
IL-1β in Neoplastic Disease and the Role of Its Tumor-Derived Form in the Progression and Treatment of Metastatic Prostate Cancer


{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. IL-1β Processing, Known Receptors, and Their Signaling
3. Immune Cell-Derived IL-1β and Its Tumor-Promoting Role
3.1. Angiogenesis and Vascularization
3.2. Immune Evasion
4. Synthesis and Secretion of IL-1β by Tumor Cells
5. Tumor-Derived IL-1β in Different Metastatic Tumors
5.1. Epithelial–Mesenchymal Transition
5.2. Invasion
5.3. Angiogenesis
5.4. Conditioning of the Metastatic Niche
6. Prostate Cancer: Clinical Course and Treatment
6.1. Localized Disease
6.2. Metastatic Disease
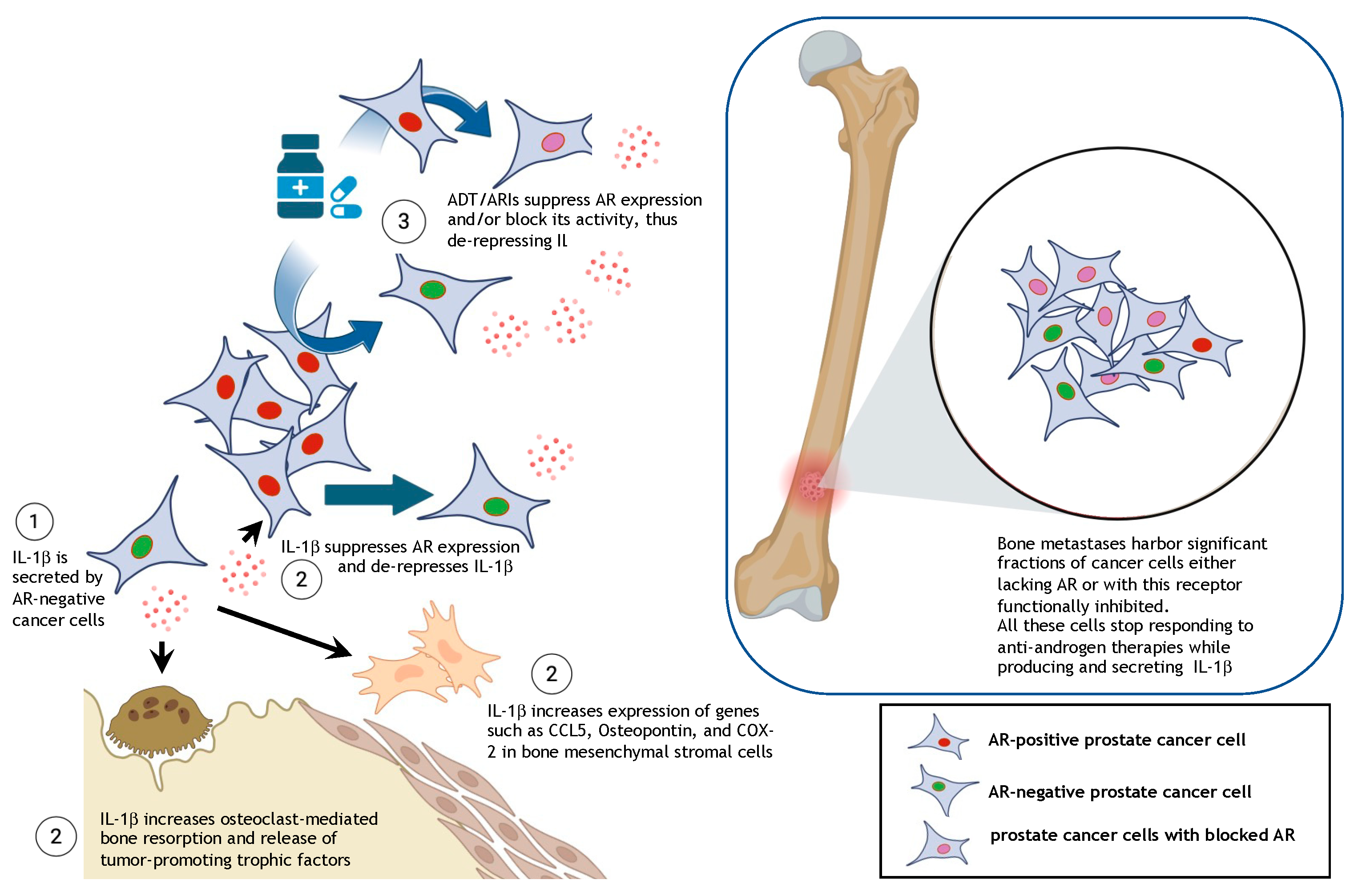
7. Tumor-Derived IL-1β in Bone Metastatic Prostate Cancer
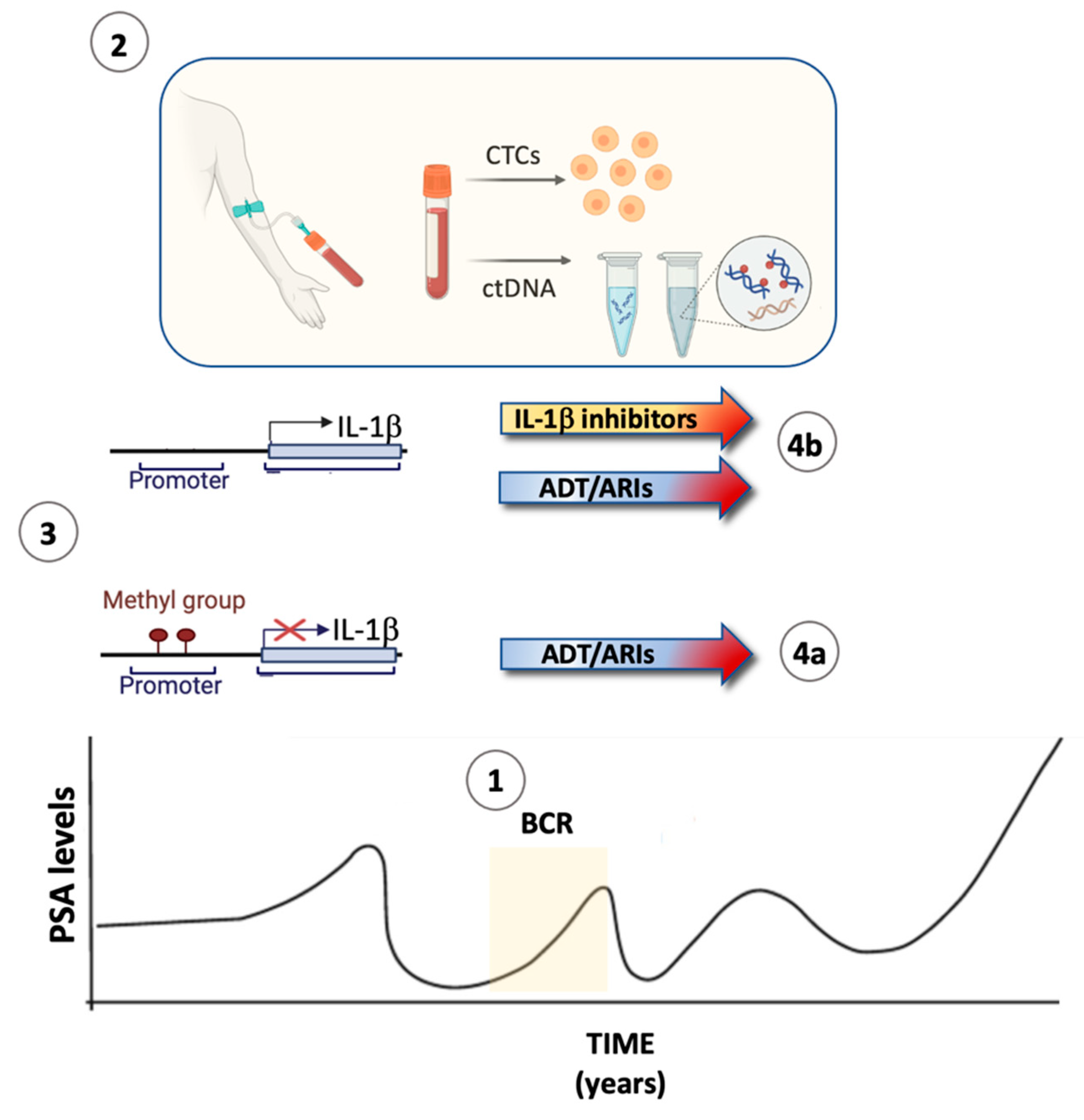
8. The Androgen Receptor Represses IL-1β Transcription
9. Rationale for Combining ADT/ARIs with Inhibitors of IL-1β Signaling
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.; Campos, T.M.; Saldanha, M.; Oliveira, S.C.; Nascimento, M.; Zamboni, D.S.; Machado, P.R.; Arruda, S.; Scott, P.; Carvalho, E.M.; et al. IL-1beta Production by Intermediate Monocytes Is Associated with Immunopathology in Cutaneous Leishmaniasis. J. Investig. Dermatol. 2018, 138, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Mahida, Y.R.; Wu, K.; Jewell, D.P. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut 1989, 30, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Irizarry-Caro, R.A.; McDaniel, M.M.; Chawla, A.S.; Carroll, K.R.; Overcast, G.R.; Philip, N.H.; Oberst, A.; Chervonsky, A.V.; Katz, J.D.; et al. T cells instruct myeloid cells to produce inflammasome-independent IL-1beta and cause autoimmunity. Nat. Immunol. 2020, 21, 65–74. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.C.; Li, S.; Liang, K.; Ting, J.P.-Y. A 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell 2023, 186, 2288–2312. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nunez, G. The NLRP3 inflammasome: Activation and regulation. Trends Biochem. Sci. 2023, 48, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Mankan, A.K.; Dau, T.; Jenne, D.; Hornung, V. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur. J. Immunol. 2012, 42, 710–715. [Google Scholar] [CrossRef]
- Pandori, W.J.; Matsuno, S.Y.; Shin, J.H.; Kim, S.C.; Kao, T.H.; Mallya, S.; Batarseh, S.N.; Lodoen, M.B. Role for Caspase-8 in the Release of IL-1beta and Active Caspase-1 from Viable Human Monocytes during Toxoplasma gondii Infection. J. Immunol. 2024, 212, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Eltom, S.; Belvisi, M.G.; Yew-Booth, L.; Dekkak, B.; Maher, S.A.; Dubuis, E.D.; Jones, V.; Fitzgerald, K.A.; Birrell, M.A. TLR4 activation induces IL-1beta release via an IPAF dependent but caspase 1/11/8 independent pathway in the lung. Respir. Res. 2014, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.B.; Park, H.H. Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis 2015, 20, 196–209. [Google Scholar] [CrossRef]
- Ito, T. PAMPs and DAMPs as triggers for DIC. J. Intensive Care 2014, 2, 67. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef]
- Flannery, S.; Bowie, A.G. The interleukin-1 receptor-associated kinases: Critical regulators of innate immune signalling. Biochem. Pharmacol. 2010, 80, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Kollewe, C.; Mackensen, A.C.; Neumann, D.; Knop, J.; Cao, P.; Li, S.; Wesche, H.; Martin, M.U. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J. Biol. Chem. 2004, 279, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kaczanowska, S.; Davila, E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front. Immunol. 2014, 5, 553. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Ikejima, T.; Warner, S.J.; Orencole, S.F.; Lonnemann, G.; Cannon, J.G.; Libby, P. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J. Immunol. 1987, 139, 1902–1910. [Google Scholar] [CrossRef]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G.; et al. Characterization of a Functional NF-κB Site in the Human Interleukin 1β Promoter: Evidence for a Positive Autoregulatory Loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [CrossRef] [PubMed]
- Streicher, K.L.; Willmarth, N.E.; Garcia, J.; Boerner, J.L.; Dewey, T.G.; Ethier, S.P. Activation of a Nuclear Factor κB/Interleukin-1 Positive Feedback Loop by Amphiregulin in Human Breast Cancer Cells. Mol. Cancer Res. 2007, 5, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P.; Malyak, M.; Guthridge, C.J.; Gabay, C. Interleukin-1 receptor antagonist: Role in biology. Annu. Rev. Immunol. 1998, 16, 27–55. [Google Scholar] [CrossRef]
- Colotta, F.; Sironi, M.; Borre, A.; Pollicino, T.; Bernasconi, S.; Boraschi, D.; Mantovani, A. Type II interleukin-1 receptor is not expressed in cultured endothelial cells and is not involved in endothelial cell activation. Blood 1993, 81, 1347–1351. [Google Scholar] [CrossRef]
- Schluter, T.; Schelmbauer, C.; Karram, K.; Mufazalov, I.A. Regulation of IL-1 signaling by the decoy receptor IL-1R2. J. Mol. Med. 2018, 96, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Garlanda, C.; Mantovani, A.; Riva, F. Cytokine decoy and scavenger receptors as key regulators of immunity and inflammation. Cytokine 2016, 87, 37–45. [Google Scholar] [CrossRef]
- Burns, K.; Janssens, S.; Brissoni, B.; Olivos, N.; Beyaert, R.; Tschopp, J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 2003, 197, 263–268. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hernandez, L.D.; Galan, J.E.; Janeway, C.A., Jr.; Medzhitov, R.; Flavell, R.A. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 2002, 110, 191–202. [Google Scholar] [CrossRef]
- Janssens, S.; Burns, K.; Tschopp, J.; Beyaert, R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr. Biol. 2002, 12, 467–471. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Jewett, A.; Kos, J.; Kaur, K.; Safaei, T.; Sutanto, C.; Chen, W.; Wong, P.; Namagerdi, A.K.; Fang, C.; Fong, Y.; et al. Natural Killer Cells: Diverse Functions in Tumor Immunity and Defects in Pre-neoplastic and Neoplastic Stages of Tumorigenesis. Mol. Ther. Oncolytics. 2020, 16, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Gunzer, M.; Janich, S.; Varga, G.; Grabbe, S. Dendritic cells and tumor immunity. Semin. Immunol. 2001, 13, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, G.; Cheng, W.A.; Dong, Z.; Sun, H.; Lee, V.Y.; Cha, S.C.; Smith, D.L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1181–1195. [Google Scholar] [CrossRef]
- Chow, M.T.; Moller, A.; Smyth, M.J. Inflammation and immune surveillance in cancer. Semin. Cancer Biol. 2012, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.; Walle, L.V.; Saavedra, P.H.V.; Lebegge, E.; Damme, H.V.; Murgaski, A.; Qian, J.; Ehling, M.; Pretto, S.; Bolli, E.; et al. IL1beta Promotes Immune Suppression in the Tumor Microenvironment Independent of the Inflammasome and Gasdermin D. Cancer Immunol. Res. 2021, 9, 309–323. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Espinoza-Sanchez, N.A.; Chimal-Ramirez, G.K.; Mantilla, A.; Fuentes-Panana, E.M. IL-1beta, IL-8, and Matrix Metalloproteinases-1, -2, and -10 Are Enriched upon Monocyte-Breast Cancer Cell Cocultivation in a Matrigel-Based Three-Dimensional System. Front. Immunol. 2017, 8, 205. [Google Scholar] [CrossRef]
- Jang, J.H.; Kim, D.H.; Lim, J.M.; Lee, J.W.; Jeong, S.J.; Kim, K.P.; Surh, Y.J. Breast Cancer Cell-Derived Soluble CD44 Promotes Tumor Progression by Triggering Macrophage IL1beta Production. Cancer Res. 2020, 80, 1342–1356. [Google Scholar] [CrossRef]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, P.; Singh, S.M. Alteration in IL-1 and arginase activity of tumor-associated macrophages: A role in the promotion of tumor growth. Cancer Lett. 1996, 107, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Voronov, E.; Dotan, S.; Lahat, N.; Rahat, M.A.; Fogel, M.; Huszar, M.; White, M.R.; Dinarello, C.A.; Apte, R.N. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J. Immunol. 2009, 183, 4705–4714. [Google Scholar] [CrossRef]
- Mantsounga, C.S.; Lee, C.; Neverson, J.; Sharma, S.; Healy, A.; Berus, J.M.; Parry, C.; Ceneri, N.M.; Lopez-Giraldez, F.; Chun, H.J.; et al. Macrophage IL-1beta promotes arteriogenesis by autocrine STAT3- and NF-kappaB-mediated transcription of pro-angiogenic VEGF-A. Cell Rep. 2022, 38, 110309. [Google Scholar] [CrossRef]
- Benner, B.; Scarberry, L.; Stiff, A.; Duggan, M.C.; Good, L.; Lapurga, G.; Butchar, J.P.; Tridandapani, S.; Carson, W.E. Evidence for interaction of the NLRP3 inflammasome and Bruton’s tyrosine kinase in tumor-associated macrophages: Implications for myeloid cell production of interleukin-1beta. OncoImmunology 2019, 8, 1659704. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Costigliolo, S.; Olcese, L.; Zocchi, M.R.; Rubartelli, A. Secretion of bioactive interleukin-1beta by dendritic cells is modulated by interaction with antigen specific T cells. Blood 2000, 95, 3809–3815. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Lotti, L.V.; Poggi, A.; Torrisi, M.R.; Zocchi, M.R.; Rubartelli, A. CD8(+) T lymphocytes induce polarized exocytosis of secretory lysosomes by dendritic cells with release of interleukin-1beta and cathepsin D. Blood 2001, 98, 2152–2159. [Google Scholar] [CrossRef]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.C.; Kang, E.; Mohamed-Hadley, A.; Buckanovich, R.J.; Holtz, D.O.; Jenkins, A.; Na, H.; Zhang, L.; et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat. Med. 2004, 10, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gebhardt, C.; Umansky, L.; Beckhove, P.; Schulze, T.J.; Utikal, J.; Umansky, V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int. J. Cancer 2015, 136, 2352–2360. [Google Scholar] [CrossRef]
- Zeisler, H.; Tempfer, C.; Joura, E.A.; Sliutz, G.; Koelbl, H.; Wagner, O.; Kainz, C. Serum interleukin 1 in ovarian cancer patients. Eur. J. Cancer 1998, 34, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Koh, Y.; Kim, D.W.; Ahn, Y.O.; Kim, T.M.; Han, S.W.; Oh, D.Y.; Lee, S.H.; Im, S.A.; Kim, T.Y.; et al. Clinical Implications of VEGF, TGF-beta1, and IL-1beta in Patients with Advanced Non-small Cell Lung Cancer. Cancer Res. Treat. 2013, 45, 325–333. [Google Scholar] [CrossRef]
- Kruger-Krasagakes, S.; Krasagakis, K.; Garbe, C.; Diamantstein, T. Production of cytokines by human melanoma cells and melanocytes. Recent. Results Cancer Res. 1995, 139, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118, e2000915118. [Google Scholar] [CrossRef] [PubMed]
- Escobar, P.; Bouclier, C.; Serret, J.; Bieche, I.; Brigitte, M.; Caicedo, A.; Sanchez, E.; Vacher, S.; Vignais, M.L.; Bourin, P.; et al. IL-1beta produced by aggressive breast cancer cells is one of the factors that dictate their interactions with mesenchymal stem cells through chemokine production. Oncotarget 2015, 6, 29034–29047. [Google Scholar] [CrossRef] [PubMed]
- Nutter, F.; Holen, I.; Brown, H.K.; Cross, S.S.; Evans, C.A.; Walker, M.; Coleman, R.E.; Westbrook, J.A.; Selby, P.J.; Brown, J.E.; et al. Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: Evidence using a novel bone-seeking cell line. Endocr.-Relat. Cancer 2014, 21, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter, F.; Wilkinson, J.M.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782. [Google Scholar] [CrossRef]
- Ricote, M.; García-Tuñón, I.; Bethencourt, F.R.; Fraile, B.; Paniagua, R.; Royuela, M. Interleukin-1 (IL-1alpha and IL-1beta) and its receptors (IL-1RI, IL-1RII, and IL-1Ra) in prostate carcinoma. Cancer 2004, 100, 1388–1396. [Google Scholar] [CrossRef]
- Liu, Q.; Russell, M.R.; Shahriari, K.; Jernigan, D.L.; Lioni, M.I.; Garcia, F.U.; Fatatis, A. Interleukin-1beta promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 2013, 73, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Shahriari, K.; Shen, F.; Worrede-Mahdi, A.; Liu, Q.; Gong, Y.; Garcia, F.U.; Fatatis, A. Cooperation among heterogeneous prostate cancer cells in the bone metastatic niche. Oncogene 2017, 36, 2846–2856. [Google Scholar] [CrossRef]
- Herroon, M.K.; Diedrich, J.D.; Rajagurubandara, E.; Martin, C.; Maddipati, K.R.; Kim, S.; Heath, E.I.; Granneman, J.; Podgorski, I. Prostate Tumor Cell–Derived IL1β Induces an Inflammatory Phenotype in Bone Marrow Adipocytes and Reduces Sensitivity to Docetaxel via Lipolysis-Dependent Mechanisms. Mol. Cancer Res. 2019, 17, 2508–2521. [Google Scholar] [CrossRef] [PubMed]
- Pretre, V.; Papadopoulos, D.; Regard, J.; Pelletier, M.; Woo, J. Interleukin-1 (IL-1) and the inflammasome in cancer. Cytokine 2022, 153, 155850. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef]
- Ahmad, I.; Muneer, K.M.; Tamimi, I.A.; Chang, M.E.; Ata, M.O.; Yusuf, N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol. Appl. Pharmacol. 2013, 270, 70–76. [Google Scholar] [CrossRef]
- Xue, Y.; Du, H.D.; Tang, D.; Zhang, D.; Zhou, J.; Zhai, C.W.; Yuan, C.C.; Hsueh, C.Y.; Li, S.J.; Heng, Y.; et al. Correlation Between the NLRP3 Inflammasome and the Prognosis of Patients With LSCC. Front. Oncol. 2019, 9, 588. [Google Scholar] [CrossRef]
- Chen, L.; Huang, C.F.; Li, Y.C.; Deng, W.W.; Mao, L.; Wu, L.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef]
- Beaupre, D.M.; Talpaz, M.; Marini, F.C., 3rd; Cristiano, R.J.; Roth, J.A.; Estrov, Z.; Albitar, M.; Freedman, M.H.; Kurzrock, R. Autocrine interleukin-1beta production in leukemia: Evidence for the involvement of mutated RAS. Cancer Res. 1999, 59, 2971–2980. [Google Scholar]
- Kang, A.R.; Cho, J.H.; Lee, N.G.; Kwon, J.H.; Song, J.Y.; Hwang, S.G.; Jung, I.S.; Kim, J.S.; Um, H.D.; Oh, S.C.; et al. Radiation-induced IL-1beta expression and secretion promote cancer cell migration/invasion via activation of the NF-kappaB-RIP1 pathway. Biochem. Biophys. Res. Commun. 2021, 534, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Ikner, A.; Ashkenazi, A. TWEAK induces apoptosis through a death-signaling complex comprising receptor-interacting protein 1 (RIP1), Fas-associated death domain (FADD), and caspase-8. J. Biol. Chem. 2011, 286, 21546–21554. [Google Scholar] [CrossRef]
- Teshima, S.; Nakanishi, H.; Nishizawa, M.; Kitagawa, K.; Kaibori, M.; Yamada, M.; Habara, K.; Kwon, A.-H.; Kamiyama, Y.; Ito, S.; et al. Up-regulation of IL-1 receptor through PI3K/Akt is essential for the induction of iNOS gene expression in hepatocytes. J. Hepatol. 2004, 40, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Sathe, S.S.; Swiatkowski, S.M.; Hampole, C.V.; Stark, G.R. Secretion of cytokines and growth factors as a general cause of constitutive NFκB activation in cancer. Oncogene 2004, 23, 2138–2145. [Google Scholar] [CrossRef]
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two Mechanistically and Temporally Distinct NF-κB Activation Pathways in IL-1 Signaling. Sci. Signal 2009, 2, ra66. [Google Scholar] [CrossRef]
- Muerkoster, S.; Arlt, A.; Sipos, B.; Witt, M.; Grossmann, M.; Kloppel, G.; Kalthoff, H.; Folsch, U.R.; Schafer, H. Increased expression of the E3-ubiquitin ligase receptor subunit betaTRCP1 relates to constitutive nuclear factor-kappaB activation and chemoresistance in pancreatic carcinoma cells. Cancer Res. 2005, 65, 1316–1324. [Google Scholar] [CrossRef]
- Chen, K.-H.; Weng, M.-S.; Lin, J.-K. Tangeretin suppresses IL-1β-induced cyclooxygenase (COX)-2 expression through inhibition of p38 MAPK, JNK, and AKT activation in human lung carcinoma cells. Biochem. Pharmacol. 2007, 73, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Elpek, K.G.; Vinjamoori, A.; Zimmerman, S.M.; Chu, G.C.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. PTEN Is a Major Tumor Suppressor in Pancreatic Ductal Adenocarcinoma and Regulates an NF-κB–Cytokine Network. Cancer Discov. 2011, 1, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; Marzo, A.M.D.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashidi, R.R.; Noraldeen, S.A.M.; Kareem, A.K.; Mahmoud, A.K.; Kadhum, W.R.; Ramírez-Coronel, A.A.; Iswanto, A.H.; Obaid, R.F.; Jalil, A.T.; Mustafa, Y.F.; et al. Malignant function of nuclear factor-kappaB axis in prostate cancer: Molecular interactions and regulation by non-coding RNAs. Pharmacol. Res. 2023, 194, 106775. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Jeon, J.M.; Na, A.Y.; Kwon, O.K.; Bang, I.H.; Ha, Y.-S.; Bae, E.J.; Park, B.-H.; Lee, E.H.; Kwon, T.G.; et al. SIRT5 Directly Inhibits the PI3K/AKT Pathway in Prostate Cancer Cell Lines. Cancer Genom. Proteom. 2022, 19, 50–59. [Google Scholar] [CrossRef]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL-1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef]
- Shadab, A.; Mahjoor, M.; Abbasi-Kolli, M.; Afkhami, H.; Moeinian, P.; Safdarian, A.-R. Divergent functions of NLRP3 inflammasomes in cancer: A review. Cell Commun. Signal 2023, 21, 232. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.R.; Peeper, D.S. Metastasis mechanisms. Biochim. Biophys. Acta (BBA) Rev. Cancer. 2009, 1796, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Bani, M.R.; Garofalo, A.; Scanziani, E.; Giavazzi, R. Effect of interleukin-1-beta on metastasis formation in different tumor systems. JNCI J. Natl. Cancer Inst. 1991, 83, 119–123. [Google Scholar] [CrossRef]
- Elaraj, D.M.; Weinreich, D.M.; Varghese, S.; Puhlmann, M.; Hewitt, S.M.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. 2006, 12, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.G.; Garofalo, A.; Padura, I.M.; Mantovani, A.; Giavazzi, R. Interleukin 1 receptor antagonist inhibits the augmentation of metastasis induced by interleukin 1 or lipopolysaccharide in a human melanoma/nude mouse system. Cancer Res. 1993, 53, 5051–5054. [Google Scholar] [PubMed]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell. 2019, 49, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Celia-Terrassa, T.; Kang, Y. How important is EMT for cancer metastasis? PLOS Biol. 2024, 22, e3002487. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Chronic IL-1beta-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 2020, 10, 377. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Huerta-Yepez, S.; Law, I.K.; Baay-Guzman, G.J.; Tirado-Rodriguez, B.; Hoffman, J.M.; Iliopoulos, D.; Hommes, D.W.; Verspaget, H.W.; Chang, L.; et al. Diminished expression of CRHR2 in human colon cancer promotes tumor growth and EMT via persistent IL-6/Stat3 signaling. CMGH Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 610–630. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Lezama, R.; Meza, I. A novel beta-catenin signaling pathway activated by IL-1beta leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett. 2014, 354, 164–171. [Google Scholar] [CrossRef]
- Jimenez-Garduno, A.M.; Mendoza-Rodriguez, M.G.; Urrutia-Cabrera, D.; Dominguez-Robles, M.C.; Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induced methylation of the estrogen receptor ERalpha gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef] [PubMed]
- Frede, S.; Freitag, P.; Otto, T.; Heilmaier, C.; Fandrey, J. The proinflammatory cytokine interleukin 1beta and hypoxia cooperatively induce the expression of adrenomedullin in ovarian carcinoma cells through hypoxia inducible factor 1 activation. Cancer Res. 2005, 65, 4690–4697. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Talia, M.; Cirillo, F.; Rigiracciolo, D.C.; Scordamaglia, D.; Guzzi, R.; Miglietta, A.M.; Francesco, E.M.D.; Belfiore, A.; Sims, A.H.; et al. The IL1beta-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2020, 39, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chu, D.; Kawamura, T.; Tanaka, K.; He, S. GRIM-19 repressed hypoxia-induced invasion and EMT of colorectal cancer by repressing autophagy through inactivation of STAT3/HIF-1alpha signaling axis. J. Cell. Physiol. 2019, 234, 12800–12808. [Google Scholar] [CrossRef]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1beta activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef]
- Xia, Y.; Yuan, M.; Li, S.; Thuan, U.T.; Nguyen, T.T.; Kang, T.W.; Liao, W.; Lian, S.; Jung, Y.D. Apigenin Suppresses the IL-1beta-Induced Expression of the Urokinase-Type Plasminogen Activator Receptor by Inhibiting MAPK-Mediated AP-1 and NF-kappaB Signaling in Human Bladder Cancer T24 Cells. J. Agric. Food Chem. 2018, 66, 7663–7673. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef]
- Petrella, B.L.; Vincenti, M.P. Interleukin-1beta mediates metalloproteinase-dependent renal cell carcinoma tumor cell invasion through the activation of CCAAT enhancer binding protein beta. Cancer Med. 2012, 1, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Przybylo, J.A.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition: Tumor progression at Snail’s pace. Int. J. Biochem. Cell Biol. 2007, 39, 1082–1088. [Google Scholar] [CrossRef]
- Sagara, A.; Miura, S.; Kobinata, A.; Naganawa, R.; Yaginuma, S.; Saito, S.; Saito, R.; Kominato, H.; Yumoto, T.; Sato, F. COL8A1 enhances the invasion/metastasis in MDA-MB-231 cells via the induction of IL1B and MMP1 expression. Biochem. Biophys. Res. Commun. 2023, 642, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hwang, Y.S. Cancer-associated fibroblast stimulates cancer cell invasion in an interleukin-1 receptor (IL-1R)-dependent manner. Oncol. Lett. 2019, 18, 4645–4650. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Padidar, S.; Farquharson, A.J.; Williams, L.M.; Hoggard, N.; Reid, M.D.; Duncan, G.J.; Drew, J.E. Impact of obesity and leptin on protein expression profiles in mouse colon. Dig. Dis. Sci. 2011, 56, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Loffreda, S.; Yang, S.Q.; Lin, H.Z.; Karp, C.L.; Brengman, M.L.; Wang, D.J.; Klein, A.S.; Bulkley, G.B.; Bao, C.; Noble, P.W.; et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12, 57–65. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Gonzalez-Perez, R.R. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 2011, 104, 128–137. [Google Scholar] [CrossRef]
- Perrier, S.; Caldefie-Chezet, F.; Vasson, M.P. IL-1 family in breast cancer: Potential interplay with leptin and other adipocytokines. FEBS Lett. 2009, 583, 259–265. [Google Scholar] [CrossRef]
- O’Brien, S.N.; Welter, B.H.; Price, T.M. Presence of leptin in breast cell lines and breast tumors. Biochem. Biophys. Res. Commun. 1999, 259, 695–698. [Google Scholar] [CrossRef]
- Faggioni, R.; Fantuzzi, G.; Fuller, J.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C. IL-1 beta mediates leptin induction during inflammation. Am. J. Physiol.-Regul., Integr. Comp. Physiol. 1998, 274, R204–R208. [Google Scholar] [CrossRef]
- Grunfeld, C.; Zhao, C.; Fuller, J.; Pollack, A.; Moser, A.; Friedman, J.; Feingold, K.R. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J. Clin. Investig. 1996, 97, 2152–2157. [Google Scholar] [CrossRef]
- Sarraf, P.; Frederich, R.C.; Turner, E.M.; Ma, G.; Jaskowiak, N.T.; Rivet, D.J., 3rd; Flier, J.S.; Lowell, B.B.; Fraker, D.L.; Alexander, H.R. Multiple cytokines and acute inflammation raise mouse leptin levels: Potential role in inflammatory anorexia. J. Exp. Med. 1997, 185, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, G.; Saarinen, N.; Abrahamsson, A.; Dabrosin, C. Tamoxifen, flaxseed, and the lignan enterolactone increase stroma- and cancer cell-derived IL-1Ra and decrease tumor angiogenesis in estrogen-dependent breast cancer. Cancer Res. 2011, 71, 51–60. [Google Scholar] [CrossRef]
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Caceres-Gutierrez, R.; Caro-Sanchez, C.H.S.; Herrera, L.A.; Diaz-Chavez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220. [Google Scholar] [CrossRef]
- Cao, Z.; Bao, M.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: A systemic review and meta-analysis. Eur. J. Cancer. 2013, 49, 3914–3923. [Google Scholar] [CrossRef] [PubMed]
- Wagenblast, E.; Soto, M.; Gutierrez-Angel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef]
- Shen, Y.; Quan, J.; Wang, M.; Li, S.; Yang, J.; Lv, M.; Chen, Z.; Zhang, L.; Zhao, X.; Yang, J. Tumor vasculogenic mimicry formation as an unfavorable prognostic indicator in patients with breast cancer. Oncotarget 2017, 8, 56408–56416. [Google Scholar] [CrossRef] [PubMed]
- Morales-Guadarrama, G.; Garcia-Becerra, R.; Mendez-Perez, E.A.; Garcia-Quiroz, J.; Avila, E.; Diaz, L. Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers. Cells 2021, 10, 1758. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Dominguez, L.; Olmedo-Nieva, L.; Munoz-Bello, J.O.; Contreras-Paredes, A.; Manzo-Merino, J.; Martinez-Ramirez, I.; Lizano, M. Mechanisms of Vasculogenic Mimicry in Ovarian Cancer. Front. Oncol. 2019, 9, 998. [Google Scholar] [CrossRef]
- Du, J.; Sun, B.; Zhao, X.; Gu, Q.; Dong, X.; Mo, J.; Sun, T.; Wang, J.; Sun, R.; Liu, Y. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol. Oncol. 2014, 133, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Do, Y.; Kwon, B.S.; Chang, W.; Lee, M.S.; Kim, J.; Cho, J.G. Angiogenesis and vasculogenic mimicry as therapeutic targets in ovarian cancer. BMB Rep. 2020, 53, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Zamudio-Martinez, E.; Fernandez-Cortes, M.; Herrera-Campos, A.B.; Olmedo-Pelayo, J.; Perez, C.J.; Exposito, J.; de Alava, E.; Amaral, A.T.; Valle, F.O.; et al. VE-Cadherin modulates beta-catenin/TCF-4 to enhance Vasculogenic Mimicry. Cell Death Dis. 2023, 14, 135. [Google Scholar] [CrossRef]
- Nisar, M.A.; Zheng, Q.; Saleem, M.Z.; Ahmmed, B.; Ramzan, M.N.; Din, S.R.U.; Tahir, N.; Liu, S.; Yan, Q. IL-1beta Promotes Vasculogenic Mimicry of Breast Cancer Cells Through p38/MAPK and PI3K/Akt Signaling Pathways. Front. Oncol. 2021, 11, 618839. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, D.; Wang, G.; Wang, D.; Zhou, H.; Liu, X.; Jiang, M.; Liao, L.; Zhou, Z.; Hu, J. Pro-Inflammatory Cytokine IL-1beta Up-Regulates CXC Chemokine Receptor 4 via Notch and ERK Signaling Pathways in Tongue Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0132677. [Google Scholar] [CrossRef]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef]
- Chen, X.; Lu, J.; Ji, Y.; Hong, A.; Xie, Q. Cytokines in osteoblast-conditioned medium promote the migration of breast cancer cells. Tumor Biol. 2014, 35, 791–798. [Google Scholar] [CrossRef]
- Zhao, C.; Cai, X.; Wang, Y.; Wang, D.; Wang, T.; Gong, H.; Sun, H.; Jia, Q.; Zhou, W.; Wu, Z.; et al. NAT1 promotes osteolytic metastasis in luminal breast cancer by regulating the bone metastatic niche via NF-kappaB/IL-1B signaling pathway. Am. J. Cancer Res. 2020, 10, 2464–2479. [Google Scholar] [PubMed]
- Templeton, Z.S.; Lie, W.R.; Wang, W.; Rosenberg-Hasson, Y.; Alluri, R.V.; Tamaresis, J.S.; Bachmann, M.H.; Lee, K.; Maloney, W.J.; Contag, C.H.; et al. Breast Cancer Cell Colonization of the Human Bone Marrow Adipose Tissue Niche. Neoplasia 2015, 17, 849–861. [Google Scholar] [CrossRef]
- Li, R.; Wen, A.; Lin, J. Pro-Inflammatory Cytokines in the Formation of the Pre-Metastatic Niche. Cancers 2020, 12, 3752. [Google Scholar] [CrossRef]
- Gouwy, M.; Struyf, S.; Proost, P.; Damme, J.V. Synergy in cytokine and chemokine networks amplifies the inflammatory response. Cytokine Growth Factor. Rev. 2005, 16, 561–580. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; der Kwast, T.V.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers. 2021, 7, 9. [Google Scholar] [CrossRef]
- Crawford, E.D. Epidemiology of prostate cancer. Urology 2003, 62, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Force, U.P.S.T.; Grossman, D.C.; Curry, S.J.; Owens, D.K.; Bibbins-Domingo, K.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Ebell, M.; Epling, J.W.; et al. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 1901–1913. [Google Scholar] [CrossRef]
- Mottet, N.; van den Bergh, R.C.N.; Briers, E.; den Broeck, T.V.; Cumberbatch, M.G.; Santis, M.D.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer—2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [CrossRef]
- Mistry, K.; Cable, G. Meta-Analysis of Prostate-Specific Antigen and Digital Rectal Examination as Screening Tests for Prostate Carcinoma. J. Am. Board. Fam. Pr. 2003, 16, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.I.; Parker, C.; Hope, T.A.; Paller, C.J. Best Approaches and Updates for Prostate Cancer Biochemical Recurrence. Am. Soc. Clin. Oncol. Educ. Book. 2022, 42, 352–359. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Karnes, R.J.; Crispen, P.L.; Rangel, L.J.; Bergstralh, E.J.; Blute, M.L. Radiation Therapy After Radical Prostatectomy: Impact on Metastasis and Survival. J. Urol. 2009, 182, 2708–2715. [Google Scholar] [CrossRef]
- Parker, C.C.; Clarke, N.W.; Cook, A.D.; Kynaston, H.G.; Petersen, P.M.; Catton, C.; Cross, W.; Logue, J.; Parulekar, W.; Payne, H.; et al. Timing of radiotherapy after radical prostatectomy (RADICALS-RT): A randomised, controlled phase 3 trial. Lancet 2020, 396, 1413–1421. [Google Scholar] [CrossRef]
- Kneebone, A.; Fraser-Browne, C.; Duchesne, G.M.; Fisher, R.; Frydenberg, M.; Herschtal, A.; Williams, S.G.; Brown, C.; Delprado, W.; Haworth, A.; et al. Adjuvant radiotherapy versus early salvage radiotherapy following radical prostatectomy (TROG 08.03/ANZUP RAVES): A randomised, controlled, phase 3, non-inferiority trial. Lancet Oncol. 2020, 21, 1331–1340. [Google Scholar] [CrossRef]
- Hoffman, A.; Amiel, G.E. The Impact of PSMA PET/CT on Modern Prostate Cancer Management and Decision Making—The Urological Perspective. Cancers 2023, 15, 3402. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Stevens, R.E.; Hodges, C.V. Studies on Prostatic Cancer: II. The Effects of Castration on Advanced Carcinoma of the Prostate Gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Lowrance, W.; Dreicer, R.; Jarrard, D.F.; Scarpato, K.R.; Kim, S.K.; Kirkby, E.; Buckley, D.I.; Griffin, J.C.; Cookson, M.S. Updates to Advanced Prostate Cancer: AUA/SUO Guideline (2023). J. Urol. 2023, 209, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.-M.; Aragon-Ching, J.B. Advances with androgen deprivation therapy for prostate cancer. Expert Opin. Pharmaco. 2022, 23, 1015–1033. [Google Scholar] [CrossRef]
- Choi, E.; Buie, J.; Camacho, J.; Sharma, P.; Riese, W.T.W. de: Evolution of Androgen Deprivation Therapy (ADT) and Its New Emerging Modalities in Prostate Cancer: An Update for Practicing Urologists, Clinicians and Medical Providers. Res. Rep. Urol. 2022, 14, 87–108. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Koochekpour, S. Therapeutic Rationales, Progresses, Failures, and Future Directions for Advanced Prostate Cancer. Int. J. Biol. Sci. 2016, 12, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R. Therapeutic targeting of the androgen receptor (AR) and AR variants in prostate cancer. Asian J. Urol. 2020, 7, 271–283. [Google Scholar] [CrossRef]
- Han, D.; Labaf, M.; Zhao, Y.; Owiredu, J.; Zhang, S.; Patel, K.; Venkataramani, K.; Steinfeld, J.S.; Han, W.; Li, M.; et al. Androgen receptor splice variants drive castration-resistant prostate cancer metastasis by activating distinct transcriptional programs. J. Clin. Investig. 2024, 134, e168649. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2018, 129, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Alumkal, J.J.; Coleman, I.M.; Nelson, P.S.; Morrissey, C. The heterogeneity of prostate cancers lacking AR activity will require diverse treatment approaches. Endocr.-Relat. Cancer. 2021, 28, T51–T66. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.; Zhang, M.; Aggarwal, R.; Li, H.; Zhang, L.; Foye, A.; Sjöström, M.; Chou, J.; Chang, K.; Moreno-Rodriguez, T.; et al. The genomic and epigenomic landscape of double-negative metastatic prostate cancer. Cancer Res. 2023, 83, 2763–2774. [Google Scholar] [CrossRef] [PubMed]
- George, D.J.; Sartor, O.; Miller, K.; Saad, F.; Tombal, B.; Kalinovský, J.; Jiao, X.; Tangirala, K.; Sternberg, C.N.; Higano, C.S. Treatment Patterns and Outcomes in Patients With Metastatic Castration-resistant Prostate Cancer in a Real-world Clinical Practice Setting in the United States. Clin. Genitourin. Cancer 2020, 18, 284–294. [Google Scholar] [CrossRef]
- Sayegh, N.; Tripathi, N.; Nussenzveig, R.H.; Thomas, V.M.; Tandar, C.; Goel, D.; Nordblad, B.; Sahu, K.K.; Li, H.; Maughan, B.L.; et al. Survival of Patients with Metastatic Prostate Cancer After Disease Progression on an Androgen Receptor Axis–Targeted Therapy Given in the Metastatic Castration-Sensitive Versus Metastatic Castration-Resistant Prostate Cancer Setting. Eur. Urol. Focus 2023, 9, 106–109. [Google Scholar] [CrossRef]
- Selvaggi, G.; Scagliotti, G.V. Management of bone metastases in cancer: A review. Crit. Rev. Oncol. Hematol. 2005, 56, 365–378. [Google Scholar] [CrossRef]
- Goode, E.A.; Wang, N.; Munkley, J. Prostate cancer bone metastases biology and clinical management (Review). Oncol. Lett. 2023, 25, 163. [Google Scholar] [CrossRef]
- Bienz, M.; Saad, F. Management of bone metastases in prostate cancer: A review. Curr. Opin. Support. Palliat. Care. 2015, 9, 261–267. [Google Scholar] [CrossRef]
- Akhtari, M.; Mansuri, J.; Newman, K.A.; Guise, T.M.; Seth, P. Biology of breast cancer bone metastasis. Cancer Biol. Ther. 2008, 7, 3–9. [Google Scholar] [CrossRef]
- Sowder, M.E.; Johnson, R.W. Bone as a Preferential Site for Metastasis. JBMR Plus. 2019, 3, e10126. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, A.; Fatatis, A. The Bone Microenvironment in Prostate Cancer Metastasis. Adv. Exp. Med. Biol. 2019, 1210, 171–184. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, H.; Dunstan, C.R.; Sutherland, R.L.; Seibel, M.J. The role of the bone microenvironment in skeletal metastasis. J. Bone Oncol. 2013, 2, 47–57. [Google Scholar] [CrossRef]
- Lucas, D. Structural organization of the bone marrow and its role in hematopoiesis. Curr. Opin. Hematol. 2020, 28, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, A.; Rucci, N. The Osteoclast in Bone Metastasis: Player and Target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef]
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.-D.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014, 74, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Karakiewicz, P.I.; Briganti, A.; Passoni, N.M.; Schiffmann, J.; Trudeau, V.; Graefen, M.; Montorsi, F.; Sun, M. Impact of the Site of Metastases on Survival in Patients with Metastatic Prostate Cancer. Eur. Urol. 2015, 68, 325–334. [Google Scholar] [CrossRef]
- Pezaro, C.; Omlin, A.; Lorente, D.; Rodrigues, D.N.; Ferraldeschi, R.; Bianchini, D.; Mukherji, D.; Riisnaes, R.; Altavilla, A.; Crespo, M.; et al. Visceral disease in castration-resistant prostate cancer. Eur. Urol. 2014, 65, 270–273. [Google Scholar] [CrossRef]
- Coleman, R.; Rubens, R. The clinical course of bone metastases from breast cancer. Br. J. Cancer 1987, 55, 61–66. [Google Scholar] [CrossRef]
- Zhang, W.; Bado, I.L.; Hu, J.; Wan, Y.-W.; Wu, L.; Wang, H.; Gao, Y.; Jeong, H.-H.; Xu, Z.; Hao, X.; et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 2021, 184, 2471–2486.e20. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.; Yue, X.; Li, X. Bone serves as a transfer station for secondary dissemination of breast cancer. Bone Res. 2023, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Aufdemorte, T.B.; Garrett, I.R.; Yates, A.J.; Mundy, G.R. Effects of interleukin-1 on bone turnover in normal mice. Endocrinology 1989, 125, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Gowen, M.; Wood, D.D.; Ihrie, E.J.; McGuire, M.K.; Russell, R.G. An interleukin 1 like factor stimulates bone resorption in vitro. Nature 1983, 306, 378–380. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Stashenko, P.P.; Mole, J.E.; Tsurumachi, T. Purification and partial sequence of human osteoclast-activating factor: Identity with interleukin 1 beta. J. Immunol. 1985, 135, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Seckinger, P.; Klein-Nulend, J.; Alander, C.; Thompson, R.C.; Dayer, J.M.; Raisz, L.G. Natural and recombinant human IL-1 receptor antagonists block the effects of IL-1 on bone resorption and prostaglandin production. J. Immunol. 1990, 145, 4181–4184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The mechanism of osteoclast differentiation induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Jules, J.; Zhang, P.; Ashley, J.W.; Wei, S.; Shi, Z.; Liu, J.; Michalek, S.M.; Feng, X. Molecular basis of requirement of receptor activator of nuclear factor kappaB signaling for interleukin 1-mediated osteoclastogenesis. J. Biol. Chem. 2012, 287, 15728–15738. [Google Scholar] [CrossRef]
- Taube, T.; Elomaa, I.; Blomqvist, C.; Beneton, M.N.; Kanis, J.A. Histomorphometric evidence for osteoclast-mediated bone resorption in metastatic breast cancer. Bone 1994, 15, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Nakamura, I.; Duong, L.T.; Ikebe, T.; Takahashi, N.; Rodan, G.A.; Suda, T. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp. Cell Res. 1999, 247, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.; Weber, K.; Baranowsky, A.; Streichert, T.; Lange, T.; Spiro, A.S.; Albers, J.; Seitz, S.; Zustin, J.; Amling, M.; et al. p65-Dependent production of interleukin-1beta by osteolytic prostate cancer cells causes an induction of chemokine expression in osteoblasts. Cancer Lett. 2012, 317, 106–113. [Google Scholar] [CrossRef]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of NF-kappa B Signaling Promotes Growth of Prostate Cancer Cells in Bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.A.; Morgado, M.; Warren, C.R.; Hinton, C.V.; Farach-Carson, M.C.; Delk, N.A. p62/SQSTM1 is required for cell survival of apoptosis-resistant bone metastatic prostate cancer cell lines. Prostate 2014, 74, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.A.; Patel, V.; Gwede, M.; Morgado, M.; Tomasevich, K.; Fong, E.L.; Farach-Carson, M.C.; Delk, N.A. IL-1β induces p62/SQSTM1 and represses androgen receptor expression in prostate cancer cells. J. Cell. Biochem. 2014, 115, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Jardin, S.E.; Kanchwala, M.S.; Jacob, J.; Merchant, S.; Meade, R.K.; Gahnim, N.M.; Nawas, A.F.; Xing, C.; Delk, N.A. Identification of an IL-1-induced gene expression pattern in AR+ PCa cells that mimics the molecular phenotype of AR- PCa cells. Prostate 2018, 78, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr. -Relat. Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Bradbury, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016, 7, 75571–75584. [Google Scholar] [CrossRef] [PubMed]
- Eyre, R.; Alférez, D.G.; Santiago-Gómez, A.; Spence, K.; McConnell, J.C.; Hart, C.; Simões, B.M.; Lefley, D.; Tulotta, C.; Storer, J.; et al. Microenvironmental IL1Î2 promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat. Commun. 2019, 10, 5016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bolton, E.C.; Jones, J.O. Androgens and androgen receptor signaling in prostate tumorigenesis. J. Mol. Endocrinol. 2015, 54, R15–R29. [Google Scholar] [CrossRef]
- Whitfield, G.K.; Jurutka, P.W.; Haussler, C.A.; Haussler, M.R. Steroid hormone receptors: Evolution, ligands, and molecular basis of biologic function. J. Cell. Biochem. 1999, 75 (Suppl. 32–33), 110–122. [Google Scholar] [CrossRef]
- Eder, I.E.; Culig, Z.; Putz, T.; Nessler-Menardi, C.; Bartsch, G.; Klocker, H. Molecular biology of the androgen receptor: From molecular understanding to the clinic. Eur. Urol. 2001, 40, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017, 32, 474–489.e6. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, A.; Worrede, A.; Iqbal, W.; Marchioli, M.; Toth, A.; Sjostrom, M.; Zhu, X.; Corey, E.; Feng, F.Y.; Zhou, W.; et al. IL-1beta expression driven by androgen receptor absence or inactivation promotes prostate cancer bone metastasis. Cancer Res. Commun. 2022, 2, 1545–1557. [Google Scholar] [CrossRef]
- ERB, H.H.H.; OSTER, M.A.; GELBRICH, N.; CAMMANN, C.; THOMAS, C.; MUSTEA, A.; STOPE, M.B. Enzalutamide-induced Proteolytic Degradation of the Androgen Receptor in Prostate Cancer Cells Is Mediated Only to a Limited Extent by the Proteasome System. Anticancer. Res. 2021, 41, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Abazid, A.; Martin, B.; Choinowski, A.; McNeill, R.V.; Brandenburg, L.; Ziegler, P.; Zimmermann, U.; Burchardt, M.; Erb, H.; Stope, M.B. The androgen receptor antagonist enzalutamide induces apoptosis, dysregulates the heat shock protein system, and diminishes the androgen receptor and estrogen receptor β1 expression in prostate cancer cells. J. Cell. Biochem. 2019, 120, 16711–16722. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Tekpli, X.; Landvik, N.E.; Anmarkud, K.H.; Skaug, V.; Haugen, A.; Zienolddiny, S. DNA methylation at promoter regions of interleukin 1B, interleukin 6, and interleukin 8 in non-small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 337–345. [Google Scholar] [CrossRef]
- Fuso, A.; Iyer, A.M.; van Scheppingen, J.; Maccarrone, M.; Scholl, T.; Hainfellner, J.A.; Feucht, M.; Jansen, F.E.; Spliet, W.G.; Krsek, P.; et al. Promoter-Specific Hypomethylation Correlates with IL-1β Overexpression in Tuberous Sclerosis Complex (TSC). J. Mol. Neurosci. 2016, 59, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Oreffo, R.O.C.; Gibson, M.B.; Goldring, M.B.; Roach, H.I. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009, 60, 3303–3313. [Google Scholar] [CrossRef]
- Arnold, D.D.; Yalamanoglu, A.; Boyman, O. Systematic Review of Safety and Efficacy of IL-1-Targeted Biologics in Treating Immune-Mediated Disorders. Front. Immunol. 2022, 13, 888392. [Google Scholar] [CrossRef] [PubMed]
- MERTENS, M.; SINGH, J.A. Anakinra for Rheumatoid Arthritis: A Systematic Review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef]
- Church, L.D.; McDermott, M.F. Canakinumab, a fully-human mAb against IL-1beta for the potential treatment of inflammatory disorders. Curr. Opin. Mol. Ther. 2009, 11, 81–89. [Google Scholar]
- Hoffman, H.M. Rilonacept for the treatment of cryopyrin-associated periodic syndromes (CAPS). Expert Opin. Biol. Ther. 2009, 9, 519–531. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J.; et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1β by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Res. 2020, 80, 5597–5605. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Lee, R.J.; Stott, S.L.; Ting, D.T.; Wittner, B.S.; Ulman, M.; Smas, M.E.; Lord, J.B.; Brannigan, B.W.; Trautwein, J.; et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discovery 2012, 2, 995–1003. [Google Scholar] [CrossRef]
- Lianidou, E. Detection and relevance of epigenetic markers on ctDNA: Recent advances and future outlook. Mol. Oncol. 2021, 15, 1683–1700. [Google Scholar] [CrossRef]
- Miller, B.F.; Petrykowska, H.M.; Elnitski, L. Assessing ZNF154 methylation in patient plasma as a multicancer marker in liquid biopsies from colon, liver, ovarian and pancreatic cancer patients. Sci. Rep. 2021, 11, 221. [Google Scholar] [CrossRef] [PubMed]
- Nassar, F.J.; Msheik, Z.S.; Nasr, R.R.; Temraz, S.N. Methylated circulating tumor DNA as a biomarker for colorectal cancer diagnosis, prognosis, and prediction. Clin. Epigenetics 2021, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Ntzifa, A.; Londra, D.; Rampias, T.; Kotsakis, A.; Georgoulias, V.; Lianidou, E. DNA Methylation Analysis in Plasma Cell-Free DNA and Paired CTCs of NSCLC Patients before and after Osimertinib Treatment. Cancers 2021, 13, 5974. [Google Scholar] [CrossRef]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and hot tumors: From molecular mechanisms to targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 274. [Google Scholar] [CrossRef]
- Wilson, B.E.; Shen, Q.; Cescon, D.W.; Reedijk, M. Exploring immune interactions in triple negative breast cancer: IL-1β inhibition and its therapeutic potential. Front. Genet. 2023, 14, 1086163. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyende, Y.; Taus, L.J.; Fatatis, A. IL-1β in Neoplastic Disease and the Role of Its Tumor-Derived Form in the Progression and Treatment of Metastatic Prostate Cancer. Cancers 2025, 17, 290. https://doi.org/10.3390/cancers17020290
Oyende Y, Taus LJ, Fatatis A. IL-1β in Neoplastic Disease and the Role of Its Tumor-Derived Form in the Progression and Treatment of Metastatic Prostate Cancer. Cancers. 2025; 17(2):290. https://doi.org/10.3390/cancers17020290
Chicago/Turabian StyleOyende, Yetunde, Luke J. Taus, and Alessandro Fatatis. 2025. "IL-1β in Neoplastic Disease and the Role of Its Tumor-Derived Form in the Progression and Treatment of Metastatic Prostate Cancer" Cancers 17, no. 2: 290. https://doi.org/10.3390/cancers17020290
APA StyleOyende, Y., Taus, L. J., & Fatatis, A. (2025). IL-1β in Neoplastic Disease and the Role of Its Tumor-Derived Form in the Progression and Treatment of Metastatic Prostate Cancer. Cancers, 17(2), 290. https://doi.org/10.3390/cancers17020290