
Adjuvant PD-1 Checkpoint Inhibition in Early Cutaneous Melanoma: Immunological Mode of Action and the Role of Ultraviolet Radiation
 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Adjuvant Anti-PD-1 Therapy: Efficacy and Potential Drawbacks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | Stage | Trial Arms (n Patients) | CLND | RFS (%) | Grade 3–5 TRAEs (%) |
|---|---|---|---|---|---|
| CheckMate-238 [14,21,27,33,34] | IIIB, IIIC, IV * | Nivolumab vs. Ipilimumab A: 3 mg/kg nivolumab; Q2W (n = 453) B: ipilimumab 10 mg/kg Q3W 4x, followed by Q12W ≤1 year (n = 453) | Yes | Nivolumab (Arm A) −12 months: 70.5% −24 months: 62.6% −36 months: 58.0% −48 months: 51.7% −60 months: 50.0% | Nivolumab (Arm A): 14.4% |
| Keynote-054 [23,24,25] | IIIA- IIIC * | Pembrolizumab vs. Placebo A: 200 mg pembrolizumab Q3W, 18 doses (~1 year) (n = 514) B: placebo (n = 505) | Yes | Pembrolizumab (Arm A) −12 months: 75.4% −24 months: 68.3% −36 months: 63.7% −60 months: 55.4% | Pembrolizumab (Arm A): 14.7% |
| SWOG S1404 [35] | IIIA- IIID, IV * | Pembrolizumab vs. Adjuvant Standard of Care A: 200 mg pembrolizumab Q3W for 1 year (n = 647) B: high-dose IFNα-2b or 10 mg/kg ipilimumab Q3W 4x, followed by up to 11 doses Q12W (n = 654) | Yes | Not reported | Pembrolizumab (Arm A): 19.5% |
| CheckMate-915 [31] | IIIB- IIID, IV # | Nivolumab/Ipilimumab vs. Nivolumab alone A: 240 mg nivolumab Q2W + ipilimumab 1 mg/kg Q6W (n = 920) B: 480 mg nivolumab Q4W (n = 924) | Yes | Nivolumab/Ipilimumab (Arm A) −24 months: 64.6% Nivolumab Arm (Arm B) −24 months: 63.2% | Nivolumab/Ipilimumab (Arm A): 33.0% Nivolumab Arm (Arm B): 12.8% |
| CheckMate-76K [16] | IIB- IIC # | Nivolumab vs. Placebo A: 480 mg nivolumab Q4W (n = 526) B: placebo (n = 264) | No | Nivolumab (Arm A) −12 months: 89% | Nivolumab (Arm A): 10.5% |
| Keynote-716 [15,36] | IIB- IIC # | Pembrolizumab vs. Placebo A: 200 mg pembrolizumab Q3W, 17 cycles (n = 487) B: placebo (n = 489) | No | Pembrolizumab (Arm A) −12 months: 90% −18 months: 86% −36 months: 76% | Pembrolizumab (Arm A): 17% |
3. Mechanisms of Cellular Response
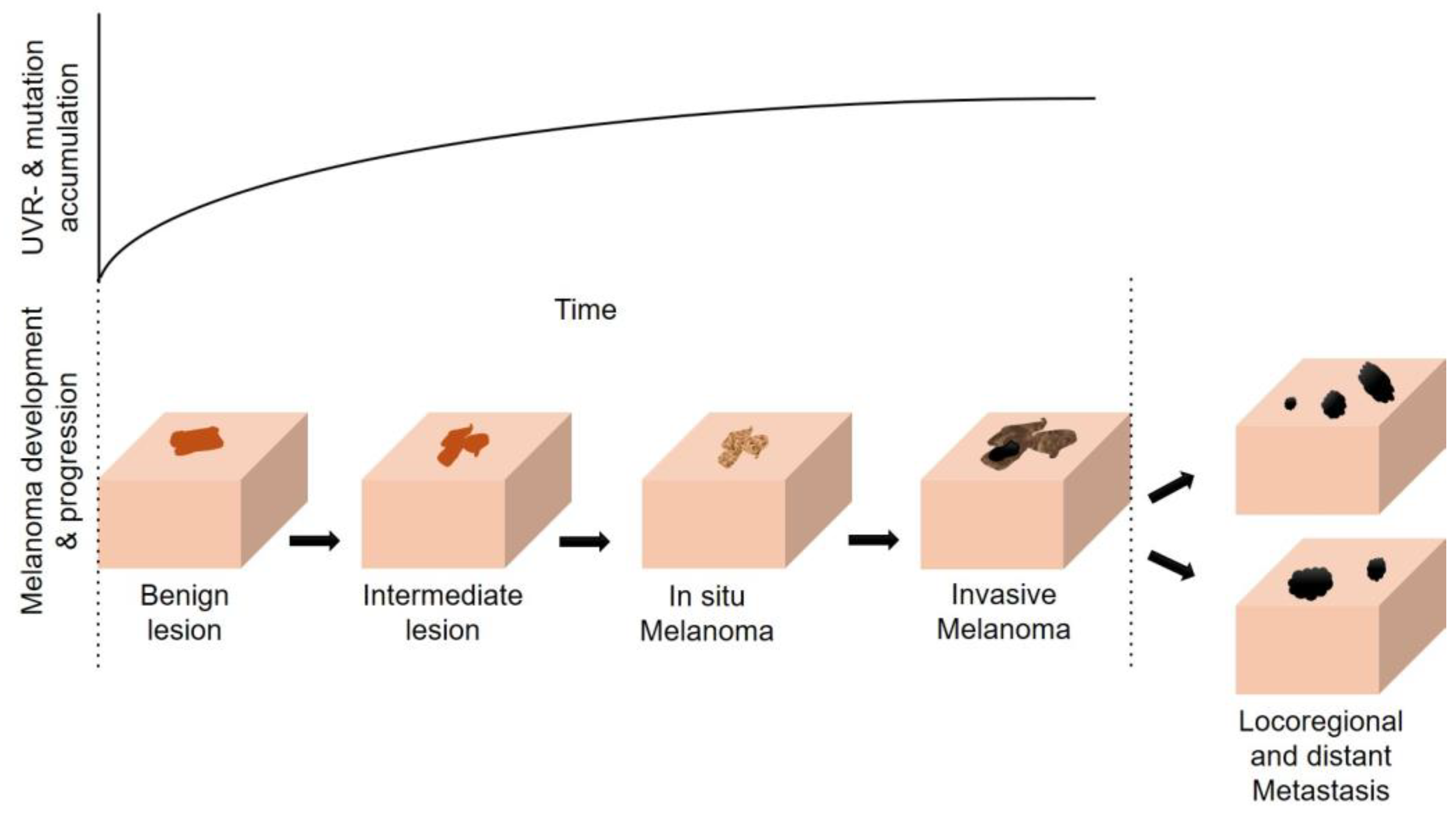
4. The Multifaceted Effects of UVR (in Melanomagenesis)
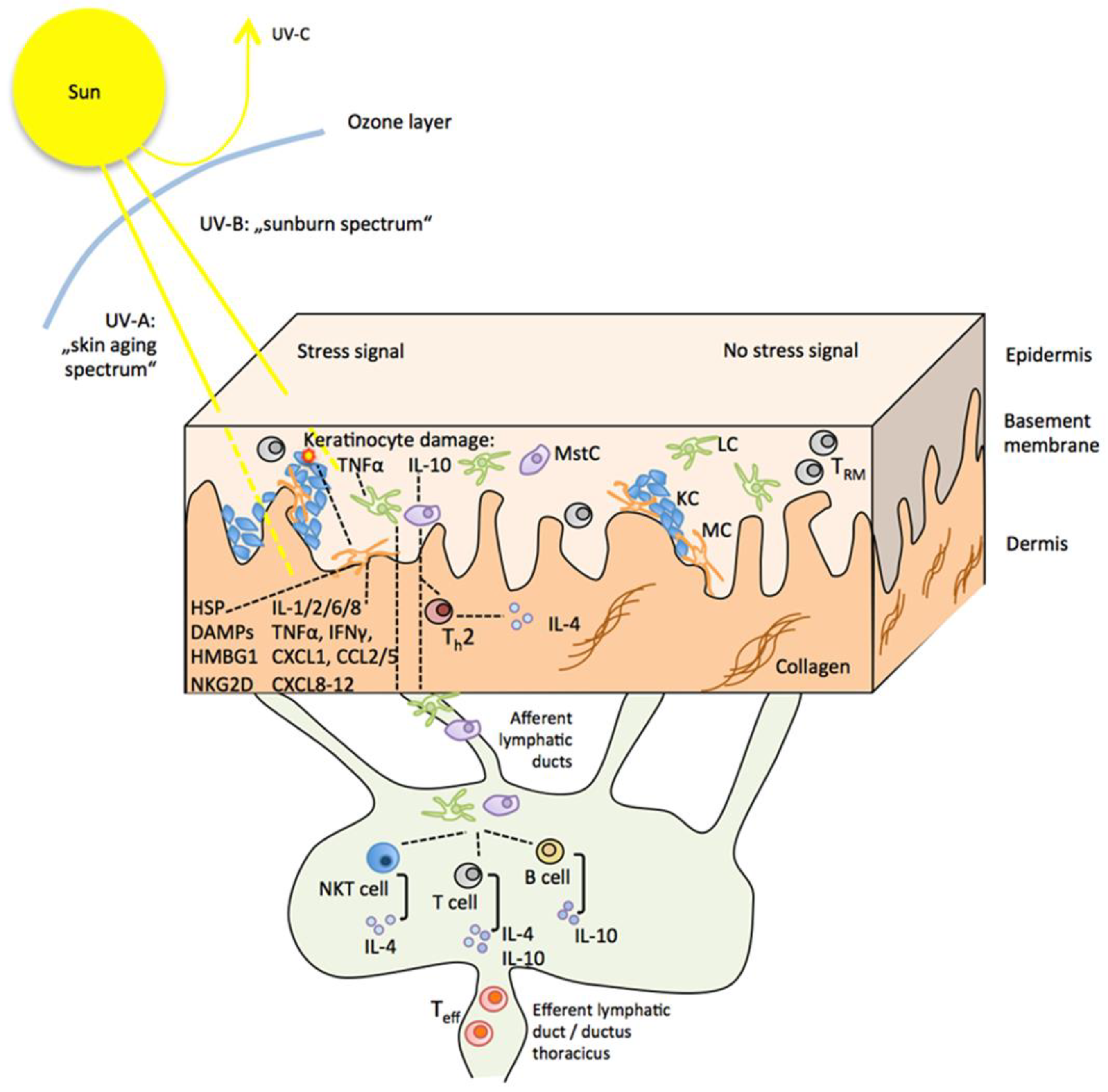
4.1. Direct and Indirect Effects of UVR on the Skin and the Sunburn Response

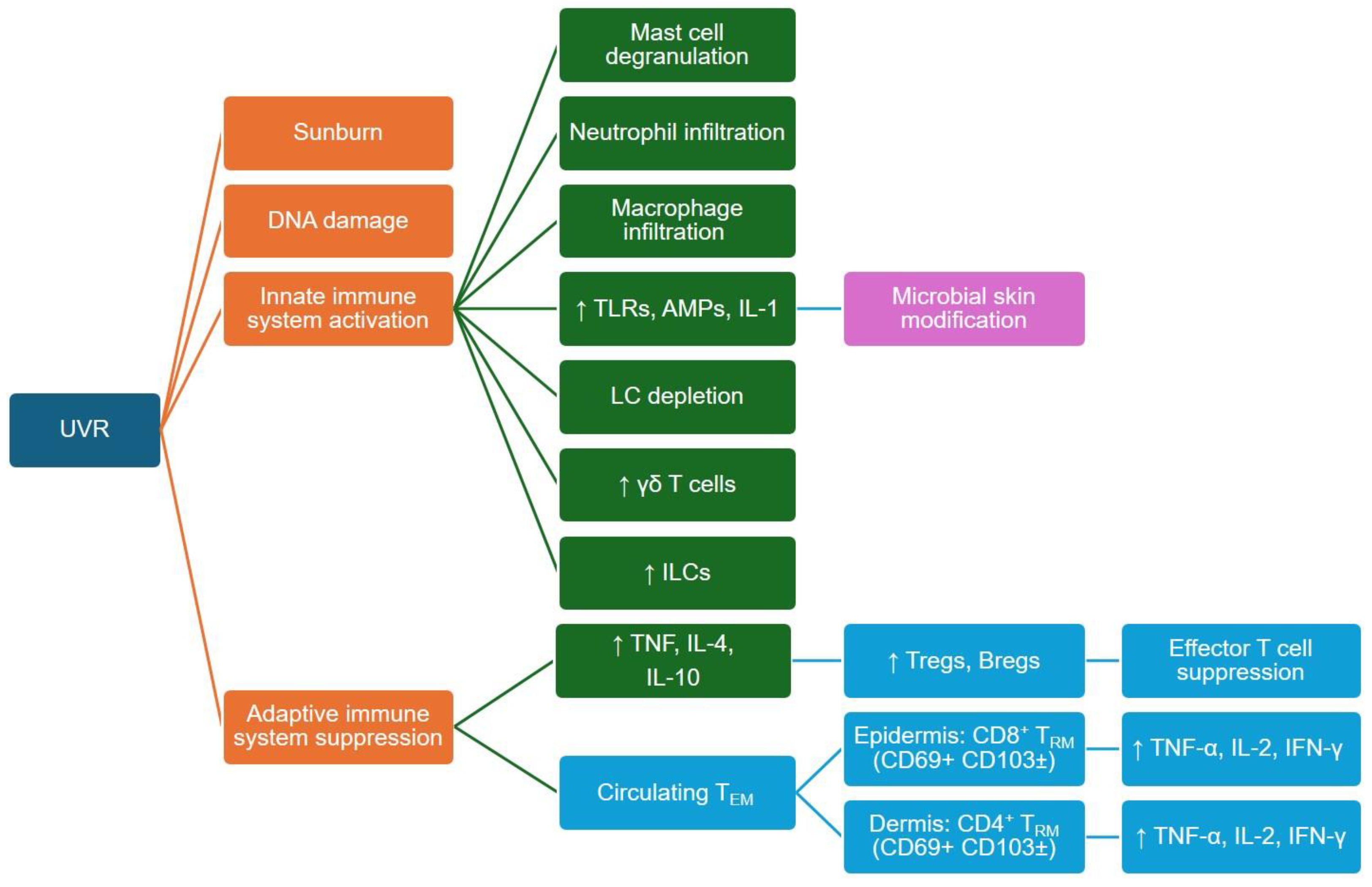
4.2. UVR Effects on Innate and Adaptive Immune Cells
4.3. UVR Effects on the Skin’s Neuroendocrine System
5. Adjuvant Anti-PD-1 Therapy—Boosting Anti-Cancer Immune Surveillance
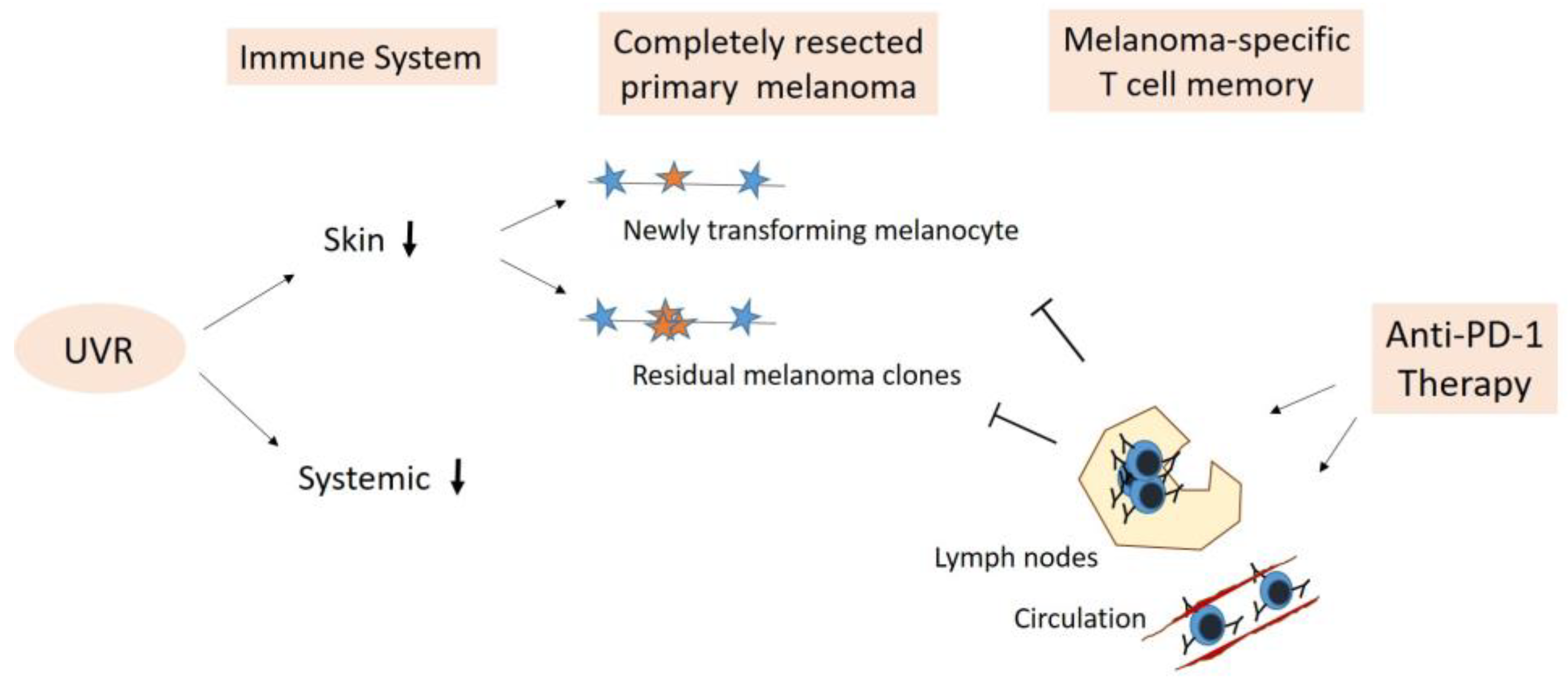
5.1. Elimination of Newly Transforming Pre-Malignant Melanocytes
5.2. Elimination of Residual Melanoma Clones/Subclinical Melanoma
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.N.; Erickson, L.A.; Rao, R.D.; McWilliams, R.R.; Kottschade, L.A.; Creagan, E.T.; Weenig, R.H.; Hand, J.L.; Pittelkow, M.R.; Pockaj, B.A.; et al. Malignant Melanoma in the 21st Century, Part 1: Epidemiology, Risk Factors, Screening, Prevention, and Diagnosis. Mayo Clin. Proc. 2007, 82, 364–380. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Rosner, B.A.; Colditz, G.A. Risk factors for melanoma by body site. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1241–1244. [Google Scholar] [CrossRef] [PubMed]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin Cancers after Organ Transplantation. N. Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 239–271. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Kaskel, P.; Sander, S.; Kron, M.; Kind, P.; Peter, R.; Krahn, G. Outdoor activities in childhood: A protective factor for cutaneous melanoma? Results of a case-control study in 271 matched pairs. Br. J. Dermatol. 2001, 145, 602–609. [Google Scholar] [CrossRef]
- Helvind, N.M.; Weitemeyer, M.B.-M.; Chakera, A.H.; Hendel, H.W.; Ellebæk, E.; Svane, I.M.; Kjærskov, M.W.; Bojesen, S.; Skyum, H.; Petersen, S.K.; et al. Stage-Specific Risk of Recurrence and Death from Melanoma in Denmark, 2008–2021. JAMA Dermatol. 2023, 159, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef]
- Garbe, C.; Keim, U.; Suciu, S.; Amaral, T.; Eigentler, T.K.; Gesierich, A.; Hauschild, A.; Heinzerling, L.; Kiecker, F.; Schadendorf, D.; et al. Prognosis of patients with stage III melanoma according to american joint committee on cancer version 8: A reassessment on the basis of 3 independent stage III melanoma cohorts. J. Clin. Oncol. 2020, 38, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Keim, U.; Amaral, T.; Berking, C.; Eigentler, T.K.; Flatz, L.; Gesierich, A.; Leiter, U.; Stadler, R.; Sunderkötter, C.; et al. Prognosis of Patients with Primary Melanoma Stage I and II According to American Joint Committee on Cancer Version 8 Validated in Two Independent Cohorts: Implications for Adjuvant Treatment. J. Clin. Oncol. 2022, 40, 3741–3749. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Weber, J.; Mandalà, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Rutkowski, P.; Queirolo, P.; Del Vecchio, M.; Mackiewicz, J.; Chiarion-Sileni, V.; Merino, L.d.l.C.; Khattak, M.A.; Schadendorf, D.; Long, G.V.; et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet 2022, 399, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Del Vecchio, M.; Weber, J.; Hoeller, C.; Grob, J.-J.; Mohr, P.; Grabbe, S.; Dutriaux, C.; Chiarion-Sileni, V.; Mackiewicz, J.; et al. Adjuvant therapy with nivolumab versus placebo in patients with resected stage IIB/C melanoma (CheckMate 76K). Ski. J. Cutan. Med. 2023, 7, s163. [Google Scholar] [CrossRef]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Sun, Z.; Fourcade, J.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Kirkwood, J.M.; Zarour, H.M. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res. 2015, 75, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.-J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB–C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Ascierto, P.A.; Middleton, M.R.; Hennicken, D.; Zoffoli, R.; Pieters, A.; Amadi, A.; Kupas, K.; Kotapati, S.; Moshyk, A.; et al. Indirect treatment comparison of nivolumab versus placebo as adjuvant treatment for resected melanoma. Eur. J. Cancer 2021, 158, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S.; et al. Longer follow-up confirms recurrence-free survival benefit of adjuvant pembrolizumab in high-risk stage III melanoma: Updated results from the EORTC 1325-MG/KEYNOTE-054 trial. J. Clin. Oncol. 2020, 38, 3925–3936. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Kicinski, M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Meshcheryakov, A.; Khattak, A.; et al. Five-Year Analysis of Adjuvant Pembrolizumab or Placebo in Stage III Melanoma. NEJM Evid. 2022, 1, EVIDoa2200214. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandalà, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Fernandez, A.M.A.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.-J.; Chiarion-Sileni, V.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III/IV Melanoma: 5-Year Efficacy and Biomarker Results from CheckMate 238. Clin. Cancer Res. 2023, 29, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.; Shoushtari, A.; Chauhan, D.; Palmieri, D.; Lee, B.; Rohaan, M.; Mangana, J.; Atkinson, V.; Zaman, F.; Young, A.; et al. Management of early melanoma recurrence despite adjuvant anti-PD-1 antibody therapy. Ann. Oncol. 2020, 31, 1075–1082. [Google Scholar] [CrossRef]
- Kluger, H.M.; Tawbi, H.A.; Ascierto, M.L.; Bowden, M.; Callahan, M.K.; Cha, E.; Chen, H.X.; Drake, C.G.; Feltquate, D.M.; Ferris, R.L.; et al. Defining tumor resistance to PD-1 pathway blockade: Recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J. Immunother. Cancer 2020, 8, e000398. [Google Scholar] [CrossRef]
- Long, G.V.; Luke, J.J.; Khattak, M.A.; Merino, L.d.l.C.; Del Vecchio, M.; Rutkowski, P.; Spagnolo, F.; Mackiewicz, J.; Chiarion-Sileni, V.; Kirkwood, J.M.; et al. Pembrolizumab versus placebo as adjuvant therapy in resected stage IIB or IIC melanoma (KEYNOTE-716): Distant metastasis-free survival results of a multicentre, double-blind, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 1378–1388. [Google Scholar] [CrossRef]
- Weber, J.S.; Schadendorf, D.; Del Vecchio, M.; Larkin, J.; Atkinson, V.; Schenker, M.; Pigozzo, J.; Gogas, H.; Dalle, S.; Meyer, N.; et al. Adjuvant Therapy of Nivolumab Combined with Ipilimumab versus Nivolumab Alone in Patients with Resected Stage IIIB-D or Stage IV Melanoma (CheckMate 915). J. Clin. Oncol. 2023, 41, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Mandala, M.; Long, G.V.; Eggermont, A.M.; van Akkooi, A.C.; Sandhu, S.; Garbe, C.; Lorigan, P. Adjuvant therapy for stage II melanoma: The need for further studies. Eur. J. Cancer 2023, 189, 112914. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Del Vecchio, M.; Mandala, M.; Gogas, H.; Arance, A.; Dalle, S.; Cowey, C.; Schenker, M.; Grob, J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab (NIVO) versus ipilimumab (IPI) in resected stage III/IV melanoma: 3-year efficacy and biomarker results from the phase III CheckMate 238 trial. Ann. Oncol. 2019, 30, v533–v534. [Google Scholar] [CrossRef]
- Weber, J.S.; Mandalà, M.; Del Vecchio, M.; Gogas, H.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Rodas, I.M.; et al. Adjuvant therapy with nivolumab (NIVO) versus ipilimumab (IPI) after complete resection of stage III/IV melanoma: Updated results from a phase III trial (CheckMate 238). J. Clin. Oncol. 2018, 36, 9502. [Google Scholar] [CrossRef]
- Grossmann, K.F.; Othus, M.; Patel, S.P.; Tarhini, A.A.; Sondak, V.K.; Knopp, M.V.; Petrella, T.M.; Truong, T.-G.; Khushalani, N.I.; Cohen, J.V.; et al. Adjuvant Pembrolizumab versus IFNα2b or Ipilimumab in Resected High-Risk Melanoma. Cancer Discov. 2022, 12, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Ascierto, P.A.; Khattak, M.A.; Merino, L.d.l.C.; Del Vecchio, M.; Rutkowski, P.; Spagnolo, F.; Mackiewicz, J.; Chiarion-Sileni, V.; Kirkwood, J.M.; et al. Pembrolizumab versus Placebo as Adjuvant Therapy in Resected Stage IIB or IIC Melanoma: Final Analysis of Distant Metastasis-Free Survival in the Phase III KEYNOTE-716 Study. J. Clin. Oncol. 2024, JCO2302355. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, H.; Wu, Z.-H. Upregulation of PD-L1 via HMGB1-activated IRF3 and NF-kB contributes to UV radiation-induced immune suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T–cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273. [Google Scholar] [PubMed]
- Sucker, A.; Zhao, F.; Real, B.; Heeke, C.; Bielefeld, N.; Maβen, S.; Horn, S.; Moll, I.; Maltaner, R.; Horn, P.A.; et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin. Cancer Res. 2014, 20, 6593–6604. [Google Scholar] [CrossRef]
- Pauken, K.E.; Torchia, J.A.; Chaudhri, A.; Sharpe, A.H.; Freeman, G.J. Emerging concepts in PD-1 checkpoint biology. Semin. Immunol. 2021, 52, 101480. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.; Camara, S.; Shakiba, M.; Scott, A.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.-C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.-A.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.-J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’gorman, W.E.; Au-Yeung, A.; et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Herbst, R.H.; Canner, D.; Li, A.; Hillman, M.; Shanahan, S.-L.; Gibbons, G.; Smith, O.C.; Kim, J.Y.; Westcott, P.; et al. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 2021, 54, 2338–2353.e6. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.A.; Kuchroo, M.; Venkat, A.; Khatun, A.; Wang, J.; William, I.; Hornick, N.I.; Fitzgerald, B.L.; Damo, M.; Kasmani, M.Y.; et al. A reservoir of stem-like CD8+ T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 2021, 6, eabg7836. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; Bosch, T.v.D.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020, 38, 685–700.e8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, X.; Wang, Z.; Chen, X.; Wang, L.; Lu, Y.; Xiong, D.; Liu, Q.; Tian, Y.; Lin, H.; et al. The primordial differentiation of tumor-specific memory CD8+ T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 2022, 185, 4049–4066.e25. [Google Scholar] [CrossRef]
- Molodtsov, A.K.; Khatwani, N.; Vella, J.L.; Lewis, K.A.; Zhao, Y.; Han, J.; Sullivan, D.E.; Searles, T.G.; Preiss, N.K.; Shabaneh, T.B.; et al. Resident memory CD8+ T cells in regional lymph nodes mediate immunity to metastatic melanoma. Immunity 2021, 54, 2117–2132.e7. [Google Scholar] [CrossRef]
- Patel, S.P.; Patel, S.P.; Othus, M.; Othus, M.; Chen, Y.; Chen, Y.; Wright, G.P.; Wright, G.P.; Yost, K.J.; Yost, K.J.; et al. Neoadjuvant–Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cho, E.; Li, W.-Q.; Weinstock, M.A.; Han, J.; Qureshi, A.A. History of Severe Sunburn and Risk of Skin Cancer among Women and Men in 2 Prospective Cohort Studies. Am. J. Epidemiol. 2016, 183, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Han, J.; Vleugels, R.A.; Puett, R.; Laden, F.; Hunter, D.J.; Qureshi, A.A. Cumulative ultraviolet radiation flux in adulthood and risk of incident skin cancers in women. Br. J. Cancer 2014, 110, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.; Willemze, R.; de Gruijl, F.R.; Bavinck, J.N.B.; Bajdik, C.D. The influence of painful sunburns and lifetime sun exposure on the risk of actinic keratoses, seborrheic warts, melanocytic nevi, atypical nevi, and skin cancer. J. Investig. Dermatol. 2003, 120, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Picconi, O.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur. J. Cancer 2005, 41, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Trucco, L.D.; Mundra, P.A.; Hogan, K.; Garcia-Martinez, P.; Viros, A.; Mandal, A.K.; Macagno, N.; Gaudy-Marqueste, C.; Allan, D.; Baenke, F.; et al. Ultraviolet radiation–induced DNA damage is prognostic for outcome in melanoma. Nat. Med. 2019, 25, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Ulisse, S.; Baldini, E.; Pironi, D.; Gagliardi, F.; Tripodi, D.; Lauro, A.; Carbotta, S.; Tarroni, D.; D’armiento, M.; Morrone, A.; et al. Is Melanoma Progression Affected by Thyroid Diseases? Int. J. Mol. Sci. 2022, 23, 10036. [Google Scholar] [CrossRef] [PubMed]
- Centeno, P.P.; Pavet, V.; Marais, R. The journey from melanocytes to melanoma. Nat. Rev. Cancer 2023, 23, 372–390. [Google Scholar] [CrossRef]
- Holman, C.D.J.; Armstrong, B.K.; Heenan, P.J. A Theory of the Etiology and Pathogenesis of Human Cutaneous Malignant Melanoma. JNCI J. Natl. Cancer Inst. 1983, 71, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Laskar, R.; Ferreiro-Iglesias, A.; Bishop, D.; Iles, M.; Kanetsky, P.; Armstrong, B.; Law, M.; Goldstein, A.; Aitken, J.; Giles, G.; et al. Risk factors for melanoma by anatomical site: An evaluation of aetiological heterogeneity. Br. J. Dermatol. 2021, 184, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Ghiasvand, R.; Robsahm, T.E.; Green, A.C.; Rueegg, C.S.; Weiderpass, E.; Lund, E.; Veierød, M.B. Association of Phenotypic Characteristics and UV Radiation Exposure with Risk of Melanoma on Different Body Sites. JAMA Dermatol. 2019, 155, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; de Vries, E.; Whiteman, D.C.; Jemal, A.; Bray, F.; Parkin, D.M.; Soerjomataram, I. Global burden of cutaneous melanoma attributable to ultraviolet radiation in 2012. Int. J. Cancer 2018, 143, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Neale, R.E.; Lucas, R.M.; Byrne, S.N.; Hollestein, L.; Rhodes, L.E.; Yazar, S.; Young, A.R.; Berwick, M.; Ireland, R.A.; Olsen, C.M. The effects of exposure to solar radiation on human health. Photochem. Photobiol. Sci. 2023, 22, 1011–1047. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Photoaging: UV radiation-induced inflammation and immunosuppression accelerate the aging process in the skin. Inflamm. Res. 2022, 71, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Norval, M. Ultraviolet radiation-induced immunosuppression and its relevance for skin carcinogenesis. Photochem. Photobiol. Sci. 2018, 17, 1872–1884. [Google Scholar] [CrossRef]
- Hughes, B.K.; Bishop, C.L. Current Understanding of the Role of Senescent Melanocytes in Skin Ageing. Biomedicines 2022, 10, 3111. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Norval, M. More Than Effects in Skin: Ultraviolet Radiation-Induced Changes in Immune Cells in Human Blood. Front. Immunol. 2021, 12, 694086. [Google Scholar] [CrossRef]
- Holick, M.F. Biological effects of sunlight, ultraviolet radiation, visible light, infrared radiation and Vitamin D for health. Anticancer Res. 2016, 36, 1345–1356. [Google Scholar] [PubMed]
- Emri, G.; Paragh, G.; Tósaki, Á.; Janka, E.; Kollár, S.; Hegedűs, C.; Gellén, E.; Horkay, I.; Koncz, G.; Remenyik, É. Ultraviolet radiation-mediated development of cutaneous melanoma: An update. J. Photochem. Photobiol. B Biol. 2018, 185, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Premi, S.; Han, L.; Mehta, S.; Knight, J.; Zhao, D.; Palmatier, M.A.; Kornacker, K.; Brash, D.E. Genomic sites hypersensitive to ultraviolet radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 24196–24205. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Hennessey, R.C.; Weiss, T.J.; Tallman, D.A.; Crawford, E.R.; Murphy, B.M.; Webb, A.; Zhang, S.; La Perle, K.M.; Burd, C.J.; et al. UVB mutagenesis differs in Nras- and Braf-mutant mouse models of melanoma. Life Sci. Alliance 2021, 4, e202101135. [Google Scholar] [CrossRef] [PubMed]
- Vicente, A.L.S.A.; Novoloaca, A.; Cahais, V.; Awada, Z.; Cuenin, C.; Spitz, N.; Carvalho, A.L.; Evangelista, A.F.; Crovador, C.S.; Reis, R.M.; et al. Cutaneous and acral melanoma cross-OMICs reveals prognostic cancer drivers associated with pathobiology and ultraviolet exposure. Nat. Commun. 2022, 13, 4115. [Google Scholar] [CrossRef]
- Strub, T.; Ballotti, R.; Bertolotto, C. The ‘ART’ of epigenetics in melanoma: From histone ‘alterations, to resistance and therapies’. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef]
- Archambault, M.; Yaar, M.; Gilchrest, B.A. Keratinocytes and fibroblasts in a human skin equivalent model enhance melanocyte survival and melanin synthesis after ultraviolet irradiation. J. Investig. Dermatol. 1995, 104, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Casalou, C.; Moreiras, H.; Mayatra, J.M.; Fabre, A.; Tobin, D.J. Loss of ‘Epidermal Melanin Unit’ Integrity in Human Skin during Melanoma-Genesis. Front. Oncol. 2022, 12, 878336. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, T.B.; Breathnach, A.S. Das epidermale melanin-einheit-system. Dermatol. Wochenschr. 1963, 147, 481–489. [Google Scholar] [PubMed]
- Corre, S.; Primot, A.; Sviderskaya, E.; Bennett, D.C.; Vaulont, S.; Goding, C.R.; Galibert, M.-D. UV-induced expression of key component of the tanning process, the POMC and MC1R genes, is dependent onp the p-38-activated upstream stimulating factor-1 (USF-1). J. Biol. Chem. 2004, 279, 51226–51233. [Google Scholar] [CrossRef]
- Visconti, A.; Duffy, D.L.; Liu, F.; Zhu, G.; Wu, W.; Chen, Y.; Hysi, P.G.; Zeng, C.; Sanna, M.; Iles, M.M.; et al. Genome-wide association study in 176,678 Europeans reveals genetic loci for tanning response to sun exposure. Nat. Commun. 2018, 9, 1684. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Fisher, D.E. MITF and UV responses in skin: From pigmentation to addiction. Pigment. Cell Melanoma Res. 2019, 32, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Guhan, S.; Klebanov, N.; Tsao, H. Melanoma genomics: A state-of-the-art review of practical clinical applications. Br. J. Dermatol. 2021, 185, 272–281. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic regulation in melanoma: Facts and hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, E.; García-Borrón, J.C.; Fargnoli, M.C.; Gandini, S.; Maisonneuve, P.; Bagnardi, V.; Specchia, C.; Liu, F.; Kayser, M.; Nijsten, T.; et al. MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: A pooled-analysis from the M-SKIP project. Int. J. Cancer 2015, 136, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.; Ghiorzo, P.; Gruis, N.; et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: A pooled analysis from the M-SKIP project. Cancer Manag. Res. 2018, 10, 1143–1154. [Google Scholar] [CrossRef]
- Cui, Y.; Miao, Y.; Cao, L.; Guo, L.; Cui, Y.; Yan, C.; Zeng, Z.; Xu, M.; Han, T. Activation of melanocortin-1 receptor signaling in melanoma cells impairs T cell infiltration to dampen antitumor immunity. Nat. Commun. 2023, 14, 5740. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Norval, M.; Byrne, S.N.; Rhodes, L.E. Exposure to Ultraviolet Radiation in the Modulation of Human Diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Patra, V.K.; Laoubi, L.; Nicolas, J.-F.; Vocanson, M.; Wolf, P. A Perspective on the interplay of ultraviolet-radiation, skin microbiome and skin resident memory TCRαβ+ cells. Front. Med. 2018, 5, 166. [Google Scholar] [CrossRef]
- Vink, A.A.; Moodycliffe, A.M.; Shreedhar, V.; Ullrich, S.E.; Roza, L.; Yarosh, D.B.; Kripke, M.L. The inhibition of antigen-presenting activity of dendritic cells resulting from UV irradiation of marine skin is restored by in vitro photorepair of cyclobutane pyrimidine dimers. Proc. Natl. Acad. Sci. USA 1997, 94, 5255–5260. [Google Scholar] [CrossRef] [PubMed]
- Vink, A.A.; Strickland, F.M.; Bucana, C.; Cox, P.A.; Roza, L.; Yarosh, D.B.; Kripke, M.L. Localization of DNA damage and its role in altered antigen-presenting cell function in ultraviolet-irradiated mice. J. Exp. Med. 1996, 183, 1491–1500. [Google Scholar] [CrossRef]
- Simon, J.C.; Tigelaar, R.E.; Bergstresser, P.R.; Edelbaum, D.; Cruz, P.D. Ultraviolet B radiation converts Langerhans cells from immunogenic to tolerogenic antigen-presenting cells. Induction of specific clonal anergy in CD4+ T helper 1 cells. J. Immunol. 1991, 146, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Renkl, A.C.; Denfeld, R.W.; de Roche, R.; Spitzlei, M.; Schöpf, E.; Simon, J.C. Low-dose UVB radiation perturbs the functional expression of B7.1 and B7.2 co-stimulatory molecules on human Langerhans cells. Eur. J. Immunol. 1995, 25, 2858–2862. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Z.-H. Immune checkpoint molecules: “New” kids on the block of skin photoimmunology. Genes Dis. 2021, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Grimbaldeston, M.A.; Swift, G.J.; Hosszu, E.K.; Finlay-Jones, J.J. A critical role for dermal mast cells in cis-urocanic acid-induced systemic suppression of contact hypersensitivity responses in mice. Photochem. Photobiol. 1999, 70, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.N.; Limón-Flores, A.Y.; Ullrich, S.E. Mast Cell Migration from the Skin to the Draining Lymph Nodes upon Ultraviolet Irradiation Represents a Key Step in the Induction of Immune Suppression. J. Immunol. 2008, 180, 4648–4655. [Google Scholar] [CrossRef]
- Byrne, S.N.; Halliday, G.M. B cells activated in lymph nodes in response to ultraviolet irradiation or by interleukin-10 inhibit dendritic cell induction of immunity. J. Investig. Dermatol. 2005, 124, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Gorman, S.; Judge, M.A.; Hart, P.H. Topical 1,25-dihydroxyvitamin D3 subverts the priming ability of draining lymph node dendritic cells. Immunology 2010, 131, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.L.X.; Scott, N.M.; Bisley, J.L.; Lambert, M.J.; Gorman, S.; Norval, M.; Hart, P.H. Characterization of regulatory dendritic cells differentiated from the bone marrow of UV-irradiated mice. Immunology 2013, 140, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.P.; Phan, T.A.; Halliday, G.M.; Barnetson, R.S.; Damian, D.L. Spectral and dose dependence of ultraviolet radiation-induced immunosuppression. Front. Biosci. 2006, 11, 394–411. [Google Scholar] [CrossRef]
- Halliday, G.M.; Rana, S. Waveband and dose dependency of sunlight-induced immunomodulation and cellular changes. Photochem. Photobiol. 2008, 84, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Grewe, M.; Gyufko, K.; Krutmann, J. Interleukin-10 production by cultured human keratinocytes: Regulation by ultraviolet B and ultraviolet A1 radiation. J. Investig. Dermatol. 1995, 104, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Salinas, R.; Limón-Flores, A.Y.; Chávez-Blanco, A.D.; Gonzalez-Estrada, A.; Ullrich, S.E. Mast Cell-Derived IL-10 Suppresses Germinal Center Formation by Affecting T Follicular Helper Cell Function. J. Immunol. 2011, 186, 25–31. [Google Scholar] [CrossRef]
- Kurimoto, I.; Streilein, J.W. cis-urocanic acid suppression of contact hypersensitivity induction is mediated via tumor necrosis factor-alpha. J. Immunol. 1992, 148, 3072–3078. [Google Scholar] [CrossRef]
- Rana, S.; Byrne, S.N.; MacDonald, L.J.; Chan, C.Y.-Y.; Halliday, G.M. Ultraviolet B suppresses immunity by inhibiting effector and memory T cells. Am. J. Pathol. 2008, 172, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Steerenberg, P.A.; Korenromp, E.L.; Loveren, H.; Mol, D.Q.; Geerse, L.; Gruijl, F.R. Natural killer cell activity during UVR-induced skin tumor formation in the Skh hairless mouse. Photochem. Photobiol. 1997, 65, 150–154. [Google Scholar] [CrossRef]
- Ortner, D.; Tripp, C.H.; Komenda, K.; Dubrac, S.; Zelger, B.; Hermann, M.; Doppler, W.; Tymoszuk, P.Z.; Boon, L.; Clausen, B.E.; et al. Langerhans cells and NK cells cooperate in the inhibition of chemical skin carcinogenesis. OncoImmunology 2017, 6, e1260215. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; e Sousa, C.R. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Slominski, A.T.; Żmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, 1–6. [Google Scholar] [CrossRef]
- Cianfarani, F.; Baldini, E.; Cavalli, A.; Marchioni, E.; Lembo, L.; Teson, M.; Persechino, S.; Zambruno, G.; Ulisse, S.; Odorisio, T.; et al. TSH receptor and thyroid-specific gene expression in human skin. J. Investig. Dermatol. 2010, 130, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Liu, L.-P.; Zhou, H.; Zheng, Y.-W.; Li, Y.-M. Recognition of Melanocytes in Immuno-Neuroendocrinology and Circadian Rhythms: Beyond the Conventional Melanin Synthesis. Cells 2022, 11, 2082. [Google Scholar] [CrossRef] [PubMed]
- Speeckaert, R.; Belpaire, A.; Speeckaert, M.; van Geel, N. The delicate relation between melanocytes and skin immunity: A game of hide and seek. Pigment. Cell Melanoma Res. 2022, 35, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Strid, J.; Tigelaar, R.E.; Hayday, A.C. Skin immune surveillance by T cells—A new order? Semin. Immunol. 2009, 21, 110–120. [Google Scholar] [CrossRef]
- Bhatia, A.; Kumar, Y. Cancer-immune equilibrium: Questions unanswered. Cancer Microenviron. 2011, 4, 209–217. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [PubMed]
- Maglio, D.H.G.; Paz, M.L.; Leoni, J. Sunlight Effects on Immune System: Is There Something Else in addition to UV-Induced Immunosuppression? BioMed Res. Int. 2016, 2016, 1934518. [Google Scholar] [CrossRef]
- Hawkshaw, N.J.; Pilkington, S.M.; Murphy, S.A.; Al-Gazaq, N.; Farrar, M.D.; Watson, R.E.; Nicolaou, A.; Rhodes, L.E. UV radiation recruits CD4+GATA3+ and CD8+GATA3+ T cells while altering the lipid microenvironment following inflammatory resolution in human skin in vivo. Clin. Transl. Immunol. 2020, 9, e01104. [Google Scholar] [CrossRef] [PubMed]
- Grichnik, J.M.; Ross, A.L.; Schneider, S.L.; Sanchez, M.I.; Eller, M.S.; Hatzistergos, K.E. How, and from which cell sources, do nevi really develop? Exp. Dermatol. 2014, 23, 310–313. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gilchrest, B.A. Photoaging. J. Investig. Dermatol. 2013, 133, E2–E6. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. Activation of immunosuppressive network in the aging process. Ageing Res. Rev. 2020, 57, 100998. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Zippelius, A.; Batard, P.; Rubio-Godoy, V.; Bioley, G.; Liénard, D.; Lejeune, F.; Rimoldi, D.; Guillaume, P.; Meidenbauer, N.; Mackensen, A.; et al. Effector Function of Human Tumor-Specific CD8 T Cells in Melanoma Lesions: A State of Local Functional Tolerance. Cancer Res. 2004, 64, 2865–2873. [Google Scholar] [CrossRef]
- Pasetto, A.; Gros, A.; Robbins, P.F.; Deniger, D.C.; Prickett, T.D.; Matus-Nicodemos, R.; Douek, D.C.; Howie, B.; Robins, H.; Parkhurst, M.R.; et al. Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol. Res. 2016, 4, 734–743. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Tran, E.; Parkhurst, M.R.; Ilyas, S.; Pasetto, A.; Groh, E.M.; Robbins, P.F.; Yossef, R.; Garcia-Garijo, A.; Fajardo, C.A.; et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Investig. 2019, 129, 4992–5004. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.P. Deciphering mechanisms of UVR-induced tumoral immune checkpoint regulation against melanoma. Cancer Res. 2019, 79, 2805–2807. [Google Scholar] [CrossRef] [PubMed]
- Dousset, L.; Poizeau, F.; Robert, C.; Mansard, S.; Mortier, L.; Caumont, C.; Routier, É.; Dupuy, A.; Rouanet, J.; Battistella, M.; et al. Positive Association btween Location of Melanoma, Ultraviolet Signature, Tumor Mutational Burden, and Response to Anti–PD-1 Therapy. JCO Precis. Oncol. 2021, 5, 1821–1829. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.-L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science (80-) 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef]
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): A randomised, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar] [CrossRef]
- Niknafs, N.; Balan, A.; Cherry, C.; Hummelink, K.; Monkhorst, K.; Shao, X.M.; Belcaid, Z.; Marrone, K.A.; Murray, J.; Smith, K.N.; et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat. Med. 2023, 29, 440–449. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S. Tumor Heterogeneity and Tumor Immunity: A Chicken-and-Egg Problem. Trends Immunol. 2016, 37, 349–351. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.M.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. npj Genom. Med. 2017, 2, 10. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, C.; He, N.; Zhao, G.; Wang, J.; Yang, Y. Integration of the Tumor Mutational Burden and Tumor Heterogeneity Identify an Immunological Subtype of Melanoma with Favorable Survival. Front. Oncol. 2020, 10, 571545. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Milo, I.; Bedora-Faure, M.; Garcia, Z.; Thibaut, R.; Périé, L.; Shakhar, G.; Deriano, L.; Bousso, P. The immune system profoundly restricts intratumor genetic heterogeneity. Sci. Immunol. 2018, 3, eaat1435. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.A.; Kawakubo, M.; Juneja, V.R.; Su, M.Y.; Erlich, T.H.; LaFleur, M.W.; Kemeny, L.V.; Rashid, M.; Malehmir, M.; Rabi, S.A.; et al. Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci. Transl. Med. 2021, 13, eabd8636. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Boussemart, L.; Mateus, C.; Routier, E.; Boutros, C.; Cazenave, H.; Viollet, R.; Thomas, M.; Roy, S.; Benannoune, N.; et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016, 152, 45–51. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandlmaier, M.; Hoellwerth, M.; Koelblinger, P.; Lang, R.; Harrer, A. Adjuvant PD-1 Checkpoint Inhibition in Early Cutaneous Melanoma: Immunological Mode of Action and the Role of Ultraviolet Radiation. Cancers 2024, 16, 1461. https://doi.org/10.3390/cancers16081461
Brandlmaier M, Hoellwerth M, Koelblinger P, Lang R, Harrer A. Adjuvant PD-1 Checkpoint Inhibition in Early Cutaneous Melanoma: Immunological Mode of Action and the Role of Ultraviolet Radiation. Cancers. 2024; 16(8):1461. https://doi.org/10.3390/cancers16081461
Chicago/Turabian StyleBrandlmaier, Matthias, Magdalena Hoellwerth, Peter Koelblinger, Roland Lang, and Andrea Harrer. 2024. "Adjuvant PD-1 Checkpoint Inhibition in Early Cutaneous Melanoma: Immunological Mode of Action and the Role of Ultraviolet Radiation" Cancers 16, no. 8: 1461. https://doi.org/10.3390/cancers16081461
APA StyleBrandlmaier, M., Hoellwerth, M., Koelblinger, P., Lang, R., & Harrer, A. (2024). Adjuvant PD-1 Checkpoint Inhibition in Early Cutaneous Melanoma: Immunological Mode of Action and the Role of Ultraviolet Radiation. Cancers, 16(8), 1461. https://doi.org/10.3390/cancers16081461