
Regulation of PD-L1 Expression by YY1 in Cancer: Therapeutic Efficacy of Targeting YY1

 ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
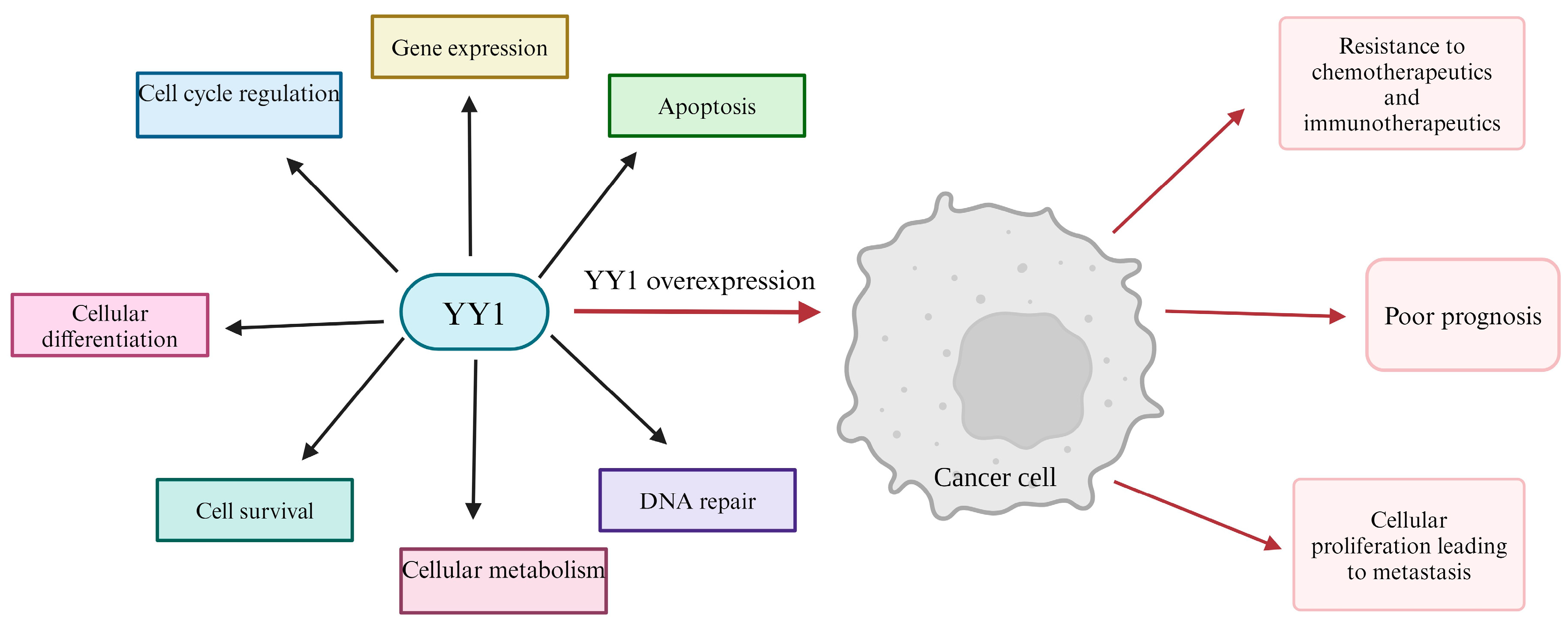
2. YY1 Overexpression in Cancer, Oncogenic and Immunosuppressive Properties
3. Role of the PD-1/PD-L1 Interaction in the Inactivation of Anti-Tumor CD8+ T Cells and Immune Escape

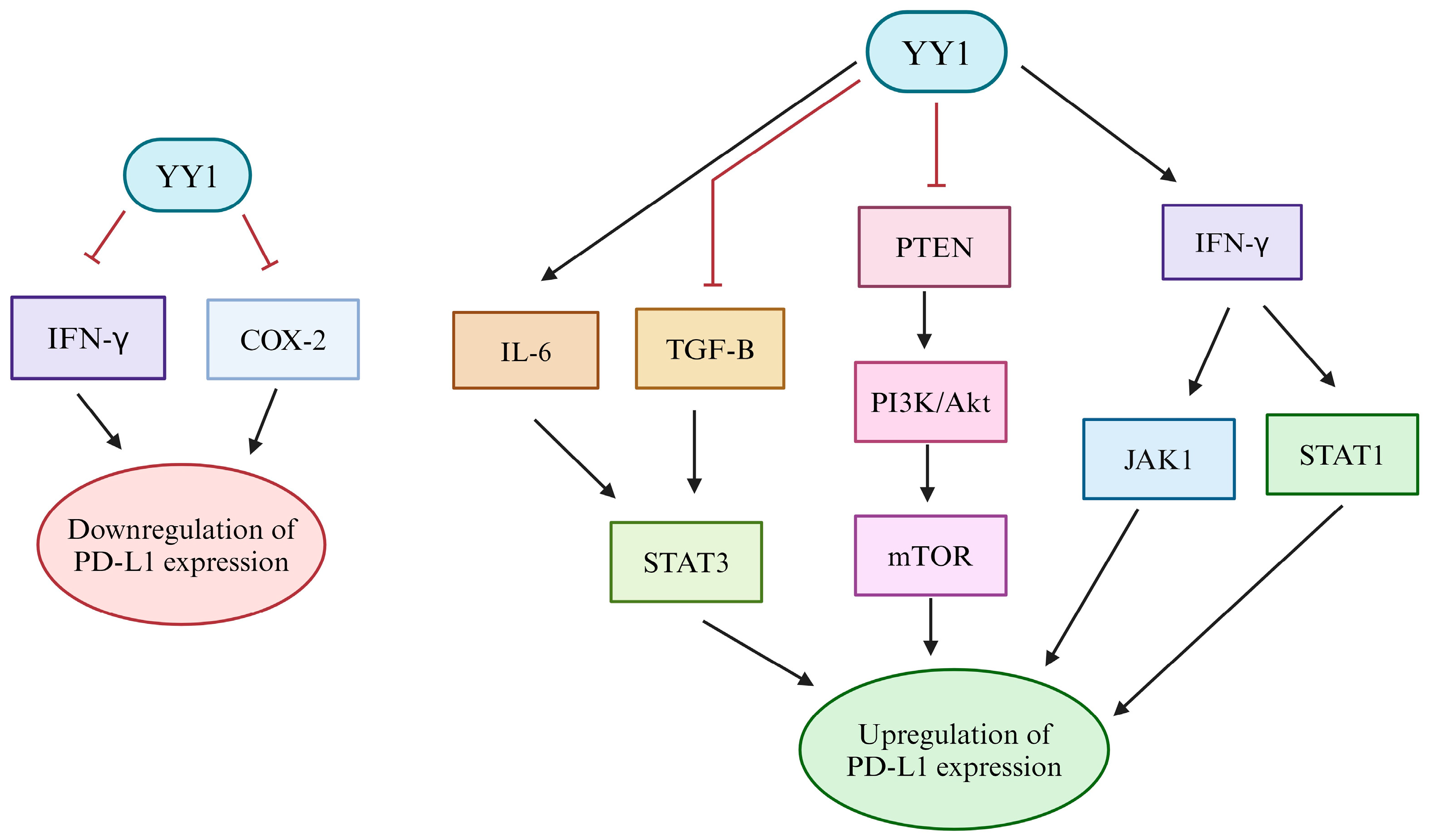
4. Regulation of PD-L1 by YY1
4.1. Transcriptional

4.2. Epigenetics

4.3. Post-Transcriptional
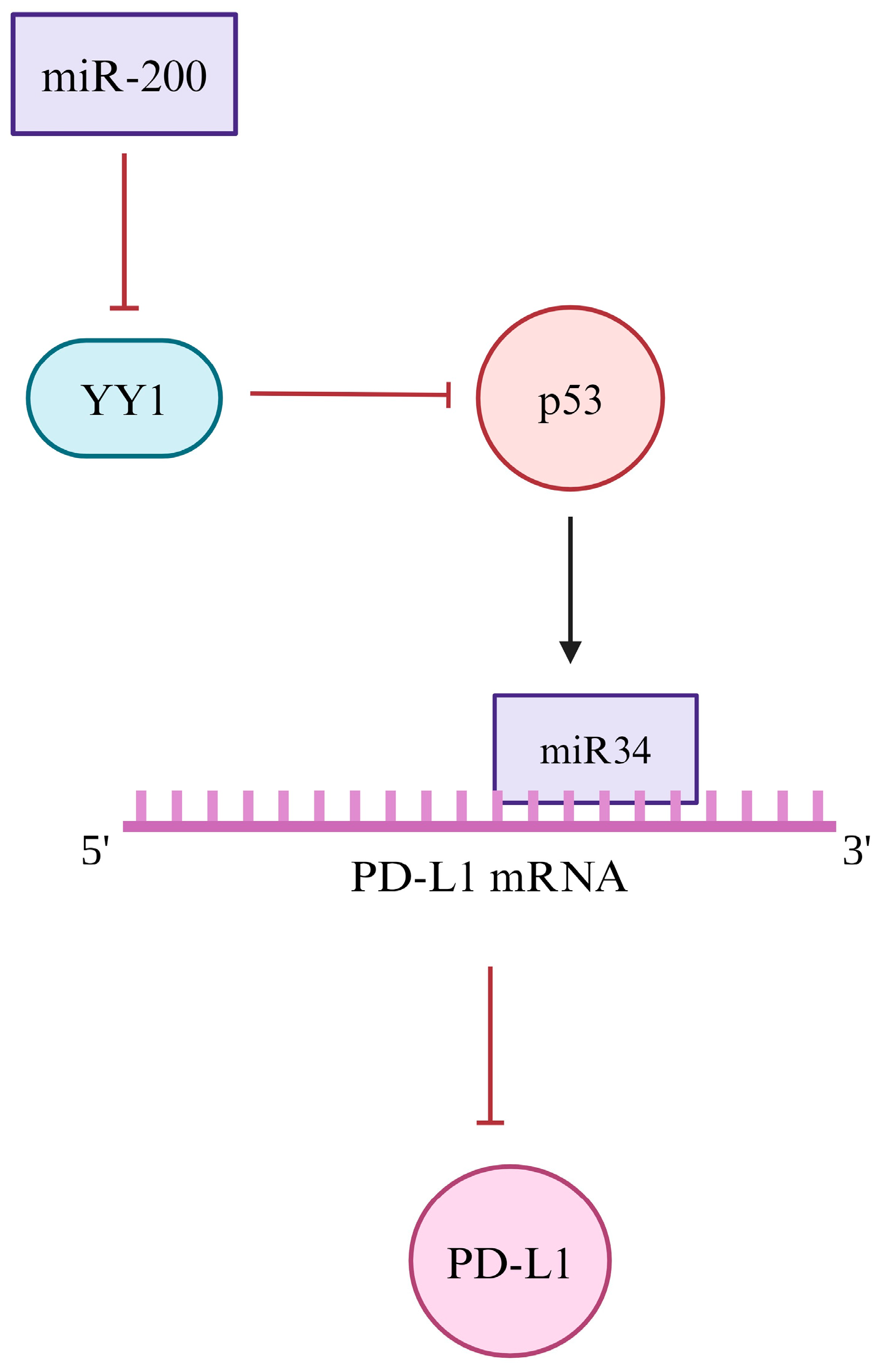
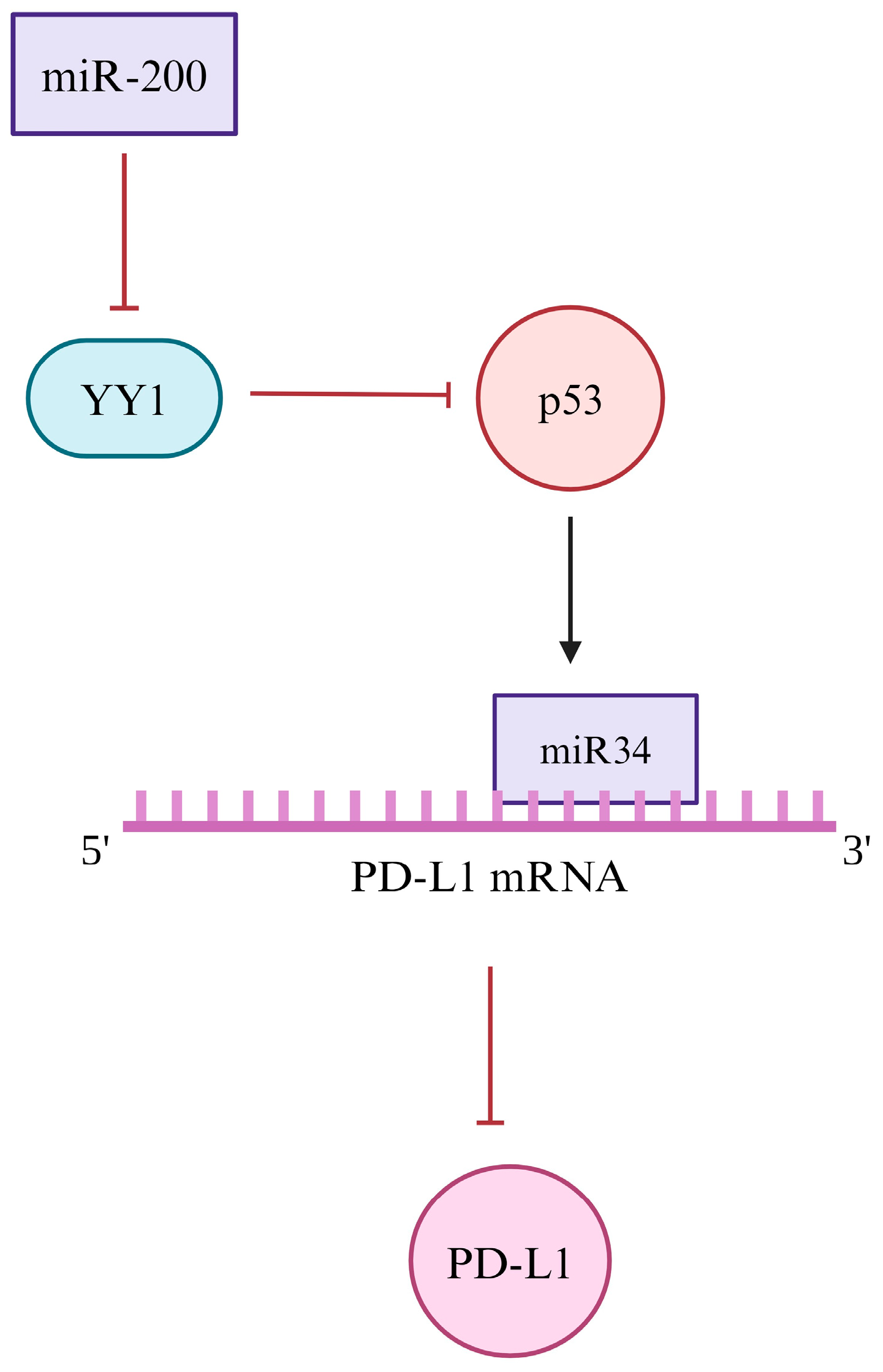
4.4. Post-Translational
5. Implications of YY1-Mediated Regulation of PD-1/PD-L1 Axis in Immune Escape

6. Bioinformatics Analysis of the Pathway Activity Scores (PASs) between High and Low YY1 and PD-L1-Expressing Tumors

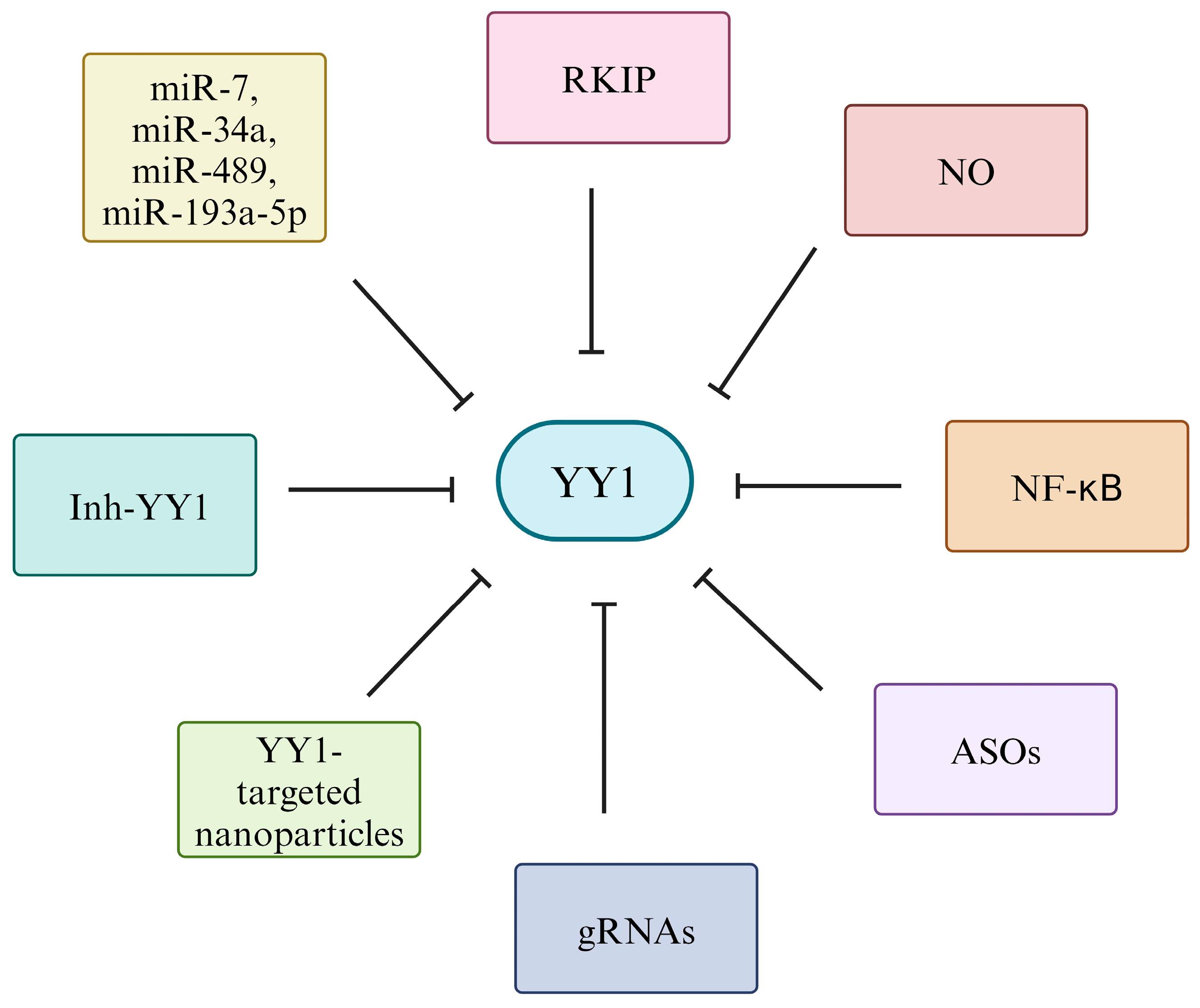
7. Therapeutic Targeting of YY1 for Immune Checkpoint Blockade
8. Overall Perspectives and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| AR | androgen receptor |
| ASO | antisense oligonucleotide |
| CAFS | cancer-associated fibroblasts |
| CD4+/CD8+ TILs | tumor-infiltrating CD4+/CD8+ T cells |
| COX-2 | cyclooxygenase 2 |
| CRC | colorectal cancer |
| CRHR2 | corticotropin-releasing hormone receptor 2 |
| CRPC | castration-resistant prostate cancer |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein-4 |
| DC | dendritic cell |
| DR5 | death receptor 5 |
| EGFR-TKI | epidermal growth factor receptor tyrosine kinase inhibitor |
| EIF4F | eukaryotic initiation factor 4F |
| EMT | epithelial–mesenchymal transition |
| EZH2 | enhancer of zeste homolog |
| FDA | Food and Drug Administration |
| FDR | false discovery rate |
| FGL1 | fibrinogen-like protein 1 |
| FP | FGL1/PD-L1 |
| GSK3β | glycogen synthase kinase 3 beta |
| GRNA | guide RNA |
| HCC | hepatocellular carcinoma |
| HDAC | histone deacetylase |
| HnRNP L | heterogeneous nuclear ribonucleoprotein L |
| Id1 | inhibitor of differentiation |
| IFN-γ | interferon gamma |
| IL-2 | interleukin 2 |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| Inh-YY1 | YY1 inhibitor |
| ISO | isorhamnetin |
| JAK | janus kinase |
| JNK | jun N-terminal |
| LUAD | MYH9 lung adenocarcinoma |
| mAbs | monoclonal antibodies |
| MDSC | myeloid-derived suppressor cell |
| MGF | mammary gland factor |
| miRNA | microRNA |
| MOR | mu opioid receptor |
| mTOR | mechanistic Target of Rapamycin |
| NF-κB | nuclear factor kappa B |
| NK | natural killer |
| NO | nitric oxide |
| NOX4 | NADPH oxidase 4 |
| NSCLC | non-small-cell lung cancer |
| PAI-1 | plasminogen activator inhibitor 1 gene |
| PARP | poly (ADP-ribose) polymerase |
| PAS | pathway activity scores |
| PD-1 | programmed death-1 |
| PD-L1 | programmed death ligand-1 |
| PD-L2 | programmed death ligand-2 |
| PI3k/AKT | phosphatidylinositol 3-kinase/protein kinase B |
| PTEN | phosphatase and tensin homolog |
| PTM | post-translational modification |
| RAS/MAPK | Ras/mitogen-activated protein kinase |
| RKIP | Raf kinase inhibitory protein |
| RPPA | reverse-phase protein array |
| RTK | receptor tyrosine kinase |
| sEVs | small extracellular vesicles |
| SDS | sodium dodecyl-sulfate |
| siRNA | small interfering RNA |
| Sp1 | specificity protein 1 |
| STAT | signal transducer and activator of transcription |
| T7-exo | T7 peptide-decorated exosome |
| TAM | tumor-associated macrophage |
| TCGA | the cancer genome atlas |
| TCPA | the cancer proteome atlas |
| TF | transcription factor |
| TGF-β | transforming growth factor-β |
| TIL | tumor-infiltrating lymphocyte |
| TME | tumor microenvironment |
| TMEM97 | transmembrane protein 97 |
| TNF-α | tumor necrosis factor-alpha |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
| Treg | regulatory T cell |
| Ucn2 | Urocortin-2 |
| YY1 | Yin Yang 1 |
References
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Deligne, C.; Gros, L. Anti-tumor monoclonal antibodies: New insights to elicit a long-term immune response. Med. Sci. 2019, 35, 982–989. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Monteleone, G.; Franzè, E.; Maresca, C.; Colella, M.; Pacifico, T.; Stolfi, C. Targeted Therapy of Interleukin-34 as a Promising Approach to Overcome Cancer Therapy Resistance. Cancers 2023, 15, 971. [Google Scholar] [CrossRef]
- Baghdadi, M.; Wada, H.; Nakanishi, S.; Abe, H.; Han, N.; Putra, W.E.; Endo, D.; Watari, H.; Sakuragi, N.; Hida, Y.; et al. Chemotherapy-Induced IL34 Enhances Immunosuppression by Tumor-Associated Macrophages and Mediates Survival of Chemoresistant Lung Cancer Cells. Cancer Res. 2016, 76, 6030–6042. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-Associated Macrophages Drive Spheroid Formation during Early Transcoelomic Metastasis of Ovarian Cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed]
- MaruYama, T.; Chen, W.; Shibata, H. TGF-β and Cancer Immunotherapy. Biol. Pharm. Bull. 2022, 45, 155–161. [Google Scholar] [CrossRef]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumor-Associated Macrophages Promote Cancer Stem Cell-like Properties via Transforming Growth Factor-Beta1-Induced Epithelial–Mesenchymal Transition in Hepatocellular Carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet VJ, A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Y.; Zhu, Y.; Shen, Y.Y.; Xu, Q.Y.; Tang, H.Y.; Cui, N.X.; Jiang, L.; Dai, X.M.; Chen, W.Q.; Lin, Q.; et al. The role of PD-1 signaling in health and immune-related diseases. Front. Immunol. 2023, 14, 1163633. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The Role of PD-1/PD-L1 and Application of Immune-Checkpoint Inhibitors in Human Cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.-J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 Antigen on the Surface of Stimulated Mouse T and B Lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Ljunggren, H.-G.; Jonsson, R.; Höglund, P. Seminal Immunologic Discoveries with Direct Clinical Implications: The 2018 Nobel Prize in Physiology or Medicine Honours Discoveries in Cancer Immunotherapy. Scand. J. Immunol. 2018, 88, e12731. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Hays, E.; Bonavida, B. YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist. 2019, 43, 10–28. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC Regulates the Antitumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Chatzinikola, A.M.; Vakis, A.F.; Soulitzis, N.; Karabetsos, D.A.; Neonakis, I.; Bonavida, B.; Spandidos, D.A. YY1 Over-Expression in Human Brain Gliomas and Meningiomas Correlates with TGF-B1, IGF-1 and FGF-2 mRNA Levels. Cancer Investig. 2009, 27, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; He, Y.; Chen, J.; Zeng, Z.; Yang, B.; Ou, Q. A critical role of transcription factor YY1 in rheumatoid arthritis by regulation of interleukin-6. J. Autoimmun. 2017, 77, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Siednienko, J.; Maratha, A.; Yang, S.; Mitkiewicz, M.; Miggin, S.M.; Moynagh, P.N. Nuclear Factor κB Subunits RelB and cRel Negatively Regulate Toll-like Receptor 3-Mediated β-Interferon Production via Induction of Transcriptional Repressor Protein YY1*. J. Biol. Chem. 2011, 286, 44750–44763. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Morton, D., Jr.; Komaragiri, S.; Millena, A.C.; Leath, C.; Khan, S.A. TGF-β Effects on Prostate Cancer Cell Migration and Invasion Are Mediated by PGE2 through Activation of PI3K/AKT/mTOR Pathway. Endocrinology 2013, 154, 1768–1779. [Google Scholar] [CrossRef]
- Hwang, S.S.; Jang, S.W.; Kim, M.K.; Kim, L.K.; Kim, B.S.; Kim, H.S.; Kim, K.; Lee, W.; Flavell, R.A.; Lee, G.R. YY1 inhibits differentiation and function of regulatory T cells by blocking Foxp3 expression and activity. Nat. Commun. 2016, 7, 10789. [Google Scholar] [CrossRef]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell Dev. Biol. 2020, 8, 592164. [Google Scholar] [CrossRef]
- Varum, S.; Baggiolini, A.; Zurkirchen, L.; Atak, Z.K.; Cantù, C.; Marzorati, E.; Bossart, R.; Wouters, J.; Häusel, J.; Tuncer, E.; et al. Yin Yang 1 Orchestrates a Metabolic Program Required for Both Neural Crest Development and Melanoma Formation. Cell Stem Cell 2019, 24, 637–653.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Li, L.; Peng, S.; Chen, F.M.; Zhang, Q.; An, Y.; Lin, X.; Li, W.; Koon, A.C.; Chan, T.; et al. Planar Cell Polarity Gene Fuz Triggers Apoptosis in Neurodegenerative Disease Models. EMBO Rep. 2018, 19, e45409. [Google Scholar] [CrossRef]
- Zurkirchen, L.; Varum, S.; Giger, S.; Klug, A.; Häusel, J.; Bossart, R.; Zemke, M.; Cantù, C.; Atak, Z.K.; Zamboni, N.; et al. Yin Yang 1 Sustains Biosynthetic Demands during Brain Development in a Stage-Specific Manner. Nat. Commun. 2019, 10, 2192. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Aziz, N.; Bonavida, B. The Forgotten YY2 in Reported YY1 Expression Levels in Human Cancers. Crit. Rev. Oncog. 2017, 22, 63–73. [Google Scholar] [CrossRef]
- Shi, Y.; Lee, J.S.; Galvin, K.M. Everything you have ever wanted to know about Yin Yang 1……. Biochim. Biophys. Acta BBA-Rev. Cancer 1997, 1332, F49–F66. [Google Scholar] [CrossRef]
- Shrivastava, A.; Calame, K. An analysis of genes regulated by the multi-functional transcriptional regulator Yin Yang-1. Nucleic Acids Res. 1994, 22, 5151–5155. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B. Chapter 2—Crosstalks between Yin-Yang 1 (YY1) and autophagy in cancer. In Autophagy in Immune Response: Impact on Cancer Immunotherapy; Chouaib, S., Ed.; Breaking Tolerance to Anti-Cancer Cell-Mediated Immunotherapy; Academic Press: Cambridge, MA, USA, 2020; Volume 1, pp. 9–27. [Google Scholar] [CrossRef]
- Meliala, I.T.S.; Hosea, R.; Kasim, V.; Wu, S. The biological implications of Yin Yang 1 in the hallmarks of cancer. Theranostics 2020, 10, 4183–4200. [Google Scholar] [CrossRef]
- Vivarelli, S.; Falzone, L.; Ligresti, G.; Candido, S.; Garozzo, A.; Magro, G.G.; Bonavida, B.; Libra, M. Role of the Transcription Factor Yin Yang 1 and Its Selectively Identified Target Survivin in High-Grade B-Cells Non-Hodgkin Lymphomas: Potential Diagnostic and Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 6446. [Google Scholar] [CrossRef]
- Cho, A.A.; Bonavida, B. Targeting the Overexpressed YY1 in Cancer Inhibits EMT and Metastasis. Crit. Rev. Oncog. 2017, 22, 49–61. [Google Scholar] [CrossRef]
- Seligson, D.; Horvath, S.; Huerta-Yepez, S.; Hanna, S.; Garban, H.; Roberts, A.; Shi, T.; Liu, X.; Chia, D.; Goodglick, L.; et al. Expression of Transcription Factor Yin Yang 1 in Prostate Cancer. Int. J. Oncol. 2005, 27, 131–141. [Google Scholar] [CrossRef]
- Chinnappan, D.; Xiao, D.; Ratnasari, A.; Andry, C.; King, T.C.; Weber, H.C. Transcription Factor YY1 Expression in Human Gastrointestinal Cancer Cells. Int. J. Oncol. 2009, 34, 1417–1423. [Google Scholar] [CrossRef]
- Thomassen, M.; Tan, Q.; Kruse, T.A. Gene Expression Meta-Analysis Identifies Metastatic Pathways and Transcription Factors in Breast Cancer. BMC Cancer 2008, 8, 394. [Google Scholar] [CrossRef] [PubMed]
- de Nigris, F.; Zanella, L.; Cacciatore, F.; De Chiara, A.; Fazioli, F.; Chiappetta, G.; Apice, G.; Infante, T.; Monaco, M.; Rossiello, R.; et al. YY1 overexpression is associated with poor prognosis and metastasis-free survival in patients suffering osteosarcoma. BMC Cancer 2011, 11, 472. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Kolapalli, S.P.; Vallabhapurapu, S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Front. Oncol. 2019, 9, 1230. [Google Scholar] [CrossRef]
- Lee, T.C.; Shi, Y.; Schwartz, R.J. Displacement of BrdUrd-Induced YY1 by Serum Response Factor Activates Skeletal Alpha-Actin Transcription in Embryonic Myoblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 9814–9818. [Google Scholar] [CrossRef]
- Rizkallah, R.; Hurt, M.M. Regulation of the Transcription Factor YY1 in Mitosis through Phosphorylation of Its DNA-binding Domain. Mol. Biol. Cell 2009, 20, 4766–4776. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Bui, I.; Bonavida, B. Role of YY1 in the Regulation of Anti-Apoptotic Gene Products in Drug-Resistant Cancer Cells. Cancers 2023, 15, 4267. [Google Scholar] [CrossRef]
- Vega, M.I.; Jazirehi, A.R.; Huerta-Yepez, S.; Bonavida, B. Rituximab-Induced Inhibition of YY1 and Bcl-xL Expression in Ramos Non-Hodgkin’s Lymphoma Cell Line via Inhibition of NF-Kappa B Activity: Role of YY1 and Bcl-xL in Fas Resistance and Chemoresistance, Respectively. J. Immunol. 2005, 175, 2174–2183. [Google Scholar] [CrossRef]
- Wu, K.; Bonavida, B. The Activated NF-κB-Snail-RKIP Circuitry in Cancer Regulates Both the Metastatic Cascade and Resistance to Apoptosis by Cytotoxic Drugs. Crit. Rev. Immunol. 2009, 29, 241–254. [Google Scholar] [CrossRef]
- Katsushima, K.; Natsume, A.; Ohka, F.; Shinjo, K.; Hatanaka, A.; Ichimura, N.; Sato, S.; Takahashi, S.; Kimura, H.; Totoki, Y.; et al. Targeting the Notch-Regulated Non-Coding RNA TUG1 for Glioma Treatment. Nat. Commun. 2016, 7, 13616. [Google Scholar] [CrossRef] [PubMed]
- Kurisaki, K.; Kurisaki, A.; Valcourt, U.; Terentiev, A.A.; Pardali, K.; Dijke, P.T.; Heldin, C.-H.; Ericsson, J.; Moustakas, A. Nuclear Factor YY1 Inhibits Transforming Growth Factor Beta- and Bone Morphogenetic Protein-Induced Cell Differentiation. Mol. Cell. Biol. 2003, 23, 4494–4510. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Huerta-Yepez, S.; Baritaki, S.; Vega, M.; Liu, H.; Chen, H.; Berenson, J. Overexpression of Yin Yang 1 in the pathogenesis of human hematopoietic malignancies. Crit. Rev. Oncog. 2011, 16, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Tsang, D.P.; Wu, W.K.; Kang, W.; Lee, Y.; Wu, F.; Yu, Z.; Xiong, L.; Chan, A.W.; Tong, J.H.; Yang, W.; et al. Yin Yang 1-mediated epigenetic silencing of tumour-suppressive microRNAs activates nuclear factor-κB in hepatocellular carcinoma. J. Pathol. 2016, 238, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Stanisavljevic, J.; Porta-de-la-Riva, M.; Batlle, R.; de Herreros, A.G.; Baulida, J. The P65 Subunit of NF-κB and PARP1 Assist Snail1 in Activating Fibronectin Transcription. J. Cell Sci. 2011, 124 Pt 24, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Vega, M.; Escoto-Chavez, S.E.; Murdock, B.; Sakai, T.; Baritaki, S.; Bonavida, B. Nitric oxide sensitizes tumor cells to TRAIL-induced apoptosis via inhibition of the DR5 transcription repressor Yin Yang 1. Nitric Oxide 2009, 20, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Vega, M.; Jazirehi, A.; Garban, H.; Hongo, F.; Cheng, G.; Bonavida, B. Nitric Oxide x Cell Lines to TRAIL-Mediated Apoptosis via Inactivation of NF-Kappa B and Inhibition of Bcl-Xl Expression. Oncogene 2004, 23, 4993–5003. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Chapman, A.; Yeung, K.; Spandidos, D.A.; Palladino, M.; Bonavida, B. Inhibition of Epithelial to Mesenchymal Transition in Metastatic Prostate Cancer Cells by the Novel Proteasome Inhibitor, NPI-0052: Pivotal Roles of Snail Repression and RKIP Induction. Oncogene 2009, 28, 3573–3585. [Google Scholar] [CrossRef]
- Baritaki, S.; Yeung, K.; Palladino, M.; Berenson, J.; Bonavida, B. Pivotal Roles of Snail Inhibition and RKIP Induction by the Proteasome Inhibitor NPI-0052 in Tumor Cell Chemoimmunosensitization. Cancer Res. 2009, 69, 8376–8385. [Google Scholar] [CrossRef]
- Burute, M.; Prioux, M.; Blin, G.; Truchet, S.; Letort, G.; Tseng, Q.; Bessy, T.; Lowell, S.; Young, J.; Filhol, O.; et al. Polarity Reversal by Centrosome Repositioning Primes Cell Scattering during Epithelial-to-Mesenchymal Transition. Dev. Cell 2017, 40, 168–184. [Google Scholar] [CrossRef]
- Li, M.; Li, M.; Xia, Y.; Li, G.; Su, X.; Wang, D.; Ye, J.; Lu, F.; Sun, T.; Ji, C. HDAC1/3-dependent moderate liquid–liquid phase separation of YY1 promotes METTL3 expression and AML cell proliferation. Cell Death Dis. 2022, 13, 992. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Wang, Z.; Liu, X.; Guo, L.; Huang, L.; Gao, L.; McNutt, M.A.; Li, G. The role of transcription factors Sp1 and YY1 in proximal promoter region in initiation of transcription of the mu opioid receptor gene in human lymphocytes. J. Cell. Biochem. 2008, 104, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Wottrich, S.; Kaufhold, S.; Chrysos, E.; Zoras, O.; Baritaki, S.; Bonavida, B. Inverse correlation between the metastasis suppressor RKIP and the metastasis inducer YY1: Contrasting roles in the regulation of chemo/immuno-resistance in cancer. Drug Resist. Updates 2017, 30, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B. Therapeutic YY1 Inhibitors in Cancer: ALL in ONE. Crit. Rev. Oncog. 2017, 22, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wan, M.; Cao, P.; Rao, A.; Cramer, S.D.; Sui, G. Yin Yang 1 regulates the transcriptional activity of androgen receptor. Oncogene 2009, 28, 3746–3757. [Google Scholar] [CrossRef]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of Transcription Factor YY1 by Acetylation and Deacetylation. Mol. Cell. Biol. 2001, 21, 5979–5991. [Google Scholar] [CrossRef]
- Lee, J.S.; Galvin, K.M.; See, R.H.; Eckner, R.; Livingston, D.; Moran, E.; Shi, Y. Relief of YY1 Transcriptional Repression by Adenovirus E1A Is Mediated by E1A-Associated Protein P300. Genes Dev. 1995, 9, 1188–1198. [Google Scholar] [CrossRef]
- Yakovleva, T.; Kolesnikova, L.; Vukojević, V.; Gileva, I.; Tan-No, K.; Austen, M.; Lüscher, B.; Ekström, T.J.; Terenius, L.; Bakalkin, G. YY1 binding to a subset of p53 DNA-target sites regulates p53-dependent transcription. Biochem. Biophys. Res. Commun. 2004, 318, 615–624. [Google Scholar] [CrossRef]
- Garbán, H.J.; Bonavida, B. Nitric Oxide Inhibits the Transcription Repressor Yin-Yang 1 Binding Activity at the Silencer Region of the Fas Promoter: A Pivotal Role for Nitric Oxide in the Up-Regulation of Fas Gene Expression in Human Tumor Cells. J. Immunol. 2001, 167, 75–81. [Google Scholar] [CrossRef]
- Pothoulakis, C.; Torre-Rojas, M.; Duran-Padilla, M.A.; Gevorkian, J.; Zoras, O.; Chrysos, E.; Chalkiadakis, G.; Baritaki, S. CRHR2/Ucn2 signaling is a novel regulator of miR-7/YY1/Fas circuitry contributing to reversal of colorectal cancer cell resistance to Fas-mediated apoptosis. Int. J. Cancer 2018, 142, 334–346. [Google Scholar] [CrossRef]
- Galloway, N.R.; Ball, K.F.; Stiff, T.; Wall, N.R. Yin Yang 1 (YY1): Regulation of Survivin and Its Role in Invasion and Metastasis. Crit. Rev. Oncog. 2017, 22, 23–36. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK Signaling Pathway Is Essential for Cancer-Immune Evasion in Human Melanoma Cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.Q.; Zhou, J.; Durflinger, K.H.; Langhan, M.M.; Shelton, T.E.; Wunderlich, J.R.; Robbins, P.F.; Rosenberg, S.A.; Dudley, M.E. Minimally Cultured Tumor-Infiltrating Lymphocytes Display Optimal Characteristics for Adoptive Cell Therapy. J. Immunother. 2008, 31, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Melino, G.; Amelio, I.; Jiang, J.; Wang, Y.; Shi, Y. Recent advances in cancer immunotherapy. Discov. Oncol. 2021, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Khoramshahi, V.; Azani, A.; Soltaninejad, E.; Aslani, S.; Zamani, M.R.; Zal, M.; Nesaei, A.; Hosseini, S.M. PD-1 and cancer: Molecular mechanisms and polymorphisms. Immunogenetics 2018, 70, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Hofmeyer, K.A.; Jeon, H.; Zang, X. The PD-1/PD-L1 (B7-H1) Pathway in Chronic Infection-Induced Cytotoxic T Lymphocyte Exhaustion. J. Biomed. Biotechnol. 2011, 2011, 451694. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Balkhi, M.Y.; Wittmann, G.; Xiong, F.; Junghans, R.P. YY1 Upregulates Checkpoint Receptors and Downregulates Type I Cytokines in Exhausted, Chronically Stimulated Human T Cells. iScience 2018, 2, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Balkhi, M.Y.; Ma, Q.; Ahmad, S.; Junghans, R.P. T cell exhaustion and Interleukin 2 downregulation. Cytokine 2015, 71, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef]
- Kwiatkowska, D.; Mazur, E.; Reich, A. YY1 Is a Key Player in Melanoma Immunotherapy/Targeted Treatment Resistance. Front. Oncol. 2022, 12, 856963. [Google Scholar] [CrossRef]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef]
- Goepfert, K.; Dinsart, C.; Rommelaere, J.; Foerster, F.; Moehler, M. Rational Combination of Parvovirus H1 With CTLA-4 and PD-1 Checkpoint Inhibitors Dampens the Tumor Induced Immune Silencing. Front. Oncol. 2019, 9, 425. [Google Scholar] [CrossRef]
- Sun, X.; Roudi, R.; Dai, T.; Chen, S.; Fan, B.; Li, H.; Zhou, Y.; Zhou, M.; Zhu, B.; Yin, C.; et al. Immune-Related Adverse Events Associated with Programmed Cell Death Protein-1 and Programmed Cell Death Ligand 1 Inhibitors for Non-Small Cell Lung Cancer: A PRISMA Systematic Review and Meta-Analysis. BMC Cancer 2019, 19, 558. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Adam, A.; Zhao, C.; Chen, H. Recent Advancements in the Mechanisms Underlying Resistance to PD-1/PD-L1 Blockade Immunotherapy. Cancers 2021, 13, 663. [Google Scholar] [CrossRef] [PubMed]
- American Association for Cancer Research. Tiragolumab Impresses in Multiple Trials. Cancer Discov. 2020, 10, 1086–1087. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Cui, B. Effect of Radiotherapy on T Cell and PD-1/PD-L1 Blocking Therapy in Tumor Microenvironment. Hum. Vaccin. Immunother. 2021, 17, 1555–1567. [Google Scholar] [CrossRef]
- Muth, S.T.; Saung, M.T.; Blair, A.B.; Henderson, M.G.; Thomas, D.L.; Zheng, L. CD137 Agonist-Based Combination Immunotherapy Enhances Activated, Effector Memory T Cells and Prolongs Survival in Pancreatic Adenocarcinoma. Cancer Lett. 2021, 499, 99–108. [Google Scholar] [CrossRef]
- Zhou, X.; Zou, L.; Liao, H.; Luo, J.; Yang, T.; Wu, J.; Chen, W.; Wu, K.; Cen, S.; Lv, D.; et al. Abrogation of HnRNP L enhances anti-PD-1 therapy efficacy via diminishing PD-L1 and promoting CD8+ T cell-mediated ferroptosis in castration-resistant prostate cancer. Acta Pharm. Sin. B 2022, 12, 692–707. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’Er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on Both Effector and Regulatory T Cell Compartments Contributes to the Antitumor Activity of Anti–CTLA-4 Antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, S.; Yao, Q.; Du, N.; Fu, X.; Lou, Y.; Wang, M.; Mao, F.; Mao, D.; Khadaroo, P.A.; et al. The efficacy and safety of combined immune checkpoint inhibitors (nivolumab plus ipilimumab): A systematic review and meta-analysis. World J. Surg. Oncol. 2020, 18, 150. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Hochster, H.; Benson, A. Perspectives on Treatment of Metastatic Colorectal Cancer with Immune Checkpoint Inhibitor Therapy. Oncologist 2020, 25, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.-J.; Gangadhar, T.C.; et al. Anti-Programmed-Death-Receptor-1 Treatment with Pembrolizumab in Ipilimumab-Refractory Advanced Melanoma: A Randomised Dose-Comparison Cohort of a Phase 1 Trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer Regression and Autoimmunity Induced by Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade in Patients with Metastatic Melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Apalla, Z.; Rapoport, B.; Sibaud, V. Dermatologic immune-related adverse events: The toxicity spectrum and recommendations for management. Int. J. Women’s Dermatol. 2021, 7, 625–635. [Google Scholar] [CrossRef]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Qiu, P.; Jiang, J.; Wu, X.; Mei, J.; Sun, H. Decidual macrophages derived NO downregulates PD-L1 in trophoblasts leading to decreased Treg cells in recurrent miscarriage. Front. Immunol. 2023, 14, 1180154. [Google Scholar] [CrossRef]
- Hsu, J.M.; Li, C.W.; Lai, Y.J.; Hung, M.C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, X.; Li, Y.; Xu, H.; Ying, B.; Qiu, J.; Huang, Z.; Shao, X. YY1 alleviates lupus nephritis-induced renal injury by reducing the Th17/Treg cell ratio via the IFN-γ/Fra2 axis. Lab. Investig. 2022, 102, 872–884. [Google Scholar] [CrossRef]
- Ye, J.; Cippitelli, M.; Dorman, L.; Ortaldo, J.R.; Young, H.A. The nuclear factor YY1 suppresses the human gamma interferon promoter through two mechanisms: Inhibition of AP1 binding and activation of a silencer element. Mol. Cell. Biol. 1996, 16, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A Novel Bifunctional Anti-PD-L1/TGF-β Trap Fusion Protein (M7824) Efficiently Reverts Mesenchymalization of Human Lung Cancer Cells. Oncoimmunology 2017, 6, e1349589. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN Loss Increases PD-L1 Protein Expression and Affects the Correlation between PD-L1 Expression and Clinical Parameters in Colorectal Cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef] [PubMed]
- Austen, M.; Cerni, C.; Lüscher-Firzlaff, J.M.; Lüscher, B. YY1 can inhibit c-Myc function through a mechanism requiring DNA binding of YY1 but neither its transactivation domain nor direct interaction with c-Myc. Oncogene 1998, 17, 511–520. [Google Scholar] [CrossRef]
- Vella, P.; Barozzi, I.; Cuomo, A.; Bonaldi, T.; Pasini, D. Yin Yang 1 extends the Myc-related transcription factors network in embryonic stem cells. Nucleic Acids Res. 2012, 40, 3403–3418. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Housley, F.; Yau, C.; Nakagawa, R.; Winkler, J.; Anttila, J.M.; Munne, P.M.; Savelius, M.; Houlahan, K.E.; Van de Mark, D.; et al. Combinatorial immunotherapies overcome MYC-driven immune evasion in triple negative breast cancer. Nat. Commun. 2022, 13, 3671. [Google Scholar] [CrossRef]
- Hosea, R.; Hillary, S.; Wu, S.; Kasim, V. Targeting Transcription Factor YY1 for Cancer Treatment: Current Strategies and Future Directions. Cancers 2023, 15, 3506. [Google Scholar] [CrossRef]
- You, J.; Tao, B.; Peng, L.; Peng, T.; He, H.; Zeng, S.; Han, J.; Chen, L.; Xia, X.; Yang, X.; et al. Transcription Factor YY1 Mediates Self-Renewal of Glioblastoma Stem Cells through Regulation of the SENP1/METTL3/MYC Axis. Cancer Gene Ther. 2023, 30, 683–693. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, T.; Yang, Y.; Gao, L.; Zhao, Q.; Zhang, W.; Xi, T.; Zheng, L. Transcriptional Factor Yin Yang 1 Promotes the Stemness of Breast Cancer Cells by Suppressing miR-873-5p Transcriptional Activity. Mol. Ther. Nucleic Acids 2020, 21, 527–541. [Google Scholar] [CrossRef]
- Zhao, L.; Li, R.; Qiu, J.Z.; Yu, J.B.; Cao, Y.; Yuan, R.T. YY1-mediated PTEN dephosphorylation antagonizes IR-induced DNA repair contributing to tongue squamous cell carcinoma radiosensitization. Mol. Cell. Probes 2020, 53, 101577. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.-L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W., III; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT–mTOR Pathway in Non–Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef]
- Lin, J.; He, Y.; Wang, B.; Xun, Z.; Chen, S.; Zeng, Z.; Ou, Q. Blocking of YY1 Reduce Neutrophil Infiltration by Inhibiting IL-8 Production via the PI3K-Akt-mTOR Signaling Pathway in Rheumatoid Arthritis. Clin. Exp. Immunol. 2019, 195, 226–236. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Wölfle, S.J.; Strebovsky, J.; Bartz, H.; Sähr, A.; Arnold, C.; Kaiser, C.; Dalpke, A.H.; Heeg, K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 2011, 41, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wu, H.Y.; Chen, S. Influence of TBX21 T-1993C variant on autoimmune hepatitis development by Yin-Yang 1 binding. World J. Gastroenterol. 2017, 23, 8500–8511. [Google Scholar] [CrossRef]
- Weill, L.; Shestakova, E.; Bonnefoy, E. Transcription Factor YY1 Binds to the Murine Beta Interferon Promoter and Regulates Its Transcriptional Capacity with a Dual Activator/Repressor Role. J. Virol. 2003, 77, 2903–2914. [Google Scholar] [CrossRef]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015, 4, e1008824. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Gaire, B.; Zou, Y.; Uddin, M.M.; Vancurova, I. IFNγ-induced PD-L1 expression in ovarian cancer cells is regulated by JAK1, STAT1 and IRF1 signaling. Cell. Signal. 2022, 97, 110400. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Wright, J.G.; Hu, N.N.; Sadikot, R.T.; Park, G.Y.; Blackwell, T.S.; Christman, J.W. Yin Yang 1 enhances cyclooxygenase-2 gene expression in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1219–L1226. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wei, J.; Xue, C.; Zhou, X.; Chen, S.; Zheng, L.; Duan, Y.; Deng, H.; Xiong, W.; Tang, F.; et al. Dissecting the roles and clinical potential of YY1 in the tumor microenvironment. Front. Oncol. 2023, 13, 1122110. [Google Scholar] [CrossRef] [PubMed]
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J. Transl. Med. 2017, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Cecil, D.L.; Gad, E.A.; Corulli, L.R.; Drovetto, N.; Lubet, R.A.; Disis, M.L. COX-2 Inhibitors Decrease Expression of PD-L1 in Colon Tumors and Increase the Influx of Type I Tumor-infiltrating Lymphocytes. Cancer Prev. Res. 2022, 15, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, N.; Chen, E.P.; Evans, R.A.; Ndong, V.; Vonderheide, R.H.; Smyth, E.M. Mammary carcinoma cell derived cyclooxygenase 2 suppresses tumor immune surveillance by enhancing intratumoral immune checkpoint activity. Breast Cancer Res. 2013, 15, R75. [Google Scholar] [CrossRef] [PubMed]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 58426. [Google Scholar] [CrossRef]
- De Cicco, P.; Panza, E.; Ercolano, G.; Armogida, C.; Sessa, G.; Pirozzi, G.; Cirino, G.; Wallace, J.L.; Ianaro, A. ATB-346, a novel hydrogen sulfide-releasing anti-inflammatory drug, induces apoptosis of human melanoma cells and inhibits melanoma development in vivo. Pharmacol. Res. 2016, 114, 67–73. [Google Scholar] [CrossRef]
- Yang, M.Y.; Lee, H.T.; Chen, C.M.; Shen, C.C.; Ma, H.I. Celecoxib Suppresses the Phosphorylation of STAT3 Protein and Can Enhance the Radiosensitivity of Medulloblastoma-Derived Cancer Stem-Like Cells. Int. J. Mol. Sci. 2014, 15, 11013–11029. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C.-K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 Activation Is Responsible for IL-6-Dependent T Cell Proliferation through Preventing Apoptosis: Generation and Characterization of T Cell-Specific Stat3-Deficient Mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar] [CrossRef]
- Raz, R.; Lee, C.K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential Role of STAT3 for Embryonic Stem Cell Pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-Renewal of Pluripotent Embryonic Stem Cells Is Mediated via Activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Xia, X.; Huang, P. The Role of Oncogenes and Redox Signaling in the Regulation of PD-L1 in Cancer. Cancers 2021, 13, 4426. [Google Scholar] [CrossRef]
- Elashi, A.A.; Sasidharan Nair, V.; Taha, R.Z.; Shaath, H.; Elkord, E. DNA Methylation of Immune Checkpoints in the Peripheral Blood of Breast and Colorectal Cancer Patients. OncoImmunology 2019, 8, e1542918. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Wu, W.; Long, H.; Huang, J.; Wang, Z.; Li, T.; Tang, S.; Zhu, B.; Chen, D. EZH2 Regulates PD-L1 Expression via HIF-1α in Non-Small Cell Lung Cancer Cells. Biochem. Biophys. Res. Commun. 2019, 517, 201–209. [Google Scholar] [CrossRef]
- Xiao, G.; Jin, L.-L.; Liu, C.-Q.; Wang, Y.-C.; Meng, Y.-M.; Zhou, Z.-G.; Chen, J.; Yu, X.-J.; Zhang, Y.-J.; Xu, J.; et al. EZH2 Negatively Regulates PD-L1 Expression in Hepatocellular Carcinoma. J. Immunother. Cancer 2019, 7, 300. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, X.; Xu, L.; Li, Y.; Zeng, C. Current insight into the regulation of PD-L1 in cancer. Exp. Hematol. Oncol. 2022, 11, 44. [Google Scholar] [CrossRef]
- Bu, L.L.; Yu, G.T.; Wu, L.; Mao, L.; Deng, W.W.; Liu, J.F.; Kulkarni, A.B.; Zhang, W.F.; Zhang, L.; Sun, Z.J. STAT3 Induces Immunosuppression by Upregulating PD-1/PD-L1 in HNSCC. J. Dent. Res. 2017, 96, 1027–1034. [Google Scholar] [CrossRef]
- Cerezo, M.; Guemiri, R.; Druillennec, S.; Girault, I.; Malka-Mahieu, H.; Shen, S.; Allard, D.; Martineau, S.; Welsch, C.; Agoussi, S.; et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat. Med. 2018, 24, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Lienlaf, M.; Perez-Villarroel, P.; Knox, T.; Pabon, M.; Sahakian, E.; Powers, J.; Woan, K.; Lee, C.; Cheng, F.; Deng, S.; et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol. Oncol. 2016, 10, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Nanamori, H.; Sawada, Y. Epigenetic Modification of PD-1/PD-L1-Mediated Cancer Immunotherapy against Melanoma. Int. J. Mol. Sci. 2022, 23, 1119. [Google Scholar] [CrossRef] [PubMed]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC Phase 2 Study of Pembrolizumab and Entinostat in Patients with Metastatic Uveal Melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef] [PubMed]
- Sah, V.R.; Karlsson, J.; Jespersen, H.; Lindberg, M.F.; Nilsson, L.M.; Ny, L.; Nilsson, J.A. Epigenetic Therapy to Enhance Therapeutic Effects of PD-1 Inhibition in Therapy-Resistant Melanoma. Melanoma Res. 2022, 32, 241–248. [Google Scholar] [CrossRef]
- Deng, S.; Hu, Q.; Zhang, H.; Yang, F.; Peng, C.; Huang, C. HDAC3 Inhibition Upregulates PD-L1 Expression in B-Cell Lymphomas and Augments the Efficacy of Anti-PD-L1 Therapy. Mol. Cancer Ther. 2019, 18, 900–908. [Google Scholar] [CrossRef]
- Shin, H.S.; Choi, J.; Lee, J.; Lee, S.Y. Histone Deacetylase as a Valuable Predictive Biomarker and Therapeutic Target in Immunotherapy for Non–Small Cell Lung Cancer. Cancer Res. Treat. 2022, 54, 458–468. [Google Scholar] [CrossRef]
- Tang, W.; Zhou, W.; Xiang, L.; Wu, X.; Zhang, P.; Wang, J.; Liu, G.; Zhang, W.; Peng, Y.; Huang, X.; et al. The p300/YY1/miR-500a-5p/HDAC2 signalling axis regulates cell proliferation in human colorectal cancer. Nat. Commun. 2019, 10, 663. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, J.G.; Jia, X.Y.; Ke, Y.; Zhang, M.K.; Stieg, D.; Liu, W.J.; Liu, L.Z.; Wang, L.; Jiang, B.H. METTL3-mediated N6-methyladenosine modification and HDAC5/YY1 promote IFFO1 downregulation in tumor development and chemo-resistance. Cancer Lett. 2023, 553, 215971. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. JNCI J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef]
- Grönroos, E.; Terentiev, A.A.; Punga, T.; Ericsson, J. YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 2004, 101, 12165–12170. [Google Scholar] [CrossRef]
- Khachigian, L.M. The Yin and Yang of YY1 in tumor growth and suppression. Int. J. Cancer 2018, 143, 460–465. [Google Scholar] [CrossRef]
- Jo, H.; Shim, K.; Jeoung, D. Potential of the miR-200 Family as a Target for Developing Anti-Cancer Therapeutics. Int. J. Mol. Sci. 2022, 23, 5881. [Google Scholar] [CrossRef]
- Hafsi, S.; Candido, S.; Maestro, R.; Falzone, L.; Soua, Z.; Bonavida, B.; Spandidos, D.A.; Libra, M. Correlation between the Overexpression of Yin Yang 1 and the Expression Levels of miRNAs in Burkitt’s Lymphoma: A Computational Study. Oncol. Lett. 2016, 11, 1021–1025. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, J.; Xiong, D.; Zheng, J.; McPherson, K.N.; Lee, S.; Huang, M.; Xu, Y.; Chen, S.-H.; Wang, Y.; et al. Aerosolized miR-138-5p and miR-200c targets PD-L1 for lung cancer prevention. Front. Immunol. 2023, 14, 1166951. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201.e10. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Mao, D.; Zhang, X.; Wang, Z.; Xu, G.; Zhang, Y. TMEM97 is transcriptionally activated by YY1 and promotes colorectal cancer progression via the GSK-3β/β-catenin signaling pathway. Hum. Cell 2022, 35, 1535–1546. [Google Scholar] [CrossRef]
- Chan, L.-C.; Li, C.-W.; Xia, W.; Hsu, J.-M.; Lee, H.-H.; Cha, J.-H.; Wang, H.-L.; Yang, W.-H.; Yen, E.-Y.; Chang, W.-C.; et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Investig. 2019, 129, 3324–3338. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring Function in Exhausted CD8 T Cells during Chronic Viral Infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Borch, T.H.; Hansen, M.; Andersen, M.H. PD-L1-specific T cells. Cancer Immunol. Immunother. 2016, 65, 797–804. [Google Scholar] [CrossRef]
- Durgan, K.; Ali, M.; Warner, P.; Latchman, Y.E. Targeting NKT cells and PD-L1 pathway results in augmented anti-tumor responses in a melanoma model. Cancer Immunol. Immunother. 2011, 60, 547–558. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Horn, L.A.; Alvarez, J.A. Novel strategies for inhibiting PD-1 pathway-mediated immune suppression while simultaneously delivering activating signals to tumor-reactive T cells. Cancer Immunol. Immunother. 2015, 64, 1287–1293. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef]
- Bonavida, B.; Kaufhold, S. Prognostic significance of YY1 protein expression and mRNA levels by bioinformatics analysis in human cancers: A therapeutic target. Pharmacol. Ther. 2015, 150, 149–168. [Google Scholar] [CrossRef]
- Palmer, M.B.; Majumder, P.; Cooper, J.C.; Yoon, H.; Wade, P.A.; Boss, J.M. Yin Yang 1 Regulates the Expression of Snail through a Distal Enhancer. Mol. Cancer Res. 2009, 7, 221–229. [Google Scholar] [CrossRef]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Meca-Cortés, O.; Mateo, F.; de Paz, A.M.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-Mesenchymal Transition Can Suppress Major Attributes of Human Epithelial Tumor-Initiating Cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef]
- Kim, S.; Koh, J.; Kim, M.Y.; Kwon, D.; Go, H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Tang, X.-Y.; Xiong, Y.-L.; Zhao, Y.-B.; Yang, J.; Shi, A.-P.; Zheng, K.-F.; Liu, Y.-J.; Shu, C.; Jiang, T.; Ma, N.; et al. Dual immunological and proliferative regulation of immune checkpoint FGL1 in lung adenocarcinoma: The pivotal role of the YY1–FGL1–MYH9 axis. Front. Immunol. 2022, 13, 1014053. [Google Scholar] [CrossRef]
- Tsai, H.; Wu, Y.; Liu, X.; Xu, Z.; Liu, L.; Wang, C.; Zhang, H.; Huang, Y.; Wang, L.; Zhang, W.; et al. Engineered Small Extracellular Vesicles as a FGL1/PD-L1 Dual-Targeting Delivery System for Alleviating Immune Rejection. Adv. Sci. 2021, 9, 2102634. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-J.; Wang, L.; Zhou, F.-M.; Liu, S.-W.; Wang, W.; Zhao, E.-J.; Yao, Q.-J.; Li, W.; Zhao, Y.-Q.; Shi, Z.; et al. Elevated NOX4 promotes tumorigenesis and acquired EGFR-TKIs resistance via enhancing IL-8/PD-L1 signaling in NSCLC. Drug Resist. Updates 2023, 70, 100987. [Google Scholar] [CrossRef]
- Hosseinahli, N.; Aghapour, M.; Duijf, P.H.G.; Baradaran, B. Treating cancer with microRNA replacement therapy: A literature review. J. Cell. Physiol. 2018, 233, 5574–5588. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Zhang, W.; Liu, H.; Jiang, Y.; Li, F.; Wu, M.; Waterhouse, G.I.N.; Sun-Waterhouse, D.; Li, D. Quercetin Induces Apoptosis in HepG2 Cells via Directly Interacting with YY1 to Disrupt YY1-P53 Interaction. Metabolites 2023, 13, 229. [Google Scholar] [CrossRef]
- Shao, Z.; Yang, W.; Meng, X.; Li, M.; Hou, P.; Li, Z.; Chu, S.; Zheng, J.; Bai, J. The Role of Transcription Factor Yin Yang-1 in Colorectal Cancer. Cancer Med. 2023, 12, 11177–11190. [Google Scholar] [CrossRef] [PubMed]
- Galloway, N.R.; Osterman CJ, D.; Reiber, K.; Jutzy JM, S.; Li, F.; Sui, G.; Soto, U.; Wall, N.R. Yin Yang 1 Regulates the Transcriptional Repression of Survivin. Biochem. Biophys. Res. Commun. 2014, 445, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Paniagua, M.; Vega, M.I.; Huerta-Yepez, S.; Hariharan, H.; Chen, H.; Berenson, J.R.; Bonavida, B. The Pivotal Role of Yin Yang 1 (YY1) Inhibition (and Downstream Bcl-2/Bclxl) by Galiximab (Anti-CD80 mAb) In the Reversal of Resistance of B-NHL Cells to Chemotherapy. Blood 2010, 116, 2887. [Google Scholar] [CrossRef]
- Notarbartolo, M.; Giannitrapani, L.; Vivona, N.; Poma, P.; Labbozzetta, M.; Florena, A.M.; Porcasi, R.; Muggeo, V.M.R.; Sandonato, L.; Cervello, M.; et al. Frequent Alteration of the Yin Yang 1/Raf-1 Kinase Inhibitory Protein Ratio in Hepatocellular Carcinoma. OMICS J. Integr. Biol. 2011, 15, 267–272. [Google Scholar] [CrossRef]
- Hays, E.; Bonavida, B. Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies. Antioxidants 2019, 8, 407. [Google Scholar] [CrossRef]
- Hongo, F.; Garban, H.; Huerta-Yepez, S.; Vega, M.; Jazirehi, A.R.; Mizutani, Y.; Miki, T.; Bonavida, B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005, 336, 692–701. [Google Scholar] [CrossRef]
- Donohoe, M.E.; Zhang, X.; McGinnis, L.; Biggers, J.; Li, E.; Shi, Y. Targeted Disruption of Mouse Yin Yang 1 Transcription Factor Results in Peri-Implantation Lethality. Mol. Cell. Biol. 1999, 19, 7237–7244. [Google Scholar] [CrossRef]
- Thomas, W.D.; Hersey, P. TNF-Related Apoptosis-Inducing Ligand (TRAIL) Induces Apoptosis in Fas Ligand-Resistant Melanoma Cells and Mediates CD4 T Cell Killing of Target Cells. J. Immunol. 1998, 161, 2195–2200. [Google Scholar] [CrossRef]
- Huerta-Yepez, S.; Vega, M.; Garban, H.; Bonavida, B. Involvement of the TNF-Alpha Autocrine-Paracrine Loop, via NF-kappaB and YY1, in the Regulation of Tumor Cell Resistance to Fas-Induced Apoptosis. Clin. Immunol. 2006, 120, 297–309. [Google Scholar] [CrossRef]
- Wang, H.; Hertlein, E.; Bakkar, N.; Sun, H.; Acharyya, S.; Wang, J.; Carathers, M.; Davuluri, R.; Guttridge, D.C. NF-κB Regulation of YY1 Inhibits Skeletal Myogenesis through Transcriptional Silencing of Myofibrillar Genes. Mol. Cell. Biol. 2007, 27, 4374–4387. [Google Scholar] [CrossRef]
- Huerta-Yepez, S.; Baritaki, S.; Baay-Guzman, G.; Hernandez-Luna, M.A.; Hernandez-Cueto, A.; Vega, M.I.; Bonavida, B. Contribution of Either YY1 or BclXL-Induced Inhibition by the NO-Donor DETANONOate in the Reversal of Drug Resistance, Both in Vitro and in Vivo. YY1 and BclXL Are Overexpressed in Prostate Cancer. Nitric Oxide 2013, 29, 17–24. [Google Scholar] [CrossRef]
- Liu, X.; Jutooru, I.; Lei, P.; Kim, K.; Lee, S.-O.; Brents, L.K.; Prather, P.L.; Safe, S. Betulinic Acid Targets YY1 and ErbB2 through Cannabinoid Receptor-Dependent Disruption of microRNA-27a:ZBTB10 in Breast Cancer. Mol. Cancer Ther. 2012, 11, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Henley, M.J.; Koehler, A.N. Advances in targeting “undruggable” transcription factors with small molecules. Nat. Rev. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Tian, X.; Wang, Y.; Yang, W.; Li, W.; Song, M.; Zhao, R.; Wang, M.; Gao, Q.; Li, T.; et al. Repurposing pentamidine for cancer immunotherapy by targeting the PD1/PD-L1 immune checkpoint. Front. Immunol. 2023, 14, 1145028. [Google Scholar] [CrossRef] [PubMed]
- Pabian-Jewuła, S.; Bragiel-Pieczonka, A.; Rylski, M. Ying Yang 1 engagement in brain pathology. J. Neurochem. 2022, 161, 236–253. [Google Scholar] [CrossRef]
- Karki, P.; Johnson, J.; Son, D.-S.; Aschner, M.; Lee, E. Transcriptional Regulation of Human Transforming Growth Factor-α in Astrocytes. Mol. Neurobiol. 2017, 54, 964–976. [Google Scholar] [CrossRef]
- Karki, P.; Kim, C.; Smith, K.; Son, D.-S.; Aschner, M.; Lee, E. Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-κB and Yin Yang 1 (YY1). J. Biol. Chem. 2015, 290, 23725–23737. [Google Scholar] [CrossRef]
- Karki, P.; Webb, A.; Smith, K.; Johnson, J.; Lee, K.; Son, D.-S.; Aschner, M.; Lee, E. Yin Yang 1 Is a Repressor of Glutamate Transporter EAAT2, and It Mediates Manganese-Induced Decrease of EAAT2 Expression in Astrocytes. Mol. Cell. Biol. 2014, 34, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Tania, L.-P.; Mariana, H.-C.; Mario, M.-M.; Johannes, T.; Till, O.; Thomas, E.; Benjamin, B.; Sara, H.-Y. A newly synthesized small chemical inhibitor of Ying Yang (YY1): Inhibition of its DNA-binding activity and sensitized drug-resistant human ALL cell lines to cell death and apoptosis by chemotherapeutic drugs. submitted for publication.
- Liu, H.; Han, J.; Lv, Y.; Zhao, Z.; Zheng, S.; Sun, Y.; Sun, T. Isorhamnetin and anti-PD-L1 antibody dual-functional mesoporous silica nanoparticles improve tumor immune microenvironment and inhibit YY1-mediated tumor progression. J. Nanobiotechnology 2023, 21, 208. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Desai, K.; Macapinlac, M.; Wadler, S.; Goldberg, G.; Fields, A.; Einstein, M.; Volterra, F.; Wong, B.; Martin, R.; et al. A Phase I Safety and Dose Escalation Trial of Docetaxel Combined with GEM®231, a Second Generation Antisense Oligonucleotide Targeting Protein Kinase A R1α in Patients with Advanced Solid Cancers. Investig. New Drugs 2006, 24, 125–134. [Google Scholar] [CrossRef]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and Efficacy of Drisapersen for the Treatment of Duchenne Muscular Dystrophy (DEMAND II): An Exploratory, Randomised, Placebo-Controlled Phase 2 Study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Chen, S.; Lu, K.; Hou, Y.; You, Z.; Shu, C.; Wei, X.; Wu, T.; Shi, N.; Zhang, G.; Wu, J.; et al. YY1 Complex in M2 Macrophage Promotes Prostate Cancer Progression by Upregulating IL-6. J. Immunother. Cancer 2023, 11, e006020. [Google Scholar] [CrossRef]
- Wójcik, P.; Berlicki, Ł. Peptide-based inhibitors of protein–protein interactions. Bioorg. Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, L.; Lu, L.; Wang, L.; Li, X.; Jiang, P.; Chan, L.K.Y.; Zhang, T.; Yu, J.; Kwong, J.; et al. A novel miR-193a-5p-YY1-APC regulatory axis in human endometrioid endometrial adenocarcinoma. Oncogene 2013, 32, 3432–3442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, X.; Wu, C.W.; Dong, Y.; Cai, M.; Mok, M.T.S.; Wang, H.; Chen, J.; Ng, S.S.M.; Chen, M.; et al. microRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene 2013, 32, 5078–5088. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Y.; Chen, J.C.; Jiao, T.T.; Hui, N.; Qi, X. MicroRNA-181 targets Yin Yang 1 expression and inhibits cervical cancer progression. Mol. Med. Rep. 2015, 11, 4541–4546. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Ge, X.; Geng, Y.; Cao, H.; Zhu, W.; Jiao, Y.; Wu, J.; Zhou, J.; Cao, J. miR-34a inhibits the migration and invasion of esophageal squamous cell carcinoma by targeting Yin Yang-1. Oncol. Rep. 2015, 34, 311–317. [Google Scholar] [CrossRef]
- Yuan, P.; He, X.-H.; Rong, Y.-F.; Cao, J.; Li, Y.; Hu, Y.-P.; Liu, Y.; Li, D.; Lou, W.; Liu, M.-F. KRAS/NF-κB/YY1/miR-489 Signaling Axis Controls Pancreatic Cancer Metastasis. Cancer Res. 2017, 77, 100–111. [Google Scholar] [CrossRef]
- Liu, X.; Cao, Z.; Liu, N.; Gao, G.; Du, M.; Wang, Y.; Cheng, B.; Zhu, M.; Jia, B.; Pan, L.; et al. Kill two birds with one stone: Engineered exosome-mediated delivery of cholesterol modified YY1-siRNA enhances chemoradiotherapy sensitivity of glioblastoma. Front. Pharmacol. 2022, 13, 975291. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Schmid, D.; Park, C.G.; Hartl, C.A.; Subedi, N.; Cartwright, A.N.; Puerto, R.B.; Zheng, Y.; Maiarana, J.; Freeman, G.J.; Wucherpfennig, K.W.; et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017, 8, 1747. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Regulation | Modifier or Pathway Downstream of YY1 | Effect on PD-L1 | Reference |
|---|---|---|---|
| Transcriptional | YY1 direct binding to the PD-L1 promoter | Promotes Transcription | [132] |
| Repression of TGF-β | Decreased expression | [63,136] | |
| Extension of MYC-related transcription factor networks | Increased expression | [80,81,82] | |
| Inhibition of MYC function | Decreased expression | [35,140,141] | |
| PI3K/Akt/mTOR activation via PTEN inhibition | Increased expression | [34,145,146,147] | |
| Positive regulation of IL-6/STAT3 | Increased expression | [37,150,161,162,163] | |
| IFN-γ-induced activation of JAK1 and STAT1 | Increased expression | [34,153,154] | |
| Increased COX-2 expression | Increased expression | [155,156,157,158,159,160] | |
| Epigenetic | Increased HDAC6 | Upregulation | [184,186] |
| Interaction with EZH2 | Upregulation | [56,65,171] | |
| Repression of STAT family proteins | Upregulation | [141,142] | |
| Post-transcriptional | Inhibition of p53 and miR-34 | Upregulation | [187,188,189] |
| Downregulation of miR-200 | Downregulation | [190,192] | |
| Post-translational | Suppression of PARP/GSK3β | Degradation | [194,195,196] |
| Stimulation of IL-6/JAK1 | Stabilization | [37,197] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dillen, A.; Bui, I.; Jung, M.; Agioti, S.; Zaravinos, A.; Bonavida, B. Regulation of PD-L1 Expression by YY1 in Cancer: Therapeutic Efficacy of Targeting YY1. Cancers 2024, 16, 1237. https://doi.org/10.3390/cancers16061237
Dillen A, Bui I, Jung M, Agioti S, Zaravinos A, Bonavida B. Regulation of PD-L1 Expression by YY1 in Cancer: Therapeutic Efficacy of Targeting YY1. Cancers. 2024; 16(6):1237. https://doi.org/10.3390/cancers16061237
Chicago/Turabian StyleDillen, Ana, Indy Bui, Megan Jung, Stephanie Agioti, Apostolos Zaravinos, and Benjamin Bonavida. 2024. "Regulation of PD-L1 Expression by YY1 in Cancer: Therapeutic Efficacy of Targeting YY1" Cancers 16, no. 6: 1237. https://doi.org/10.3390/cancers16061237
APA StyleDillen, A., Bui, I., Jung, M., Agioti, S., Zaravinos, A., & Bonavida, B. (2024). Regulation of PD-L1 Expression by YY1 in Cancer: Therapeutic Efficacy of Targeting YY1. Cancers, 16(6), 1237. https://doi.org/10.3390/cancers16061237