
Targeted Therapy for Highly Desmoplastic and Immunosuppressive Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma

 ,
,  , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
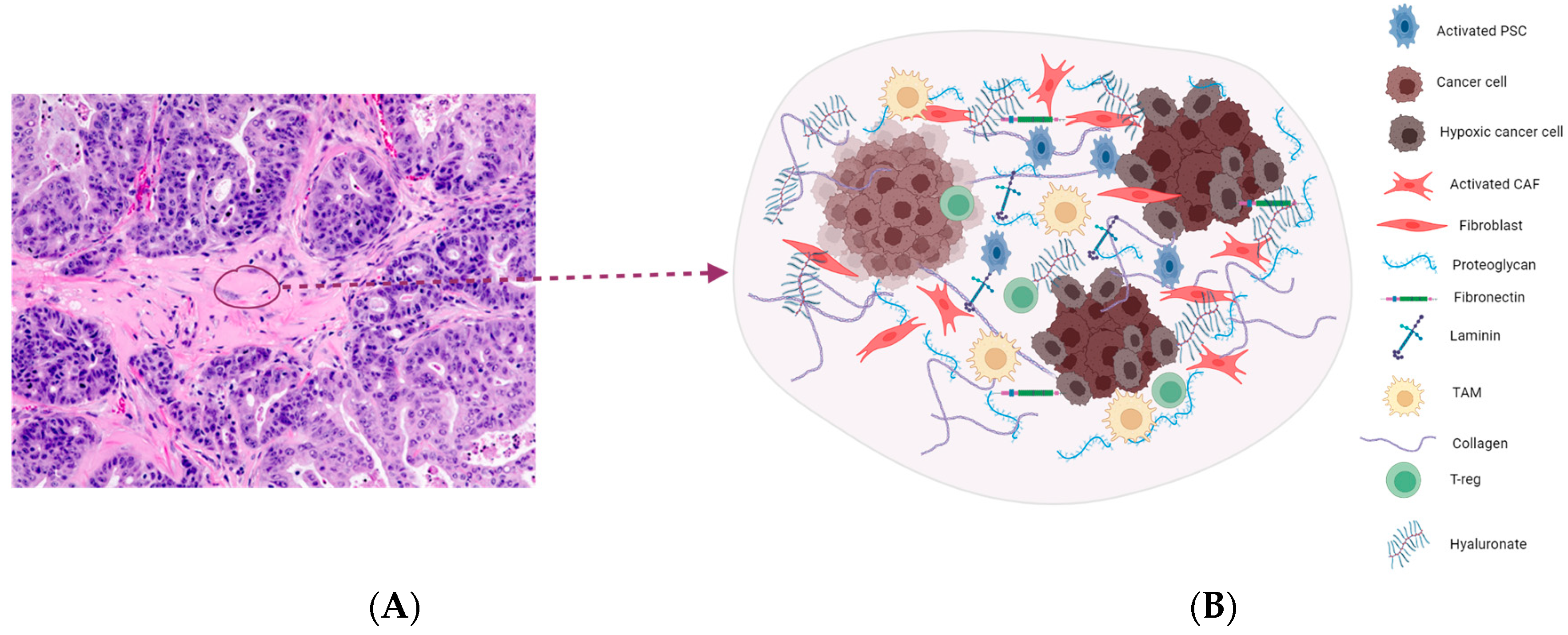
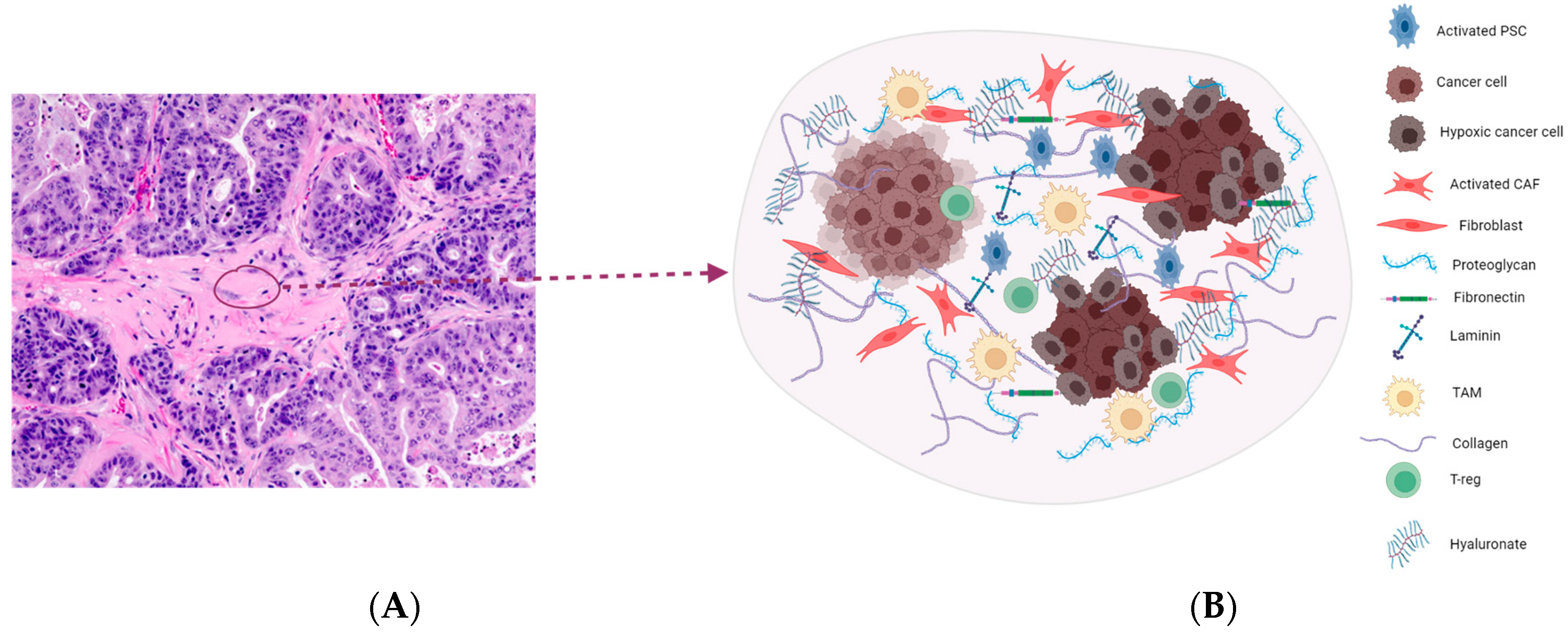
2. Highly Desmoplastic Tumor Microenvironment in PDAC
2.1. Targeting the ECM Components
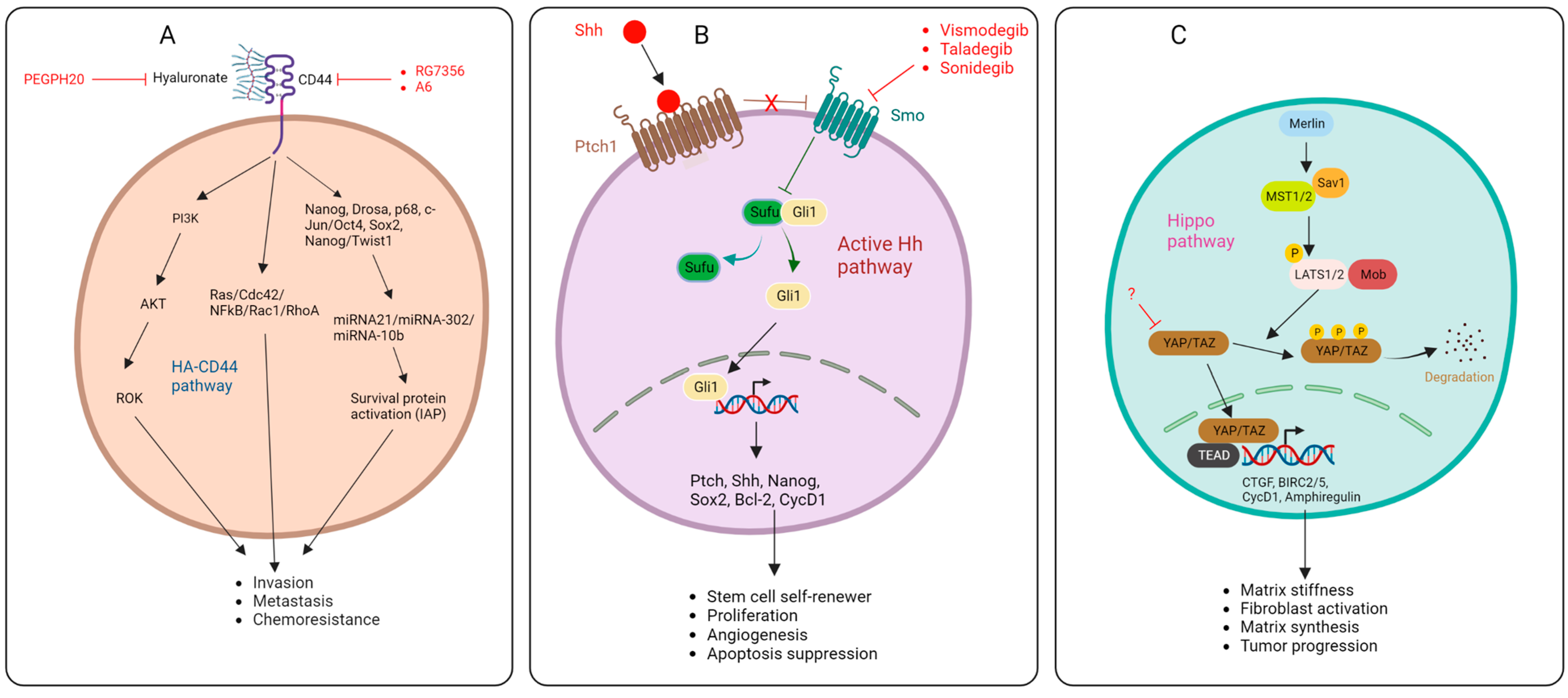
2.2. Hyaluronic Acid
2.3. Integrins
2.4. Sonic Hedgehog Pathway
2.5. Hippo Pathway
2.6. Cancer-Associated Fibroblasts (CAFs)

{kind=link}
{kind=link}
{kind=link}
| Target | Agent(s) | Phase | Disease(s) | Mechanism | Reference(s) |
|---|---|---|---|---|---|
| Hyaluronic Acid (HA) | Hymecromone (4-MU) | Bile duct obstruction and COVID-19 | Inhibit HA synthesis and impede migration | [10,11] | |
| Hyaluronic Acid (HA) | RG7356 | I | High CD44-expressing solid tumors | Selective binding on CD44 inhibits growth | [13] |
| Hyaluronic Acid (HA) | A6 | II | Recurrent EOC/FTC/PPC | Binds to CD44, reducing CD44 bonding and activity | [14] |
| Hyaluronic Acid (HA) | Bivatuzumab + Mertansine | I | Squamous cell carcinoma of head, neck, or esophagus | Binding to CD44v6 to enable intracellular release and induce miotic arrest and cell death | [15] |
| Hyaluronic Acid (HA) | RHAMM peptide (CD168) | I/II | Chronic lymphocytic leukemia | Hinders miosis | [17,18] |
| Hyaluronic Acid (HA) | PEGH20 | I/II | Degrades HA in the stroma | [20,21,22,23] | |
| Hyaluronic Acid (HA) | PEGH20 + atezolizumab | Ib/II | PDAC | Degrades HA in the stroma | NCT03193190 NCT03281369 |
| Hyaluronic Acid (HA) | PEGH20 | III | HA-high stage IV pancreatic cancer | Increases tumor profusion by decreasing tumor pressure and increasing tumor plasticity | [25] |
| Hyaluronic Acid (HA) | PEGH20 + Nab-paclitaxel/gemcitabine | III | HA-high metastatic PDAC | Degrades HA in the stroma | [26] |
| αV Integrins and neurophilin-1 | CEND-1 + Nab-paclitaxel/gemcitabine | I | PDAC | Interacts with αV integrins and activates drug transport via neurophilin-1 | NCT03517176 |
| α2 Integrins | E7820 | I | Malignant solid tumors or lymphomas | Prevents mRNA expression of α2 integrins | [29] |
| SHH | Vismodegib (GDC-0449) | I/II |
PDAC and medulloblastoma | Inhibits SMO to inhibit Hh signaling pathway | [35,36] |
| SHH | Taladegib | I | Advanced solid tumors | Inhibition of Hh signaling pathway mediated by protein SMO | [37] |
| SHH | Sonidegib (LDE225) | II | Medulloblastoma | Inhibitor of Smoothened, preventing downstream activation | [38] |
| Cancer-associated fibroblasts | Ruxolitinib + capecitabine | III | Pancreatic cancer | Inhibition of JAK1 and JAK2, impeding cell signaling | [66] |
| Cancer-associated fibroblasts | Anakinra | II | PDAC | IL-1R antagonist | NCT04926467 |
| Cancer-associated fibroblasts | Futibatinib | I/II | FGFR-aberrant tumors | Anti-FGFR inhibits FGFR signaling pathways | [52] |
| Cancer-associated fibroblasts | Pemigatinib | II/III | Refractory advanced malignancies | Anti-FGFR inhibits FGFR signaling pathways | [53] |
| Cancer-associated fibroblasts | Dovitinib + gemcitabine and capecitabine | Ib | Pancreatic cancer | Anti-FGFR inhibits FGFR signaling pathways | [54] |
| Cancer-associated fibroblasts | Lenvatinib | II | Biliary tract cancer | Anti-FGFR inhibits FGFR signaling pathways | [55] |
| Cancer-associated fibroblasts | Plerixafor (AMD3100) | II | Metastatic pancreatic cancer | CXCR4 inhibition, activating intratumoral immunity | [57] |
| Cancer-associated fibroblasts | Motixafortide (BL-8040) + pembrolizumab | II COMBAT trial | Pancreatic cancer | Inhibition of CXCR4 activation, increasing intratumoral immunity | [58] NCT04543071 |
| Cancer-associated fibroblasts | Losartan + FLOFIRNOX and 9-Ing-41 | II | PDAC | Inhibit collagen I synthesis | NCT05077800 |
| Cancer-associated fibroblasts | SMOi sonidegib + docetaxel | I | Triple-negative breast cancer | Inhibition of signaling and inhibition of microtubule assembly | [61] |
| Cancer-associated fibroblasts | ProAgio | Preclinical | Triple-negative breast cancer | CAF depletion by targeting integrin αvβ3-expressing cells and inducing apoptosis | [62] |
| Cancer-associated fibroblasts | ST316 | I/II | Advanced solid tumors | Suppress transcription of Wnt genes | NCT05848739 |
| Cancer-associated fibroblasts | Dual-targeting CART-cells | Preclinical | Multiple myeloma | CART signaling, targeting malignant plasma cells and CAFs | [65] |
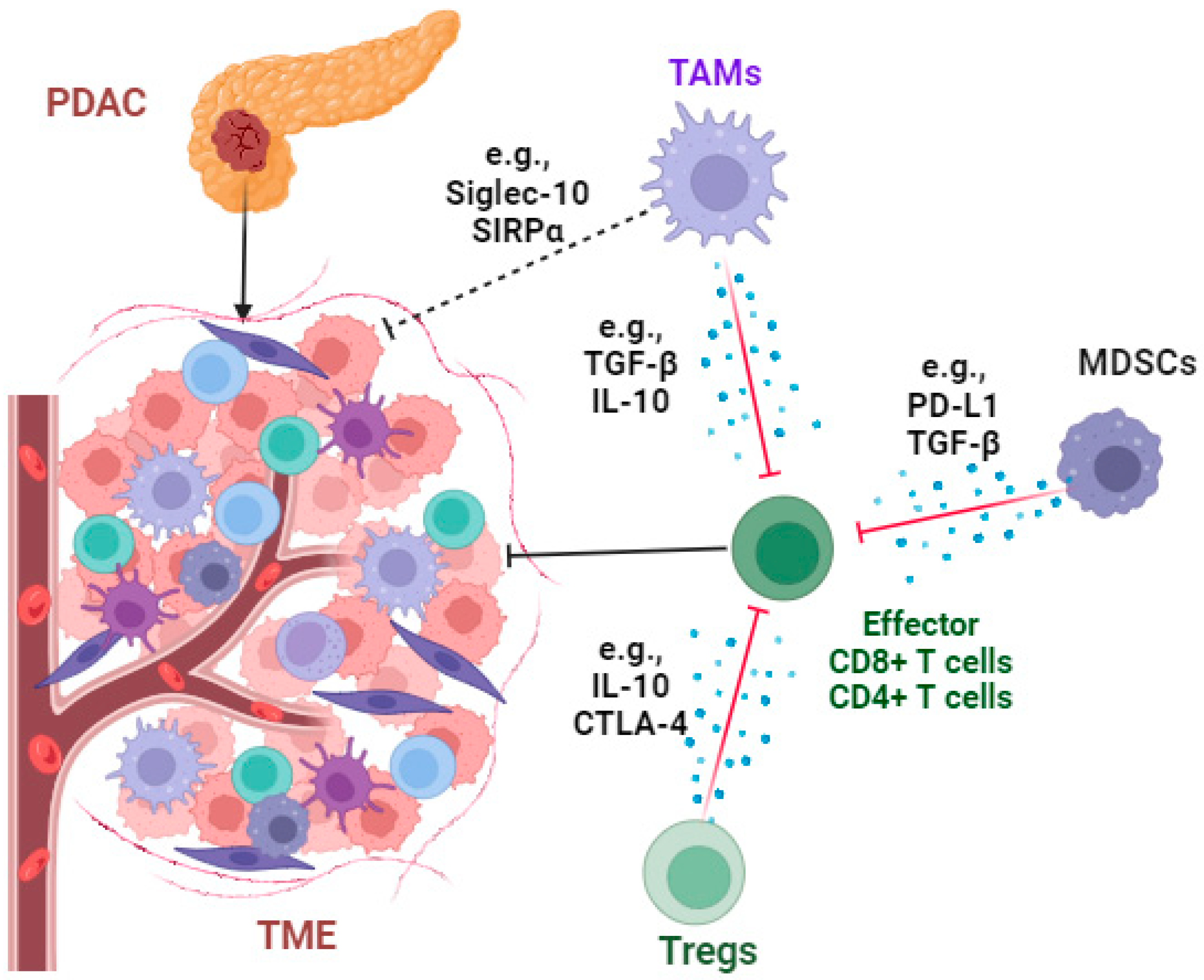
3. Profound Immunosuppressive TME and Immunotherapy in PDAC
3.1. Targeting Immunosuppressive Cells
3.1.1. Tumor-Associated Macrophages (TAMs)
3.1.2. Regulatory T Cells (Tregs)
3.1.3. Myeloid-Derived Suppressor Cells (MDSCs)
3.2. Targeting Immunosuppressive Molecules
3.2.1. Immune Checkpoints
3.2.2. TGF-β Pathway
3.2.3. IL-10
3.2.4. Focal Adhesion Kinase (FAK)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ma, S.; Sokale, I.O.; Thrift, A.P. Trends and Variations in Pancreatic Cancer Mortality Among US Metro and Nonmetro Adults, 1999–2020. J Clin Gastroenterol 2023. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Semwal, R.; Sharma, A.; Varadwaj, P.K. A systematic assessment of statistics, risk factors, and underlying features involved in pancreatic cancer. Cancer Epidemiol. 2019, 58, 104–110. [Google Scholar] [CrossRef]
- Sherman, M.H.; Beatty, G.L. Tumor Microenvironment in Pancreatic Cancer Pathogenesis and Therapeutic Resistance. Annu. Rev. Pathol. 2023, 18, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, C.; Nagalo, B.M.; Chabu, C.Y.; Tesfay, M.Z.; Coleman-Barnett, J.; West, J.T.; Moaven, O. Pancreatic cancer tumor microenvironment is a major therapeutic barrier and target. Front. Immunol. 2024, 15, 1287459. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Posner, R.G.; Von Hoff, D.D.; Han, H. Desmoplasia and chemoresistance in pancreatic cancer. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Edwards, P.; Kang, B.W.; Chau, I. Targeting the Stroma in the Management of Pancreatic Cancer. Front. Oncol. 2021, 11, 691185. [Google Scholar] [CrossRef]
- Sato, N.; Cheng, X.B.; Kohi, S.; Koga, A.; Hirata, K. Targeting hyaluronan for the treatment of pancreatic ductal adenocarcinoma. Acta Pharm. Sin. B 2016, 6, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Halbrook, C.J.; Kerk, S.A.; Radyk, M.; Wisner, S.; Kremer, D.M.; Sajjakulnukit, P.; Andren, A.; Hou, S.W.; Trivedi, A.; et al. Hyaluronic acid fuels pancreatic cancer cell growth. eLife 2021, 10, e62645. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.B.; Sato, N.; Kohi, S.; Koga, A.; Hirata, K. 4-Methylumbelliferone inhibits enhanced hyaluronan synthesis and cell migration in pancreatic cancer cells in response to tumor-stromal interactions. Oncol. Lett. 2018, 15, 6297–6301. [Google Scholar] [CrossRef]
- Yoshida, E.; Kudo, D.; Nagase, H.; Suto, A.; Shimoda, H.; Suto, S.; Kakizaki, I.; Endo, M.; Hakamada, K. 4-Methylumbelliferone Decreases the Hyaluronan-rich Extracellular Matrix and Increases the Effectiveness of 5-Fluorouracil. Anticancer Res. 2018, 38, 5799–5804. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Y.; Kane, K.T.; Collins, M.A.; Simeone, D.M.; Di Magliano, M.P.; Nguyen, K.T. CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol. Cancer Res. 2015, 13, 9–15. [Google Scholar] [CrossRef]
- Menke-van der Houven van Oordt, C.W.; Gomez-Roca, C.; Van Herpen, C.; Coveler, A.L.; Mahalingam, D.; Verheul, H.M.; Van der Graaf, W.T.; Christen, R.; Rüttinger, D.; Weigand, S.; et al. First-in-human phase I clinical trial of RG7356, an anti-CD44 humanized antibody, in patients with advanced, CD44-expressing solid tumors. Oncotarget 2016, 7, 80046–80058. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.A.; Brady, W.E.; Lankes, H.A.; Rose, P.G.; Kelley, J.L.; De Geest, K.; Crispens, M.A.; Resnick, K.E.; Howell, S.B. A phase II study of a urokinase-derived peptide (A6) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Tijink, B.M.; Buter, J.; De Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; Van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [PubMed]
- Gouda, H.M.; Abdel Mohsen, M.M. Frequency of expression of RHAMM/CD168 in Egyptian patients with CML. J. Egypt. Natl. Canc Inst. 2009, 21, 93–99. [Google Scholar]
- Giannopoulos, K.; Własiuk, P.; Dmoszyńska, A.; Roliński, J.; Schmitt, M. Peptide vaccination induces profound changes in the immune system in patients with B-cell chronic lymphocytic leukemia. Folia Histochem. Cytobiol. 2011, 49, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Reiman, T.; Li, W.; Maxwell, C.A.; Sen, S.; Pilarski, L.; Daniels, T.R.; Penichet, M.L.; Feldman, R.; Lichtenstein, A. Targeting aurora kinases as therapy in multiple myeloma. Blood 2007, 109, 3915–3921. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Heineman, T.; Baumgart, M.; Nanavati, C.; Gabrail, N.; Van Wart, S.A.; Mager, D.E.; Maneval, D.C.; Fathallah, A.M.; Sekulovich, R.E. Safety and pharmacokinetics of docetaxel in combination with pegvorhyaluronidase alfa in patients with non-small cell lung cancer. Clin. Transl. Sci. 2021, 14, 1875–1885. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef]
- Infante, J.R.; Korn, R.L.; Rosen, L.S.; LoRusso, P.; Dychter, S.S.; Zhu, J.; Maneval, D.C.; Jiang, P.; Shepard, H.M.; Frost, G.; et al. Phase 1 trials of PEGylated recombinant human hyaluronidase PH20 in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Kim, K.P.; Siveke, J.T.; Lopez, C.D.; Lacy, J.; O’Reilly, E.M.; Macarulla, T.; Manji, G.A.; Lee, J.; Ajani, J.; et al. Atezolizumab Plus PEGPH20 Versus Chemotherapy in Advanced Pancreatic Ductal Adenocarcinoma and Gastric Cancer: MORPHEUS Phase Ib/II Umbrella Randomized Study Platform. Oncologist 2023, 28, 553–e472. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109-301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 2018, 14, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Dean, A.; Gill, S.; McGregor, M.; Broadbridge, V.; Järveläinen, H.A.; Price, T. Dual αV-integrin and neuropilin-1 targeting peptide CEND-1 plus nab-paclitaxel and gemcitabine for the treatment of metastatic pancreatic ductal adenocarcinoma: A first-in-human, open-label, multicentre, phase 1 study. Lancet Gastroenterol. Hepatol. 2022, 7, 943–951. [Google Scholar] [CrossRef]
- Keizer, R.J.; Funahashi, Y.; Semba, T.; Wanders, J.; Beijnen, J.H.; Schellens, J.H.; Huitema, A.D. Evaluation of α2-integrin expression as a biomarker for tumor growth inhibition for the investigational integrin inhibitor E7820 in preclinical and clinical studies. Aaps J. 2011, 13, 230–239. [Google Scholar] [CrossRef]
- Damhofer, H.; Medema, J.P.; Veenstra, V.L.; Badea, L.; Popescu, I.; Roelink, H.; Bijlsma, M.F. Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 1031–1042. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients with Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed]
- Frappaz, D.; Barritault, M.; Montané, L.; Laigle-Donadey, F.; Chinot, O.; Le Rhun, E.; Bonneville-Levard, A.; Hottinger, A.F.; Meyronnet, D.; Bidaux, A.S.; et al. MEVITEM-a phase I/II trial of vismodegib + temozolomide vs temozolomide in patients with recurrent/refractory medulloblastoma with Sonic Hedgehog pathway activation. Neuro-Oncology 2021, 23, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Kondo, S.; Yoshikawa, S.; Inoue, K.; Andre, V.; Tajimi, M.; Murakami, H. A phase I and pharmacokinetic study of taladegib, a Smoothened inhibitor, in Japanese patients with advanced solid tumors. Investig. New Drugs 2018, 36, 647–656. [Google Scholar] [CrossRef]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncology 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Ansari, D.; Ohlsson, H.; Althini, C.; Bauden, M.; Zhou, Q.; Hu, D.; Andersson, R. The Hippo Signaling Pathway in Pancreatic Cancer. Anticancer Res. 2019, 39, 3317–3321. [Google Scholar] [CrossRef]
- Higashi, T.; Hayashi, H.; Kitano, Y.; Yamamura, K.; Kaida, T.; Arima, K.; Taki, K.; Nakagawa, S.; Okabe, H.; Nitta, H.; et al. Statin attenuates cell proliferative ability via TAZ (WWTR1) in hepatocellular carcinoma. Med. Oncol. 2016, 33, 123. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. Semin. Cancer Biol. 2022, 82, 184–196. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; Correa de Sampaio, P.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Hurwitz, H.; Van Cutsem, E.; Bendell, J.; Hidalgo, M.; Li, C.P.; Salvo, M.G.; Macarulla, T.; Sahai, V.; Sama, A.; Greeno, E.; et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. New Drugs 2018, 36, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Ju, H.Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-κB Activation. Clin. Cancer Res. 2016, 22, 1432–1444. [Google Scholar] [CrossRef]
- Hu, C.; Xia, R.; Zhang, X.; Li, T.; Ye, Y.; Li, G.; He, R.; Li, Z.; Lin, Q.; Zheng, S.; et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol. Cancer 2022, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Lin, Z.; Xu, M.; Pan, J.; Wang, Z.W. Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. 2019, 52, e12605. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an Irreversible FGFR1-4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef]
- Subbiah, V.; Iannotti, N.O.; Gutierrez, M.; Smith, D.C.; Féliz, L.; Lihou, C.F.; Tian, C.; Silverman, I.M.; Ji, T.; Saleh, M. FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann. Oncol. 2022, 33, 522–533. [Google Scholar] [CrossRef]
- Ma, W.W.; Xie, H.; Fetterly, G.; Pitzonka, L.; Whitworth, A.; LeVea, C.; Wilton, J.; Mantione, K.; Schihl, S.; Dy, G.K.; et al. A Phase Ib Study of the FGFR/VEGFR Inhibitor Dovitinib With Gemcitabine and Capecitabine in Advanced Solid Tumor and Pancreatic Cancer Patients. Am. J. Clin. Oncol. 2019, 42, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Ikeda, M.; Sasaki, T.; Nagashima, F.; Mizuno, N.; Shimizu, S.; Ikezawa, H.; Hayata, N.; Nakajima, R.; Morizane, C. Phase 2 study of lenvatinib monotherapy as second-line treatment in unresectable biliary tract cancer: Primary analysis results. BMC Cancer 2020, 20, 1105. [Google Scholar] [CrossRef]
- Izumi, D.; Ishimoto, T.; Miyake, K.; Sugihara, H.; Eto, K.; Sawayama, H.; Yasuda, T.; Kiyozumi, Y.; Kaida, T.; Kurashige, J.; et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin β1 clustering and invasiveness in gastric cancer. Int. J. Cancer 2016, 138, 1207–1219. [Google Scholar] [CrossRef]
- Fearon, D.T.; Janowitz, T. AMD3100/Plerixafor overcomes immune inhibition by the CXCL12-KRT19 coating on pancreatic and colorectal cancer cells. Br. J. Cancer 2021, 125, 149–151. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Takabatake, K.; Shimo, T.; Murakami, J.; Anqi, C.; Kawai, H.; Yoshida, S.; Wathone Oo, M.; Haruka, O.; Sukegawa, S.; Tsujigiwa, H.; et al. The Role of Sonic Hedgehog Signaling in the Tumor Microenvironment of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5779. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Turaga, R.C.; Yuan, Y.; Satyanarayana, G.; Mishra, F.; Bian, Z.; Liu, W.; Sun, L.; Yang, J.; Liu, Z.R. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J. Exp. Med. 2021, 218, e20200712. [Google Scholar] [CrossRef]
- Huang, T.X.; Tan, X.Y.; Huang, H.S.; Li, Y.T.; Liu, B.L.; Liu, K.S.; Chen, X.; Chen, Z.; Guan, X.Y.; Zou, C.; et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut 2022, 71, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Sakemura, R.; Hefazi, M.; Siegler, E.L.; Cox, M.J.; Larson, D.P.; Hansen, M.J.; Manriquez Roman, C.; Schick, K.J.; Can, I.; Tapper, E.E.; et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood 2022, 139, 3708–3721. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.C.; Goel, V.K.; Xiong, W.; Sham, J.G.; Pollack, S.M.; Leahy, A.M.; Whiting, S.H.; Yeh, M.M.; Yee, C.; Riddell, S.R.; et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS ONE 2014, 9, e96565. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, M.; Ericsson, A.C. Function of Macrophages in Disease: Current Understanding on Molecular Mechanisms. Front. Immunol. 2021, 12, 620510. [Google Scholar] [CrossRef]
- Xia, Q.; Jia, J.; Hu, C.; Lu, J.; Li, J.; Xu, H.; Fang, J.; Feng, D.; Wang, L.; Chen, Y. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene 2022, 41, 865–877. [Google Scholar] [CrossRef]
- Alausa, A.; Lawal, K.A.; Babatunde, O.A.; Obiwulu, E.N.O.; Oladokun, O.C.; Fadahunsi, O.S.; Celestine, U.O.; Moses, E.U.; Akaniro, I.R.; Adegbola, P.I. Overcoming immunotherapeutic resistance in PDAC: SIRPα-CD47 blockade. Pharmacol. Res. 2022, 181, 106264. [Google Scholar] [CrossRef]
- Xi, Q.; Zhang, J.; Yang, G.; Zhang, L.; Chen, Y.; Wang, C.; Zhang, Z.; Guo, X.; Zhao, J.; Xue, Z.; et al. Restoration of miR-340 controls pancreatic cancer cell CD47 expression to promote macrophage phagocytosis and enhance antitumor immunity. J. Immunother. Cancer 2020, 8, e000253. [Google Scholar] [CrossRef] [PubMed]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Deng, W.; Zhang, Y.; Chang, Y.; Shelat, V.G.; Tsuchida, K.; Lino-Silva, L.S.; Wang, Z. NLRP3 activation in tumor-associated macrophages enhances lung metastasis of pancreatic ductal adenocarcinoma. Transl. Lung Cancer Res. 2022, 11, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Benveniste, E.N. Involvement of STAT-1 and ets family members in interferon-gamma induction of CD40 transcription in microglia/macrophages. J. Biol. Chem. 2000, 275, 23674–23684. [Google Scholar] [CrossRef]
- Soong, R.S.; Song, L.; Trieu, J.; Lee, S.Y.; He, L.; Tsai, Y.C.; Wu, T.C.; Hung, C.F. Direct T cell activation via CD40 ligand generates high avidity CD8+ T cells capable of breaking immunological tolerance for the control of tumors. PLoS ONE 2014, 9, e93162. [Google Scholar] [CrossRef]
- Van Essen, D.; Kikutani, H.; Gray, D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature 1995, 378, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Morrison, A.H.; Diamond, M.S.; Hay, C.A.; Byrne, K.T.; Vonderheide, R.H. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 8022–8031. [Google Scholar] [CrossRef]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Linehan, D.; Noel, M.S.; Hezel, A.F.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.M.E.; Erdkamp, F.; Wilmink, J.; Diehl, J.; et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J. Clin. Oncol. 2018, 36, 92. [Google Scholar] [CrossRef]
- Cassier, P.A.; Garin, G.; Eberst, L.; Delord, J.-P.; Chabaud, S.; Terret, C.; Montane, L.; Bidaux, A.-S.; Laurent, S.; Jaubert, L.; et al. MEDIPLEX: A phase 1 study of durvalumab (D) combined with pexidartinib (P) in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC). J. Clin. Oncol. 2019, 37, 2579. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, Q.; Zhang, X.; Song, Y. Advance of T regulatory cells in tumor microenvironment remodeling and immunotherapy in pancreatic cancer. Eur. J. Inflamm. 2022, 20, 1721727X221092900. [Google Scholar] [CrossRef]
- Yano, H.; Thakur, A.; Tomaszewski, E.N.; Choi, M.; Deol, A.; Lum, L.G. Ipilimumab augments antitumor activity of bispecific antibody-armed T cells. J. Transl. Med. 2014, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Peng, H.; James, C.A.; Cullinan, D.R.; Hogg, G.D.; Mudd, J.L.; Zuo, C.; Takchi, R.; Caldwell, K.E.; Liu, J.; DeNardo, D.G.; et al. Neoadjuvant FOLFIRINOX Therapy Is Associated with Increased Effector T Cells and Reduced Suppressor Cells in Patients with Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 6761–6771. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, T.; Ijsselsteijn, M.; Oosting, J.; Ruano, D.; Van der Ploeg, M.; Dijk, F.; Bonsing, B.; Fariña, A.; Morreau, H.; Vahrmeijer, A.; et al. A Paradoxical Role for Regulatory T Cells in the Tumor Microenvironment of Pancreatic Cancer. Cancers 2022, 14, 3862. [Google Scholar] [CrossRef]
- Aida, K.; Miyakawa, R.; Suzuki, K.; Narumi, K.; Udagawa, T.; Yamamoto, Y.; Chikaraishi, T.; Yoshida, T.; Aoki, K. Suppression of Tregs by anti-glucocorticoid induced TNF receptor antibody enhances the antitumor immunity of interferon-α gene therapy for pancreatic cancer. Cancer Sci. 2014, 105, 159–167. [Google Scholar] [CrossRef]
- Seifert, L.; Plesca, I.; Müller, L.; Sommer, U.; Heiduk, M.; Von Renesse, J.; Digomann, D.; Glück, J.; Klimova, A.; Weitz, J.; et al. LAG-3-Expressing Tumor-Infiltrating T Cells Are Associated with Reduced Disease-Free Survival in Pancreatic Cancer. Cancers 2021, 13, 1297. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Schalck, A.; Jiang, S.; Shang, X.; Wu, C.-J.; Hou, P.; Ruiz, S.H.; Soto, L.S.; Parra, E.; Ying, H.; et al. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat. Cancer 2023, 4, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Pu, N.; Zhao, G.; Yin, H.; Li, J.A.; Nuerxiati, A.; Wang, D.; Xu, X.; Kuang, T.; Jin, D.; Lou, W.; et al. CD25 and TGF-β blockade based on predictive integrated immune ratio inhibits tumor growth in pancreatic cancer. J. Transl. Med. 2018, 16, 294. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 3048–3058. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Wei, X.; Jiang, H.; Lan, C.; Yang, S.; Wang, H.; Yang, Y.; Tian, C.; Xu, Z.; et al. PD-L1 is a direct target of cancer-FOXP3 in pancreatic ductal adenocarcinoma (PDAC), and combined immunotherapy with antibodies against PD-L1 and CCL5 is effective in the treatment of PDAC. Signal Transduct. Target. Ther. 2020, 5, 38. [Google Scholar] [CrossRef]
- Piper, M.; Hoen, M.; Darragh, L.B.; Knitz, M.W.; Nguyen, D.; Gadwa, J.; Durini, G.; Karakoc, I.; Grier, A.; Neupert, B.; et al. Simultaneous targeting of PD-1 and IL-2Rβγ with radiation therapy inhibits pancreatic cancer growth and metastasis. Cancer Cell 2023, 41, 950–969.e956. [Google Scholar] [CrossRef] [PubMed]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.S.; Lee, H.M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Kumar, S.; Ross, K.A.; Gautam, S.; Poelaert, B.; Nasser, M.W.; Aithal, A.; Bhatia, R.; Wannemuehler, M.J.; Narasimhan, B.; et al. Emerging trends in the immunotherapy of pancreatic cancer. Cancer Lett. 2018, 417, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, Y.; Tazawa, H.; Yamada, M.; Kanaya, N.; Fushimi, T.; Kikuchi, S.; Kuroda, S.; Ohara, T.; Noma, K.; Yoshida, R.; et al. Oncolytic virus-mediated reducing of myeloid-derived suppressor cells enhances the efficacy of PD-L1 blockade in gemcitabine-resistant pancreatic cancer. Cancer Immunol. Immunother. 2023, 72, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Araki, H.; Tazawa, H.; Kanaya, N.; Kajiwara, Y.; Yamada, M.; Hashimoto, M.; Kikuchi, S.; Kuroda, S.; Yoshida, R.; Umeda, Y.; et al. Oncolytic virus-mediated p53 overexpression promotes immunogenic cell death and efficacy of PD-1 blockade in pancreatic cancer. Mol. Ther. Oncolytics 2022, 27, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Christenson, E.; Lim, S.J.; Wang, H.; Ferguson, A.; Parkinson, R.; Cetasaan, Y.; Rodriguez, C.; Burkhart, R.; De Jesus-Acosta, A.; He, J.; et al. Nivolumab and a CCR2/CCR5 dual antagonist (BMS-813160) with or without GVAX for locally advanced pancreatic ductal adenocarcinomas: Results of phase I study. J. Clin. Oncol. 2023, 41, 730. [Google Scholar] [CrossRef]
- Loch, F.N.; Kamphues, C.; Beyer, K.; Schineis, C.; Rayya, W.; Lauscher, J.C.; Horst, D.; Dragomir, M.P.; Schallenberg, S. The Immune Checkpoint Landscape in Tumor Cells of Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 2160. [Google Scholar] [CrossRef]
- Seifert, A.M.; Eymer, A.; Heiduk, M.; Wehner, R.; Tunger, A.; Von Renesse, J.; Decker, R.; Aust, D.E.; Welsch, T.; Reissfelder, C.; et al. PD-1 Expression by Lymph Node and Intratumoral Regulatory T Cells Is Associated with Lymph Node Metastasis in Pancreatic Cancer. Cancers 2020, 12, 2756. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Almhanna, K. Pancreatic cancer and immune checkpoint inhibitors-still a long way to go. Transl. Gastroenterol. Hepatol. 2021, 6, 6. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Lim, K.H.; McWilliams, R.; Suresh, R.; Lockhart, A.C.; Brown, A.; Breden, M.; Belle, J.I.; Herndon, J.; Bogner, S.J.; et al. Defactinib, Pembrolizumab, and Gemcitabine in Patients with Advanced Treatment Refractory Pancreatic Cancer: A Phase I Dose Escalation and Expansion Study. Clin. Cancer Res. 2022, 28, 5254–5262. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Duffy, A.G.; Brar, G.; Fioravanti, S.; Mabry-Hrones, D.; Walker, M.; Bonilla, C.M.; Wood, B.J.; Citrin, D.E.; Gil Ramirez, E.M.; et al. Immune Checkpoint Blockade in Combination with Stereotactic Body Radiotherapy in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Shui, L.; Cheng, K.; Li, X.; Shui, P.; Zhou, X.; Li, J.; Yi, C.; Cao, D. Study protocol for an open-label, single-arm, phase Ib/II study of combination of toripalimab, nab-paclitaxel, and gemcitabine as the first-line treatment for patients with unresectable pancreatic ductal adenocarcinoma. BMC Cancer 2020, 20, 636. [Google Scholar] [CrossRef] [PubMed]
- Trebska-McGowan, K.; Chaib, M.; Alvarez, M.A.; Kansal, R.; Pingili, A.K.; Shibata, D.; Makowski, L.; Glazer, E.S. TGF-β Alters the Proportion of Infiltrating Immune Cells in a Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Surg. 2022, 26, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.M.; Kansal, R.G.; Alvarez, M.A.; Hollingsworth, T.J.; Elahi, A.; Miranda-Carboni, G.; Hendrick, L.E.; Pingili, A.K.; Albritton, L.M.; Dickson, P.V.; et al. Role of TGF-β in pancreatic ductal adenocarcinoma progression and PD-L1 expression. Cell Oncol. 2021, 44, 673–687. [Google Scholar] [CrossRef]
- Lee, J.E.; Lee, P.; Yoon, Y.C.; Han, B.S.; Ko, S.; Park, M.S.; Lee, Y.J.; Kim, S.E.; Cho, Y.J.; Lim, J.H.; et al. Vactosertib, TGF-β receptor I inhibitor, augments the sensitization of the anti-cancer activity of gemcitabine in pancreatic cancer. Biomed. Pharmacother. 2023, 162, 114716. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFβ receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; George, V.; Shrestha, N.; Wang, M.; Dee, M.J.; Zhu, X.; Liu, B.; Egan, J.; D’Eramo, F.; Spanoudis, C.; et al. Immunotherapeutic HCW9218 augments anti-tumor activity of chemotherapy via NK cell-mediated reduction of therapy-induced senescent cells. Mol. Ther. 2022, 30, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-B.; Yang, Z.-H.; Guo, Q.-Q. Immune checkpoint inhibition for pancreatic ductal adenocarcinoma: Limitations and prospects: A systematic review. Cell Commun. Signal. 2021, 19, 117. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Eisinger, S.; He, F.; Bergsland, M.; Pelicano, C.; Driescher, C.; Westberg, K.; Benitez, I.I.; Humoud, R.; Palano, G.; et al. Targeting myeloid suppressive cells revives cytotoxic anti-tumor responses in pancreatic cancer. iScience 2022, 25, 105317. [Google Scholar] [CrossRef] [PubMed]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Papadopoulos, K.P.; Falchook, G.S.; Patel, M.R.; Infante, J.R.; Aljumaily, R.; Wong, D.J.; Autio, K.A.; Wainberg, Z.A.; Bauer, T.M.; et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Invest. New Drugs 2021, 39, 182–192. [Google Scholar] [CrossRef]
- Hecht, J.R.; Lonardi, S.; Bendell, J.; Sim, H.W.; Macarulla, T.; Lopez, C.D.; Van Cutsem, E.; Muñoz Martin, A.J.; Park, J.O.; Greil, R.; et al. Randomized Phase III Study of FOLFOX Alone or With Pegilodecakin as Second-Line Therapy in Patients With Metastatic Pancreatic Cancer That Progressed After Gemcitabine (SEQUOIA). J. Clin. Oncol. 2021, 39, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions With Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Davidson, C.; Taggart, D.; Sims, A.H.; Lonergan, D.W.; Canel, M.; Serrels, A. FAK promotes stromal PD-L2 expression associated with poor survival in pancreatic cancer. Br. J. Cancer 2022, 127, 1893–1905. [Google Scholar] [CrossRef]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Liu, S.; Yang, M. Clinical diagnosis and management of pancreatic cancer: Markers, molecular mechanisms, and treatment options. World J. Gastroenterol. 2022, 28, 6827–6845. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Liu, X.; Knolhoff, B.L.; Hegde, S.; Lee, K.B.; Jiang, H.; Fields, R.C.; Pachter, J.A.; Lim, K.H.; DeNardo, D.G. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 2020, 69, 122–132. [Google Scholar] [CrossRef]
- Zhang, J.; He, D.H.; Zajac-Kaye, M.; Hochwald, S.N. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014, 13, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.; Murphy, K.; Cock, T.-A.; Devlin, M.; Herrmann, D.; Timpson, P. The effect of adding a selective FAK inhibitor AMP945 to FOLFIRINOX in a model of pancreatic cancer. J. Clin. Oncol. 2023, 41, e15128. [Google Scholar] [CrossRef]
- Springfeld, C.; Ferrone, C.R.; Katz, M.H.G.; Philip, P.A.; Hong, T.S.; Hackert, T.; Büchler, M.W.; Neoptolemos, J. Neoadjuvant therapy for pancreatic cancer. Nat. Rev. Clin. Oncol. 2023, 20, 318–337. [Google Scholar] [CrossRef] [PubMed]


| Class of Therapies | Targets | Treatments | Clinical Trials | Phase |
|---|---|---|---|---|
| Immune checkpoint inhibitors | CTLA-4 | Ipilimumab, an anti-CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) antibody. | NCT00112580 | 2 |
| PD-1 | Anti-PD-1 antibody pembrolizumab in combination with macrophage activator BXCL701. | NCT05558982 | 2 | |
| PD-L1 | Anti-PD-L1 antibody Durvalumab with a tyrosine kinase inhibitor of colony-stimulating factor 1 receptor (CSF1R) (pexidartinib). | NCT02777710 | 1 | |
| PD-L1 | Anti-PD-L1 antibody durvalumab co-administration with galunisertib. | NCT02734160 | 1 | |
| Cytokines/cytokine receptors | IL-10 | Pegilodecakin, a pegylated recombinant human IL-10, in combination with FOLFOX (folinic acid, fluorouracil, and oxaliplatin). | NCT02009449 | 1 |
| TGF-β+IL-15 | HCW9218, a bifunctional TGF-β antagonist comprising IL-15 immunostimulatory activity. | NCT05304936 | 1/2 | |
| TGF-β receptor | TGF-β receptor I kinase inhibitor galunisertib plus gemcitabine. | NCT01373164 | 1/2 | |
| TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab. | NCT02734160 | 1 | ||
| Chemokines/ chemokine receptors | CCR2/CCR5 | CCR2/CCR5 inhibitor BMS-813160 in combination with nivolumab and gemcitabine and nab-paclitaxel. | NCT03496662 | 1/2 |
| CCR2 | CCR2 inhibitor PF-04136309 in combination with chemotherapy FOLFIRINOX (oxaliplatin and irinotecan plus leucovorin and fluorouracil). | NCT01413022 | 1 | |
| CCR2 | CCR2-specific antagonist CCX872-B plus FOLFIRINOX. | NCT02345408 | 1 | |
| CCR2 | CCR2 inhibitor PF-04136309 in combination with chemotherapy FOLFIRINOX (oxaliplatin and irinotecan plus leucovorin and fluorouracil). | NCT01413022 | 1 | |
| Co-stimulatory molecules | CD25 | ADCT-301, a monoclonal antibody that binds to CD25 conjugated to a PBD dimer toxin, in combination with or without anti-PD-1 therapy (pembrolizamab). | NCT03621982 | 1 |
| Kinase inhibitors | CSF1R | Pexidartinib, a tyrosine kinase inhibitor of colony-stimulating factor 1 receptor (CSF1R). | NCT02777710 | 1 |
| FAK | Focal adhesion kinase (FAK) inhibitor defactinib with pembrolizumab. | NCT03727880 | 2 | |
| FAK | Defactinib with stereotactic body radiotherapy (SBRT). | NCT04331041 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olaoba, O.T.; Yang, M.; Adelusi, T.I.; Maidens, T.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Li, G. Targeted Therapy for Highly Desmoplastic and Immunosuppressive Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 1470. https://doi.org/10.3390/cancers16081470
Olaoba OT, Yang M, Adelusi TI, Maidens T, Kimchi ET, Staveley-O’Carroll KF, Li G. Targeted Therapy for Highly Desmoplastic and Immunosuppressive Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma. Cancers. 2024; 16(8):1470. https://doi.org/10.3390/cancers16081470
Chicago/Turabian StyleOlaoba, Olamide T., Ming Yang, Temitope I. Adelusi, Tessa Maidens, Eric T. Kimchi, Kevin F. Staveley-O’Carroll, and Guangfu Li. 2024. "Targeted Therapy for Highly Desmoplastic and Immunosuppressive Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma" Cancers 16, no. 8: 1470. https://doi.org/10.3390/cancers16081470
APA StyleOlaoba, O. T., Yang, M., Adelusi, T. I., Maidens, T., Kimchi, E. T., Staveley-O’Carroll, K. F., & Li, G. (2024). Targeted Therapy for Highly Desmoplastic and Immunosuppressive Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma. Cancers, 16(8), 1470. https://doi.org/10.3390/cancers16081470