter, B.W.; Godoy, M.C.B.; Feldman, H.; William, W.N., Jr.; Xi, Y.; Basu, S.; et al. Neoadjuvant chemotherapy plus nivolumab with or without ipilimumab in operable non-small cell lung cancer: The phase 2 platform NEOSTR trial. Nat. Med. 2023, 29, 593–604. [Google Scholar] [CrossRef]
- O’Byrne, K.; Popoff, E.; Badin, F.; Lee, A.; Yuan, Y.; Lozano-Ortega, G.; Eccles, L.J.; Varol, N.; Waser, N.; Penrod, J.R.; et al. Long-term comparative efficacy and safety of nivolumab plus ipilimumab relative to other first-line therapies for advanced non-small cell lung cancer: A systemic literature review and network meta-analysis. Lung Cancer 2023, 177, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Arru, C.; De Miglio, M.R.; Cossu, A.; Muroni, M.R.; Carru, C.; Zinellu, A.; Paliogiannis, P. Durvalumab plus tremelimumab in solid tumors: A systematic review. Adv. Ther. 2021, 38, 3674–3693. [Google Scholar] [CrossRef] [PubMed]
- Rabbie, R.; Ferguson, P.; Molina-Aguilar, C.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2019, 247, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringner, M.; Lillehaug, J.R.; Borg, A.; Lonning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef]
- Harbst, K.; Staaf, J.; Lauss, M.; Karlsson, A.; Masback, A.; Johansson, I.; Bendahl, P.O.; Vallon-Christersson, J.; Torngren, T.; Ekedahl, H.; et al. Molecular profiling reveals low- and high-grade forms of primary melanoma. Clin. Cancer Res. 2012, 18, 4026–4036. [Google Scholar] [CrossRef]
- Lauss, M.; Nsengimana, J.; Staaf, J.; Newton-Bishop, J.; Jonsson, G. Consensus of melanoma gene expression subtypes converges on biological entities. J. Invest. Dermatol. 2016, 136, 2502–2505. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Kong, Y.; Si, L.; Zhu, Y.; Xu, X.; Corless, C.L.; Flaherty, K.T.; Li, L.; Li, H.; Sheng, X.; Cui, C.; et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin. Cancer Res. 2011, 17, 1684–1691. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Kuk, D.; Shoushtari, A.N.; Barker, C.A.; Panageas, K.S.; Munhoz, R.R.; Momtaz, P.; Ariyan, C.E.; Brady, M.S.; Coit, D.G.; Bogatch, K.; et al. Prognosis of mucosal, uveal, acral, Nonacral cutaneous, and unknown primary melanoma from the time of first metastasis. Oncologist 2016, 21, 848–854. [Google Scholar] [CrossRef]
- Klemen, N.D.; Wang, M.; Rubinstein, J.C.; Olino, K.; Clune, J.; Ariyan, S.; Cha, C.; Weiss, S.A.; Kluger, H.M.; Sznol, M. Survival after checkpoint inhibitors for metastatic acral, mucosal, and uveal melanoma. J. Immunother. Cancer 2020, 8, e000341. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Munhoz, R.R.; Kuk, D.; Ott, P.A.; Johnson, D.B.; Tsai, K.K.; Rapisuwon, S.; Eroglu, Z.; Sullivan, R.J.; Luke, J.J.; et al. The efficacy of anti-PD-1 agents in acral and mucosal melanoma. Cancer 2016, 122, 3354–3362. [Google Scholar] [CrossRef]
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I.; et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016, 122, 3344–3353. [Google Scholar] [CrossRef]
- Verbiest, A.; Van Hoef, V.; Rodriguez-Antona, C.; Gracia-Donas, J.; Grana-Castro, O.; Albersen, M.; Baldewijns, M.; Laenen, A.; Roussel, E.; Schoffski, P.; et al. MicroRNA expression profiles in molecular subtypes of clear-cell renal cell carcinoma are associated with clinical outcome and repression of specific mRNA targets. PLoS ONE 2020, 15, e0238809. [Google Scholar] [CrossRef]
- Hahn, A.W.; Lebenthal, J.; Genovese, G.; Sircar, K.; Tannir, N.M.; Msaouel, P. The significance of sarcomatoid and rhabdoid dedifferentiation in renal cell carcinoma. Cancer Treat. Res. Comm. 2022, 33, 100640. [Google Scholar] [CrossRef]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Al-Alwan, M.; Ghebeh, H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Parra Cuentas, E.R.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef]
- Joseph, R.W.; Millis, S.Z.; Carballido, E.M.; Bryant, D.; Gatalica, Z.; Reddy, S.; Bryce, A.H.; Vogelzang, N.J.; Stanton, M.L.; Castle, E.P.; et al. PD-1 and PD-L1 Expression in Renal Cell Carcinoma with Sarcomatoid Differentiation. Cancer Immunol. Res. 2015, 3, 1303–1307. [Google Scholar] [CrossRef]
- Pichler, R.; Comperat, E.; Klatte, T.; Pichler, M.; Loidl, W.; Lusuardi, L.; Schmidinger, M. Renal cell carcinoma with sarcomatoid features: Finally new therapeutic hope? Cancers 2019, 11, 422. [Google Scholar] [CrossRef]
- Malouf, G.G.; Ali, S.M.; Wang, K.; Balasubramanian, S.; Ross, J.S.; Miller, V.A.; Stephens, P.J.; Khayat, D.; Pal, S.K.; Su, X.; et al. Genomic Characterization of Renal Cell Carcinoma with Sarcomatoid Dedifferentiation Pinpoints Recurrent Genomic Alterations. Eur. Urol. 2016, 70, 348–357. [Google Scholar] [CrossRef]
- Wang, Z.; Kim, T.B.; Peng, B.; Karam, J.; Creighton, C.; Joon, A.; Kawakami, F.; Trevisan, P.; Jonasch, E.; Chow, C.W.; et al. Sarcomatoid Renal Cell Carcinoma Has a Distinct Molecular Pathogenesis, Driver Mutation Profile, and Transcriptional Landscape. Clin. Cancer Res. 2017, 23, 6686–6696. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2018, 174, 1033. [Google Scholar] [CrossRef]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomized, open-label, phase III trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Timer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase III trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burlington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef]
- Feng, X.; Guo, J.; An, G.; Wu, Y.; Liu, Z.; Meng, B.; He, N.; Zhao, X.; Chen, S.; Zhu, Y.; et al. Genetic Aberrations and Interaction of NEK2 and TP53 Accelerate Aggressiveness of Multiple Myeloma. Adv. Sci. 2022, 9, e2104491. [Google Scholar] [CrossRef]
- Rivera-Rivera, Y.; Marina, M.; Jusino, S.; Lee, M.; Velazquez, J.V.; Chardon-Colon, C.; Vargas, G.; Padmanabhan, J.; Chellappan, S.P.; Saavedra, H.I. The Nek2 centrosome-mitotic kinase contributes to the mesenchymal state, cell invasion, and migration of triple-negative breast cancer cells. Sci. Rep. 2021, 11, 9016. [Google Scholar] [CrossRef]
- Gu, Z.; Xia, J.; Xu, H.; Frech, I.; Tricot, G.; Zhan, F. NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate kinase. J. Hem. Oncol. 2017, 10, 17. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zheng, Z.; et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, W.; Xia, J.; Gu, Z.; Wendlandt, E.; Zhan, X.; Janz, S.; Tricot, G.; Zhan, F. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget 2014, 5, 11986–11997. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Graf, R.P.; Fisher, V.; Weberpals, J.; Gjoerup, O.; Tierno, M.B.; Huang, R.S.P.; Sayegh, N.; Lin, D.I.; Raskina, K.; Schrock, A.B.; et al. Comparative effectiveness of immune checkpoint inhibitors vs chemotherapy by tumor mutational burden in metastatic castration-resistant prostate cancer. JAMA Netw. Open 2022, 5, e225394. [Google Scholar] [CrossRef]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E., 3rd; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: A randomized phase 3 trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef]
- Tang, F.; Xu, D.; Wang, S.; Wong, C.K.; Martinez-Fundichely, A.; Lee, C.J.; Cohen, S.; Park, J.; Hill, C.E.; Eng, K.; et al. Chromatin profiles classify castration-resistant prostate cancers suggesting therapeutic targets. Science 2022, 376, eabe1505. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; Nelson, P.S.; Yu, E.Y.; Montgomery, B.; et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget 2016, 7, 82504–82510. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Antonarakis, E.S.; Bismar, T.A.; Guedes, L.B.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; et al. Genomic characterization of prostatic ductal adenocarcinoma identifies a high prevalence of DNA repair gene mutations. JCO Precis. Oncol. 2019, 3, PO.18.00327. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 130, 4492–4505. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Singh, S.; Arnaoutakis, K.; Malapati, S.; Bhatti, S.A.; Joon, A.Y.; Atiq, O.T.; Posters, L.L. Stem cell theory of cancer: Implications for translational research from bedside to bench. Cancers 2022, 14, 3345. [Google Scholar] [CrossRef]
- Silk, A.W.; Kaufman, H.L.; Curti, B.; Mehnert, J.M.; Margolin, K.; McDermott, D.; Clark, J.; Newman, J.; Bommareddy, P.K.; Denzin, L.; et al. High-dose ipilimumab and high-dose interleukin-2 for patients with advanced melanoma. Front. Oncol. 2020, 9, 1483. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Othus, M.; Chen, Y.; Wright, G.P., Jr.; Yost, K.J.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.D.; Fecher, L.A.; Truong, T.G.; et al. Neoadjuvant-adjuvant or adjuvant-only pembrolizumab in advanced melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef]
- Blank, C.U.; Rozeman, E.A.; Menzes, A.M.; van de Wiel, B.A.; Adhikari, C.; Silkorska, K.; Krijgsman, O.; Eriksson, H.; Bierman, C.; Grijpink-Ongering, L.; et al. OpACIN-neo: A multicenter phase II study to identify the optimal neo-adjuvant combination scheme of ipilimumab and nivolumab. Ann. Oncol. 2018, 29 (Suppl. S8), viii734. [Google Scholar] [CrossRef]
- Schuchter, L.M. Adjuvant interferon therapy for melanoma: High-dose, low-dose, no dose, which dose? J. Clin. Oncol. 2004, 22, 7–10. [Google Scholar] [CrossRef]
- Najjar, Y.G.; McCurry, D.; Lin, H.; Lin, Y.; Zang, Y.; Davar, D.; Karunamurthy, A.; Drabick, J.J.; Neves, R.I.; Butterfield, L.H.; et al. Neoadjuvant pembrolizumab and high-dose IFNbeta-2b in resectable regionally advanced melanoma. Clin. Cancer Res. 2021, 27, 4195–4204. [Google Scholar] [CrossRef]
- Long, G.V.; Spillane, A.J.; Pennington, T.E.; Shannon, K.F.; Stretch, J.; Gonzalez, M.; Saw, R.P.M.; Low, S.N.; Scolyer, R.A.; Menzies, A.M. A phase II trial of neoadjuvant pembrolizumab combined with Lenvatinib in resectable stage III melanoma. Ann. Oncol. 2022, 33, S906–S907. [Google Scholar] [CrossRef]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated overall survival and PD-L1 subgroup analysis of patients with extensive-stage small cell lung cancer treated with atezolizumab, carboplatin, and etoposide (Impower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef]
- Gelmi, M.C.; Houtzagers, L.E.; Strub, T.; Krossa, I.; Jager, M.J. MITF in normal melanocytes, cutaneous, and uveal melanomas: A delicate balance. Int. J. Mol. Sci. 2022, 23, 6001. [Google Scholar] [CrossRef]
- Parreno, V.; Martinez, A.M.; Cavalli, G. Mechanisms of polycomb group protein function in cancer. Cell Res. 2022, 32, 231–253. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Korkaya, H.; Wicha, M.S. Breast cancer stem cells: We have got them surrounded. Clin. Cancer Res. 2013, 19, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Fan, M.; Candas, D.; Menaa, C.; Liu, H.C.; Nantajit, D.; Wen, Y.; Xiao, K.; Eldridge, A.; Chromy, B.A.; et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin. Cancer Res. 2012, 18, 6634–6647. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.; Braybrooke, J.; Gray, R.; Hills, R.; Liu, Z.; Peto, R.; Davies, L.; Dodwell, D.; McGale, P.; Pan, H.; et al. Trastuzumab for early-stage, HER2-positive breast cancer: A meta-analysis of 13,864 women in seven randomised trials. Lancet Oncol. 2021, 22, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Hoff, P.M.; Shen, L.; Ohtsu, A.; Shah, M.A.; Siddiqui, A.; Heeson, S.; Kiermaier, A.; Macharia, H.; Restuccia, E.; et al. Pertuzumab, trastuzumab, and chemotherapy in HER2-positive gastric/gastroesophageal junction cancer: End-of-study analysis of the JACOB phase III randomized clinical trial. Gastric Cancer 2023, 26, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Reches, A.; Ophir, Y.; Stein, N.; Kol, I.; Isaacson, B.; Amikam, Y.C.; Elnekave, A.; Tsukerman, P.; Brlic, P.K.; Lenac, T.; et al. Nectin4 is a novel TIGIT ligand which combines checkpoint inhibition and tumor specificity. J. Immunother. Cancer 2020, 8, e000266. [Google Scholar] [CrossRef] [PubMed]
- Siddarth, S.; Goutam, K.; Das, S.; Nayak, A.; Nayak, D.; Sethy, C.; Wyatt, M.D.; Kundu, C.N. Nectin-4 is a breast cancer stem cell marker that induces WNT/beta-catenin signaling via Pi3k/Akt axis. Int. J. Biochem. Cell Biol. 2017, 89, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Kedashiro, S.; Kameyama, T.; Mizutani, K.; Takai, Y. Nectin-4 and p95-ErbB2 cooperatively regulate Hippo signaling-dependent SOX2 gene expression, enhancing anchorage-independent T47D cell proliferation. Sci. Rep. 2021, 11, 7344. [Google Scholar] [CrossRef] [PubMed]
- Hoimes, C.J.; Flaig, T.W.; Milowsky, M.I.; Friedlander, T.W.; Bilen, M.A.; Gupta, S.; Srinivas, S.; Merchan, J.R.; McKay, R.R.; Petrylak, D.P.; et al. Enfortumab vedotin plus pembrolizumab in previously untreated advanced urothelial cancer. J. Clin. Oncol. 2023, 41, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Aydin, A.M.; Maraboyina, S.; Chen, Z.; Singh, S.; Gokden, N.; Langford, T. Stem cell origin of cancer: Implications of oncogenesis recapitulating embryogenesis in cancer care. Cancers 2023, 15, 2516. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juarez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal cell carcinoma. NEJM 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grunwald, V.; Hutson, T.E.; Kopyltsov, E.; Mendez-Vidal, M.J.; et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimuab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Cattrini, C.; Messina, C.; Airoldi, C.; Buti, S.; Roviello, G.; Mennitto, A.; Caffo, O.; Gennari, A.; Bersanelli, M. Is there a preferred first-line therapy for metastatic renal cell carcinoma? A network meta-analysis. Ther. Adv. Urol. 2021, 13, 17562872211053189. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.; et al. Atezolizumab plus bevacizumab in unresctable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyas, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immune-metabolic adjuvant for cancer immunotherapy. OncoImmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Ciccarese, C.; Iacovelli, R.; Buti, S.; Primi, F.; Astore, S.; Massari, F.; Grazia Ferrara, M.; Palermo, G.; Foschi, N.; Iacovelli, V.; et al. Concurrent nivolumab and metformin in diabetic cancer patients: Is it safe and more active? Anticancer Res. 2022, 42, 1487–1493. [Google Scholar] [CrossRef]
- Pietras, H.; Xu, H.; Hu, X.; Matheny, C.; Sandler, A.; Patel, M. Retrospective descriptive analysis of metformin with atezolizumab in advanced non-small cell lung cancer in the OAK trial. J. Thorac. Oncol. 2018, 13, S538–S539. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef]
- Jiang, Z.; He, J.; Zhang, B.; Wang, L.; Long, C.; Zhao, B.; Yang, Y.; Du, L.; Luo, W.; Hu, J.; et al. A potential “anti-Warburg effect” in circulating tumor cell-mediated metastatic progression? Aging Dis. 2024, 16, 1–14. [Google Scholar] [CrossRef]
- Paul, S.; Sa, G. Curcumin as an adjuvant to cancer immunotherapy. Front. Oncol. 2021, 11, 675923. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Kang, L.; Zhang, J.; Wu, Z.; Wang, H.; Huang, M.; Lan, P.; Wu, X.; Wang, C.; Cao, W.; et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): A single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol. Hepatol. 2022, 7, 38–48. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cancer Type Stage Trial | Treatments | PFS or DFS (Months) or pCR (%) | OS (Months) | References (Randomized Phase II or III Trial) |
|---|---|---|---|---|
| Melanoma Advanced, metastatic untreated CHECKMATE 067 | Nivo + ipi Nivo Ipi | 11.5 6.9 2.9 | 72.1 36.9 19.9 | Wolchok, 2022 [39] (III) |
| Melanoma Advanced untreated RELATIVITY 047 | Nivo + relatlimab Nivo | 10.1 4.6 | Tawbi, 2022 [27] (II–III) | |
| Melanoma, metastatic Anti-IDO1 ECHO-301/KEYNOTE 252 | Pembro + epacadostat Pembro + placebo | 4.7 4.9 | Long, 2019 [40] (III) | |
| SCLC-ED IMPower-133 | Atezo + chemo Chemo | 5.2 4.3 | 12.3 10.3 | Horn, 2018 [41] (III) |
| SCLC-ED CASPIAN | Durva + chemo Chemo | 13.0 10.3 | Paz-Ares, 2019 [42] (III) | |
| SCLC-ED CASPIAN | Durva + treme + chemo Durva + chemo Chemo | 10.4 12.9 10.5 | Goldman, 2021 [43] (III) | |
| SCLC-ED untreated Anti-TIGIT SKYSCRAPER-02 | Atezo + chemo + tiragolumab Atezo + chemo | 5.4 5.6 | 13.1 13.1 | Rudin, 2024 [44] (III) |
| NSCLC Unresectable, stage III PACIFIC | Durva after chemoRT ChemoRT | 12.2 5.6 | 48 29 | Antonia, 2018 [45] (III) |
| NSCLC Metastatic Non-squamous 1st-line KEYNOTE-189 | Pembro + chemo Chemo | 8.8 4.9 | NR 11.3 | Gandhi, 2018 [46] (III) |
| NSCLC Metastatic Non-squamous 1st-line POSEIDON | Durva + chemo + treme Durva + chemo | 6.8 6.4 | 17.2 14.8 | Johnson, 2023 [47] (III) |
| NSCLC Metastatic Squamous 1st-line KEYNOTE-407 | Pembro + chemo Chemo | 6.4 4.8 | 15.9 11.3 | Paz-Ares, 2018 [48] (III) |
| NSCLC Metastatic Squamous 1st-line POSEIDON | Durva + chemo + treme Durva + chemo | 4.6 4.7 | 10.4 11.5 | Johnson, 2023 [47] (III) |
| NSCLC Advanced or metastatic 1st-line PDL1-high Anti-TIGIT SKYSCRAPER-01 | Atezo+ tiragolumab Atezo | 22.9 16.7 | *** | |
| NSCLC Metastatic 1st-line ≥ PDL1 > 25% MYSTIC | Durva + treme Durva Chemo | 3.9 4.7 5.4 | 11.9 16.3 12.9 | Rizvi, 2020 [49] (III) |
| NSCLC Metastatic 1st-line PDL1 TPS > 50 KEYNOTE 598 | Pembro + ipi Pembro | 8.2 8.4 | 21.4 21.9 | Boyer, 2021 [50] (III) |
| Breast cancer TNBC Neoadjuvant stage II/III KEYNOTE-522 | Pembro + chemo Chemo | 65% 51% | Schmid, 2022 [51] (III) | |
| Breast cancer TNBC, CPS > 10 Metastatic or unresectable 1st line KEYNOTE-355 | Pembro + chemo Chemo | 9.7 5.6 | 23.0 16.1 | Cortes, 2020 [52] (III) |
| Breast cancer TNBC PDL1+ Metastatic or unresectable 1st line IMpassion 130 | Atezo + chemo Chemo | 7.2 5.5 | 25.0 15.5 | Schmid, 2018 [53] (III) |
| Breast cancer, HER2+, metastatic 2nd-line KATE2 | Atezo + T-DM1 T-DM1 | 8.2 6.8 | Emens 2021 [54] (II) | |
| Breast cancer, HER2+, neoadjuvant Impassion050 | Atezo + chemo + trastu + pertu chemo + trastu + pertu | 62.4% (ITT) 64.2% (PDL1+) 62.7% (ITT) 72.5% (PDL1+) | Huober 2021 [55] (III) |
| Cancer Type Stage Trial | Treatments | PFS or DFS (Months) or pCR (%) | OS (Months) | References (Randomized Phase II or III Trial) |
|---|---|---|---|---|
| RCC 1st line, advanced KEYNOTE-426 | Pembro + axitinib Sunitinib | 15.7 11.1 | 47.2 40.8 | Plimack, 2023 [56] (III) |
| RCC 1st line, advanced CEAR | Pembro + Lenvatinib Sunitinib | 23.3 9.2 | NR, HR 0.72 NR | Choueiri, 2023 [57] (III) |
| RCC 1st line, advanced CheckMate 9ER | Nivo + cabozantinib Sunitinib | 16.6 8.4 | 49.5 35.5 | Burotto, 2023 [58] (III) |
| RCC 1st line, advanced CheckMate 214 | Ipi + nivo Sunitinib | 12.3, HR 0.86 12.3 | 55.7 38.4 | Motzer, 2022 [59] (III) |
| RCC 1st line, advanced COSMIC-313 | Ipi + nivo + cabozantinib Ipi + nivo | 15.3 11.3 | Choueiri, 2023 [60] (III) | |
| RCC 1st line Advanced or metastatic Pegylated IL-2 PIVOT-09 | Nivo + BEMPEG Tyrosine kinase inhibitor | 29.0 NR | Tannir, 2022 [61] (III) | |
| HCC HIMALAYA | Treme + durva Durva Sorafenib | 3.8 3.7 4.1 | 16.4 16.6 13.8 | Abou-Alfa, 2022 [62] (III) |
| HCC IMbrave 150 | Atezo +bevacizum Sorafenib | 6.8 4.3 | 19.2 13.4 | Finn, 2020 [63] (III) |
| GEJ adenoca Adjuvant Checkmate 577 | Nivo after preop chemoRT Preop chemoRT | 22.4 11.0 | Kelly, 2021 [64] (III) | |
| Gastric or GEJ adenoca HER2+ metastatic KEYNOTE-811 | Pembro + trastuzumab + chemo Trastuzumab + chemo | 10.0 8.1 | 20.0 16.8 | Janjigian, 2023 [65] (III) |
| Esophageal Squamous cell ca Metastatic KEYNOTE-590 | Pembro + chemo Chemo | 6.3 5.8 | 12.4 9.8 | Sun, 2021 [66] (III) |
| Esophageal Squamous cell ca Metastatic PDL1 ≥ 1% Checkmate-648 | Nivo + chemo Nivo + ipi Chemo | 6.9 4.0 4.4 | 15.4 13.7 9.1 | Doki, 2022 [67] (III) |
| Biliary tract cancer Metastatic TOPAZ-1 | Durva + chemo Chemo | 7.2 5.7 | 12.8 11.5 | Oh, 2022 [68] (III) |
| Pancreatic cancer Metastatic | Durva + treme Durva | 1.5 1.5 | 3.1 3.6 | O’Reilly, 2019 [69] (II) |
| HNSCC Previously treated PDL1 ≥ 25% EAGLE | Durva + treme Durva SOC | 2.0 2.1 3.7 | 4.8 9.8 9.0 | Ferris, 2020 [70] (III) |
| HNSCC PDL1-low/neg CONDOR | Durva + treme Durva Treme | 7.6 6.0 5.5 | Siu, 2019 [71] (II) | |
| HNSCC Recurrent or metastatic CHECKMATE 714 | Nivo + ipi Nivo (platinum-refractory) | 2.6 2.6 | 10.0 9.6 | Harrington, 2023 [72] (II) |
| HNSCC Recurrent or metastatic CHECKMATE 714 | Nivo + ipi Nivo (platinum-eligible) | 2.8 2.9 | 10.0 12.9 | Harrington, 2023 [72] (II) |
| Urothelial Untreated advanced or metastatic DANUBE | Dur + treme Durva Chemo | 15.1 14.4 12.1 | Powles, 2020 [73] (III) | |
| Ovarian cancer Recurrent or persistent NRG | Nivo + ip Nivo | 3.9 2.0 | 28.1, p = 0.43 21.8 | Zamarin, 2020 [74] (II) |
| CRC Neoadjuvant Locally advanced | Toripa + celecoxib Toripa | 88% 65% | Hu, 2022 [75] (II) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.-M.; Trikannad, A.K.; Vellanki, S.; Hussain, M.; Malik, N.; Singh, S.R.; Jillella, A.; Obulareddy, S.; Malapati, S.; Bhatti, S.A.; et al. Stem Cell Origin of Cancer: Clinical Implications beyond Immunotherapy for Drug versus Therapy Development in Cancer Care. Cancers 2024, 16, 1151. https://doi.org/10.3390/cancers16061151
Tu S-M, Trikannad AK, Vellanki S, Hussain M, Malik N, Singh SR, Jillella A, Obulareddy S, Malapati S, Bhatti SA, et al. Stem Cell Origin of Cancer: Clinical Implications beyond Immunotherapy for Drug versus Therapy Development in Cancer Care. Cancers. 2024; 16(6):1151. https://doi.org/10.3390/cancers16061151
Chicago/Turabian StyleTu, Shi-Ming, Anup K. Trikannad, Sruthi Vellanki, Munawwar Hussain, Nazish Malik, Sunny R. Singh, Anusha Jillella, Sri Obulareddy, Sindhu Malapati, Sajjad A. Bhatti, and et al. 2024. "Stem Cell Origin of Cancer: Clinical Implications beyond Immunotherapy for Drug versus Therapy Development in Cancer Care" Cancers 16, no. 6: 1151. https://doi.org/10.3390/cancers16061151