
Calreticulin—From the Endoplasmic Reticulum to the Plasma Membrane—Adventures of a Wandering Protein




Simple Summary
Abstract

1. Introduction
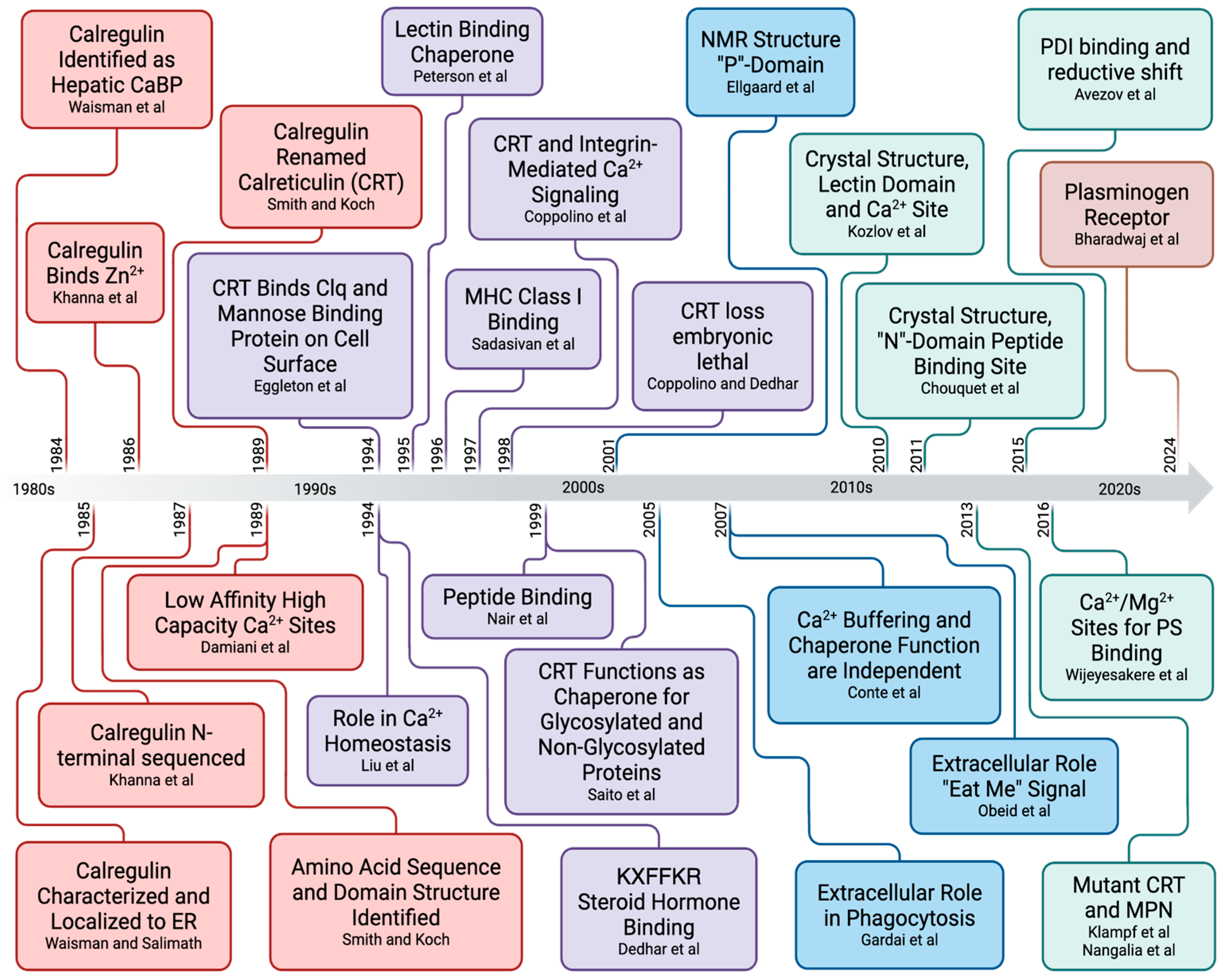
2. Calreticulin-Enigmatic Discovery
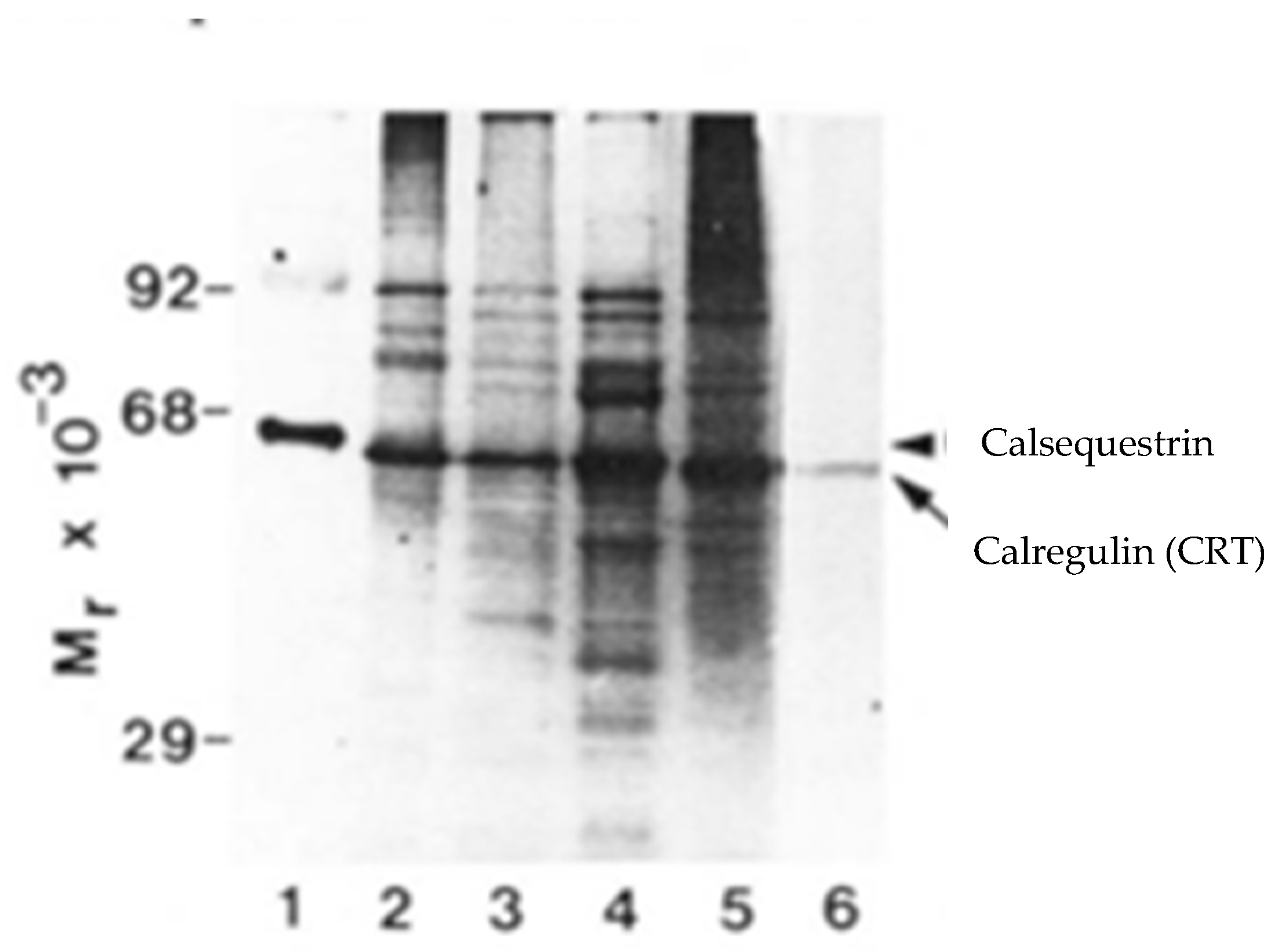
2.1. Identification of the HACBP in Skeletal Muscle SR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Study Year | CSQ | HACBP | 53 kDa Glycoprotein | Method of Detection | Reference |
|---|---|---|---|---|---|---|
| Ostwald and MacLennan | 1974 | + | + | ND | Coomassie Blue Stain 1 | [13] |
| Michalak et al. | 1980 | + | + | + | Coomassie Blue Stain 2,3 | [3] |
| Campbell et al. | 1980 | + | + 4 | + | Coomassie Blue Stain 3 | [12] |
| Campbell and MacLennan | 1981 | + | ND | + | Coomassie Blue Stain 3 | [4] |
| Campbell and MacLennan | 1982 | + | ND | + | Coomassie Blue Stain 3 | [24] |
| Campbell et al. | 1983 | + | ND | + | Coomassie Blue Stain 2, Stains-All | [25] |
| Campbell et al. | 1983 | + | ND | + | Coomassie Blue Stain 2, Stains-All | [17] |
| Macer and Koch | 1988 | + | ND | + | Coomassie Blue Stain 2, 45Ca2+ Autoradiography | [26] |
| Damiani et al. | 1988 | + | ND | 45Ca2+ Autoradiography | [27] | |
| Leberer et al. | 1989 | + | ND | + | Coomassie Blue Stain 2, 45Ca2+ Autoradiography | [28] |
| Hofmann et al. | 1989 | + | ND | + | 45Ca2+ Autoradiography, Stains-All | [29] |
| Van et al. | 1989 | + | ND | ND | Stains-All | [16] |
| Leberer et al. | 1990 | + | ND | + | Coomassie Blue Stain 2 | [30] |
| Cala et al. | 1990 | + | ND | ND | Stains-All | [31] |
| Damiani and Margret | 1991 | + | ND | + | Stains-All, 45Ca2+ Autoradiography, Ponceau Red 3 | [32] |
| Raeymaekers et al. | 1993 | ND | ND | ND | Immunodetection with CRT Antibody | [23] |
| Treves | 2009 | + | ND | + | Coomassie Blue | [33] |
| Staunton | 2012 | + | ND | + | Mass Spectrometry | [34] |
2.2. Does the HACBP Actually Exist?
2.3. Are the HACBP and CRT the Same Protein?
2.4. Discovery of CRT as Calregulin
3. Calreticulin–Gene Structure
4. CRT Domain Structure
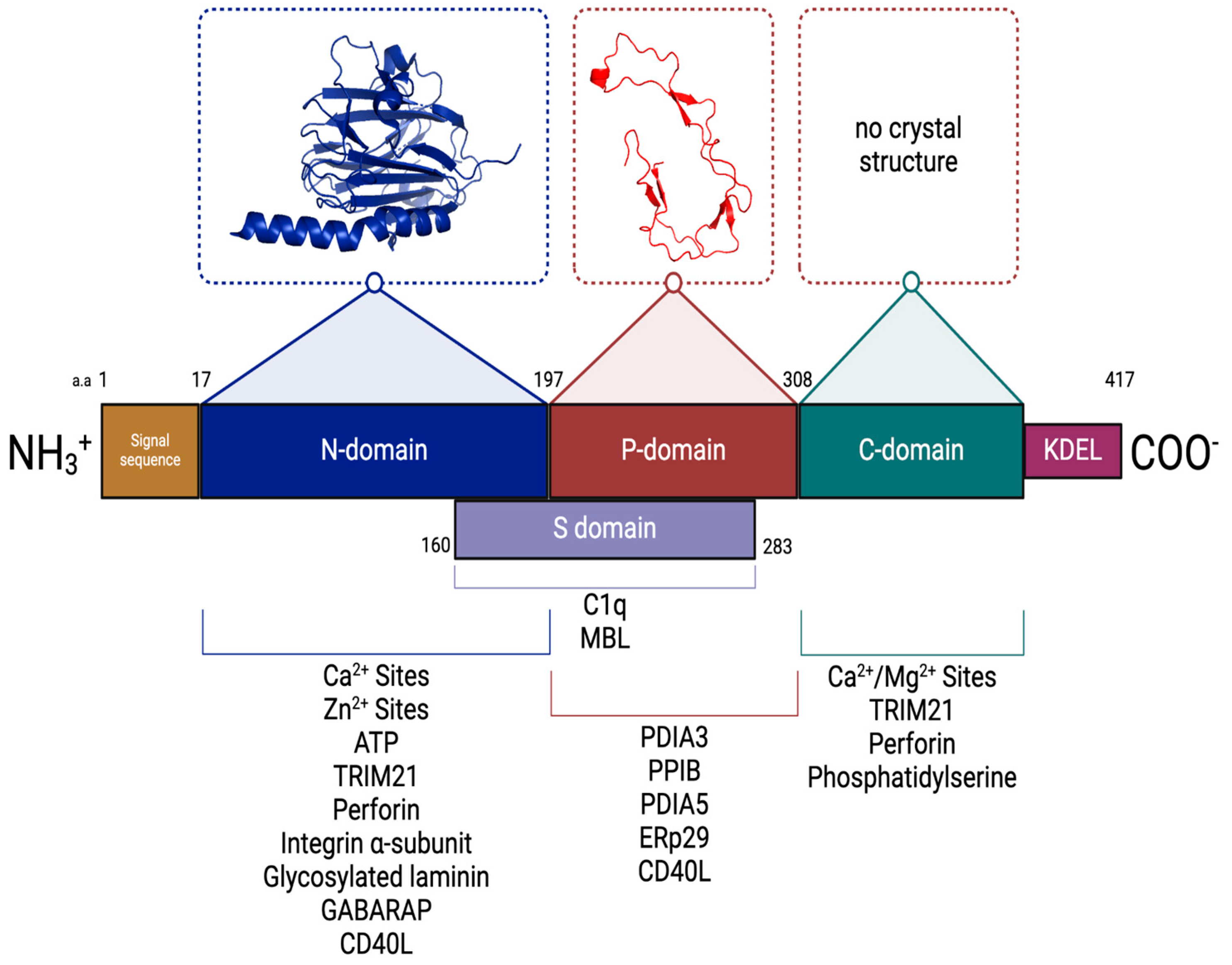
| Protein | Binding Affinity (Kd) | CRT Domain | Reference(s) |
|---|---|---|---|
| TRIM21 (tripartite motif-containing protein 21) | NR | N- and C-terminal domains | [90] |
| PDIA3 (protein disulfide–isomerase A3, ERp57) | 5–9 μM | P-domain | [80,91,92] |
| Perforin | 1.2 μM | N- and C-domains | [93,94,95] |
| Integrin α-subunit | 0.25 μM | N-domain | [96,97] |
| PPIB (peptidyl–prolyl cis–trans isomerase B) | 10 μM | P-domain | [75] |
| PDIA5 (protein disulfide–isomerase A5) | 0.16 μM | P-domain | [98] |
| Glycosylated laminin | 0.5 μM | N-domain | [99] |
| GABARAP (Gamma-aminobutyric acid receptor-associated protein) | 64 nM | N-domain | [100,101] |
| SPACA9 (sperm acrosome-associated protein 9) | NR | NR | [102] |
| CLCC1 (chloride channel CLIC-like 1) | NR | NR | [103] |
| C1q | 100 nM | N- and P-domains | [88,104,105,106] |
| MBL (mannose-binding lectin) | NR | N- and P-domains | [107] |
| ERp29 | 13 μM | P-domain | [74] |
| CD40L | 50 nM | N- and P-domains | [108] |
| LDL receptor-related protein 1 (CD-91) | NR | [109] | |
| Thrombospondin (TSP-1) | NR | N-domain | [110,111] |
| Phosphatidylserine | 12 μM | C-domain | [87] |
5. CRT–Metal Ion Binding Properties
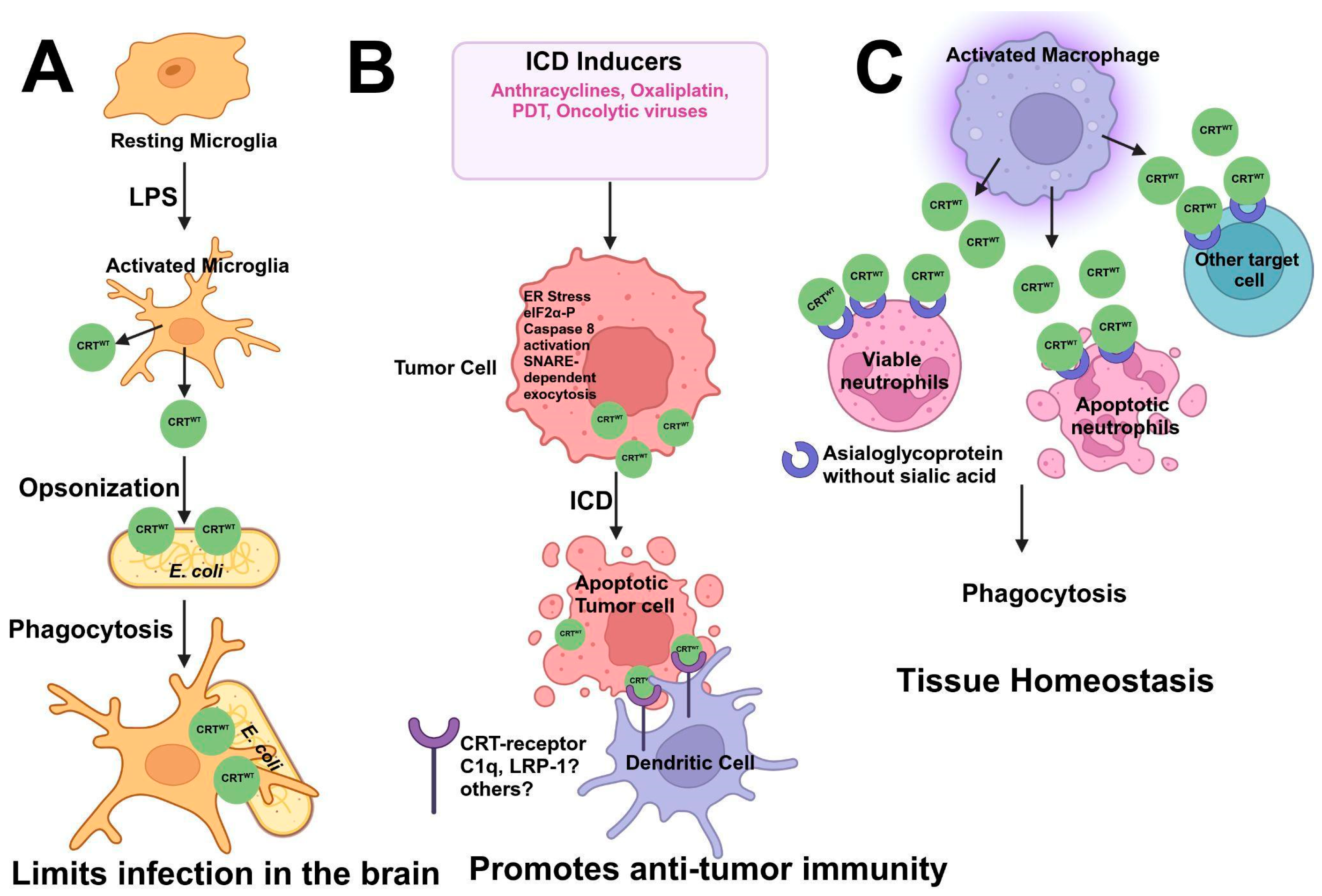
6. Calreticulin Functions: From the Endoplasmic Reticulum to the Extracellular Surface
| Function | References |
|---|---|
| Protein Folding | [58,63,64,115,133] |
| Calcium Homeostasis | [124,140,141,142] |
| Cell Adhesion | [99,111,143,144,145] |
| Wound Healing | [146,147] |
| Phagocytosis | [107,138,148,149] |
| Immunogenic Cell Death | [148,150,151,152] |
| RNA Stability | [153,154] |
| Plasminogen Receptor | [155] |
6.1. CRT and Ca2+ Homeostasis
6.2. CRT and Integrin Binding
6.3. CRT Chaperone Function
6.4. CRT in MHC Class I Assembly and Antigen Presentation
6.5. CRT in Immunogenic Cell Death
7. Calreticulin Mutants and Cancer: Impacts on Biological Functions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fliegel, L.; Burns, K.; MacLennan, D.H.; Reithmeier, R.A.; Michalak, M. Molecular Cloning of the High Affinity Calcium-Binding Protein (Calreticulin) of Skeletal Muscle Sarcoplasmic Reticulum. J. Biol. Chem. 1989, 264, 21522–21528. [Google Scholar] [CrossRef] [PubMed]
- Okura, G.C.; Bharadwaj, A.G.; Waisman, D.M. Calreticulin-Enigmatic Discovery. Biomolecules 2024, 14, 866. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Campbell, K.P.; MacLennan, D.H. Localization of the High Affinity Calcium Binding Protein and an Intrinsic Glycoprotein in Sarcoplasmic Reticulum Membranes. J. Biol. Chem. 1980, 255, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; MacLennan, D.H. Purification and Characterization of the 53,000-Dalton Glycoprotein from the Sarcoplasmic Reticulum. J. Biol. Chem. 1981, 256, 4626–4632. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P. Protein Components and Their Roles in Sarcoplasmic Reticulum Function. In Sarcoplasmic Reticulum in Muscle Physiology; Sarcoplasmic Reticulum in Muscle Physiology; CRC-Press: Boca Raton, FL, USA, 1986; Volume 1, pp. 65–99. ISBN 978-0-8493-6181-4. [Google Scholar]
- MacLennan, D.H.; Ostwald, T.J.; Stewart, P.S. Structural Components of the Sarcoplasmic Reticulum Membrane. Ann. N. Y. Acad. Sci. 1974, 227, 527–536. [Google Scholar] [CrossRef]
- Ostwald, T.J.; MacLennan, D.H. Isolation of a High Affinity Calcium-Binding Protein from Sarcoplasmic Reticulum. J. Biol. Chem. 1974, 249, 974–979. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H. Purification and Properties of an Adenosine Triphosphatase from Sarcoplasmic Reticulum. J. Biol. Chem. 1970, 245, 4508–4518. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Osborn, M. The Reliability of Molecular Weight Determinations by Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis. J. Biol. Chem. 1969, 244, 4406–4412. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.; Klip, A.; Reithmeier, R.; Michalak, M.; Campbell, K. Possible Sites of Ion Flow in the Sarcoplasmic Reticulum Membrane. In Membrane Bioenergetics; Addison-Wesley Publishing Company: Boston, MA, USA, 1979. [Google Scholar]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; Franzini-Armstrong, C.; Shamoo, A.E. Further Characterization of Light and Heavy Sarcoplasmic Reticulum Vesicles. Identification of the ‘Sarcoplasmic Reticulum Feet’ Associated with Heavy Sarcoplasmic Reticulum Vesicles. Biochim. Biophys. Acta BBA—Biomembr. 1980, 602, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Ostwald, T.J.; MacLennan, D.H.; Dorrington, K.J. Effects of Cation Binding on the Conformation of Calsequestrin and the High Affinity Calcium-Binding Protein of Sarcoplasmic Reticulum. J. Biol. Chem. 1974, 249, 5867–5871. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Yip, C.C.; Iles, G.H.; Seeman, P. Isolation of Sarcoplasmic Reticulum Proteins. Cold Spring Harb. Symp. Quant. Biol. 1973, 37, 469–477. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Wong, P.T. Isolation of a Calcium-Sequestering Protein from Sarcoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 1971, 68, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Van, P.N.; Peter, F.; Söling, H.D. Four Intracisternal Calcium-Binding Glycoproteins from Rat Liver Microsomes with High Affinity for Calcium. No Indication for Calsequestrin-like Proteins in Inositol 1,4,5-Trisphosphate-Sensitive Calcium Sequestering Rat Liver Vesicles. J. Biol. Chem. 1989, 264, 17494–17501. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; MacLennan, D.H.; Jorgensen, A.O. Staining of the Ca2+-Binding Proteins, Calsequestrin, Calmodulin, Troponin C, and S-100, with the Cationic Carbocyanine Dye “Stains-All”. J. Biol. Chem. 1983, 258, 11267–11273. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.C.; Tokuda, M.; Waisman, D.M. Calregulin: Purification, Cellular Localization, and Tissue Distribution. Methods Enzym. 1987, 139, 36–50. [Google Scholar]
- Baksh, S.; Burns, K.; Busaan, J.; Michalak, M. Expression and Purification of Recombinant and Native Calreticulin. Protein Expr. Purif. 1992, 3, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Treves, S.; De Mattei, M.; Landfredi, M.; Villa, A.; Green, N.M.; MacLennan, D.H.; Meldolesi, J.; Pozzan, T. Calreticulin Is a Candidate for a Calsequestrin-like Function in Ca2(+)-Storage Compartments (Calciosomes) of Liver and Brain. Biochem. J. 1990, 271, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.C.; Tokuda, M.; Waisman, D.M. Conformational Changes Induced by Binding of Divalent Cations to Calregulin. J. Biol. Chem. 1986, 261, 8883–8887. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, K.; Maeshima, M. Equilibrium Dialysis Measurements of the Ca2+-Binding Properties of Recombinant Radish Vacuolar Ca2+-Binding Protein Expressed in Escherichia Coli. Biosci. Biotechnol. Biochem. 2002, 66, 2382–2387. [Google Scholar] [CrossRef]
- Raeymaekers, L.; Verbist, J.; Wuytack, F.; Plessers, L.; Casteels, R. Expression of Ca2+ Binding Proteins of the Sarcoplasmic Reticulum of Striated Muscle in the Endoplasmic Reticulum of Pig Smooth Muscles. Cell Calcium 1993, 14, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; MacLennan, D.H. A Calmodulin-Dependent Protein Kinase System from Skeletal Muscle Sarcoplasmic Reticulum. Phosphorylation of a 60,000-Dalton Protein. J. Biol. Chem. 1982, 257, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; MacLennan, D.H.; Jorgensen, A.O.; Mintzer, M.C. Purification and Characterization of Calsequestrin from Canine Cardiac Sarcoplasmic Reticulum and Identification of the 53,000 Dalton Glycoprotein. J. Biol. Chem. 1983, 258, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Macer, D.R.; Koch, G.L. Identification of a Set of Calcium-Binding Proteins in Reticuloplasm, the Luminal Content of the Endoplasmic Reticulum. J. Cell Sci. 1988, 91, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Damiani, E.; Spamer, C.; Heilmann, C.; Salvatori, S.; Margreth, A. Endoplasmic Reticulum of Rat Liver Contains Two Proteins Closely Related to Skeletal Sarcoplasmic Reticulum Ca-ATPase and Calsequestrin. J. Biol. Chem. 1988, 263, 340–343. [Google Scholar] [CrossRef]
- Leberer, E.; Charuk, J.H.; Green, N.M.; MacLennan, D.H. Molecular Cloning and Expression of cDNA Encoding a Lumenal Calcium Binding Glycoprotein from Sarcoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 1989, 86, 6047–6051. [Google Scholar] [CrossRef]
- Hofmann, S.L.; Brown, M.S.; Lee, E.; Pathak, R.K.; Anderson, R.G.; Goldstein, J.L. Purification of a Sarcoplasmic Reticulum Protein That Binds Ca2+ and Plasma Lipoproteins. J. Biol. Chem. 1989, 264, 8260–8270. [Google Scholar] [CrossRef] [PubMed]
- Leberer, E.; Timms, B.G.; Campbell, K.P.; MacLennan, D.H. Purification, Calcium Binding Properties, and Ultrastructural Localization of the 53,000- and 160,000 (Sarcalumenin)-Dalton Glycoproteins of the Sarcoplasmic Reticulum. J. Biol. Chem. 1990, 265, 10118–10124. [Google Scholar] [CrossRef]
- Cala, S.E.; Scott, B.T.; Jones, L.R. Intralumenal Sarcoplasmic Reticulum Ca(2+)-Binding Proteins. Semin. Cell Biol. 1990, 1, 265–275. [Google Scholar] [PubMed]
- Damiani, E.; Margreth, A. Subcellular Fractionation to Junctional Sarcoplasmic Reticulum and Biochemical Characterization of 170 kDa Ca(2+)- and Low-Density-Lipoprotein-Binding Protein in Rabbit Skeletal Muscle. Biochem. J. 1991, 277, 825–832. [Google Scholar] [CrossRef]
- Treves, S.; Vukcevic, M.; Maj, M.; Thurnheer, R.; Mosca, B.; Zorzato, F. Minor Sarcoplasmic Reticulum Membrane Components That Modulate Excitation–Contraction Coupling in Striated Muscles. J. Physiol. 2009, 587, 3071–3079. [Google Scholar] [CrossRef]
- Staunton, L.; Ohlendieck, K. Mass Spectrometric Characterization of the Sarcoplasmic Reticulum from Rabbit Skeletal Muscle by On-Membrane Digestion. Protein Pept. Lett. 2012, 19, 252–263. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H. Isolation of Proteins of the Sarcoplasmic Reticulum. Methods Enzymol. 1974, 32, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Fliegel, L.; Newton, E.; Burns, K.; Michalak, M. Molecular Cloning of cDNA Encoding a 55-kDa Multifunctional Thyroid Hormone Binding Protein of Skeletal Muscle Sarcoplasmic Reticulum. J. Biol. Chem. 1990, 265, 15496–15502. [Google Scholar] [CrossRef] [PubMed]
- Waisman, D.M.; Smallwood, J.; Lafreniere, D.; Rasmussen, H. Identification of a Novel Hepatic Calcium Binding Protein. Biochem. Biophys. Res. Commun. 1984, 119, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Waisman, D.M.; Salimath, B.P.; Anderson, M.J. Isolation and Characterization of CAB-63, a Novel Calcium- Binding Protein. J. Biol. Chem. 1985, 260, 1652–1660. [Google Scholar] [CrossRef]
- Khanna, N.C.; Tokuda, M.; Waisman, D.M. Comparison of Calregulins from Vertebrate Livers. Biochem. J. 1987, 242, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Koch, G.L. Multiple Zones in the Sequence of Calreticulin (CRP55, Calregulin, HACBP), a Major Calcium Binding ER/SR Protein. EMBO J. 1989, 8, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- McCauliffe, D.P.; Lux, F.A.; Lieu, T.S.; Sanz, I.; Hanke, J.; Newkirk, M.M.; Bachinski, L.L.; Itoh, Y.; Siciliano, M.J.; Reichlin, M. Molecular Cloning, Expression, and Chromosome 19 Localization of a Human Ro/SS-A Autoantigen. J. Clin. Investig. 1990, 85, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- McCauliffe, D.P.; Zappi, E.; Lieu, T.S.; Michalak, M.; Sontheimer, R.D.; Capra, J.D. A Human Ro/SS-A Autoantigen Is the Homologue of Calreticulin and Is Highly Homologous with Onchocercal RAL-1 Antigen and an Aplysia “Memory Molecule”. J. Clin. Investig. 1990, 86, 332–335. [Google Scholar] [CrossRef] [PubMed]
- McCauliffe, D.P.; Yang, Y.S.; Wilson, J.; Sontheimer, R.D.; Capra, J.D. The 5’-Flanking Region of the Human Calreticulin Gene Shares Homology with the Human GRP78, GRP94, and Protein Disulfide Isomerase Promoters. J. Biol. Chem. 1992, 267, 2557–2562. [Google Scholar] [CrossRef]
- Guo, L.; Lynch, J.; Nakamura, K.; Fliegel, L.; Kasahara, H.; Izumo, S.; Komuro, I.; Agellon, L.B.; Michalak, M. COUP-TF1 Antagonizes Nkx2.5-Mediated Activation of the Calreticulin Gene during Cardiac Development. J. Biol. Chem. 2001, 276, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Guo, L.; Gelebart, P.; Chilibeck, K.; Xu, J.; Molkentin, J.D.; Agellon, L.B.; Michalak, M. Calreticulin Signals Upstream of Calcineurin and MEF2C in a Critical Ca(2+)-Dependent Signaling Cascade. J. Cell Biol. 2005, 170, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Lynch, J.; Guo, L.; Yatsula, B.; Perkins, A.S.; Michalak, M. Regulation of the Calreticulin Gene by GATA6 and Evi-1 Transcription Factors. Biochemistry 2008, 47, 3697–3704. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Michalak, M. Transcriptional Control of the Calreticulin Gene in Health and Disease. Int. J. Biochem. Cell Biol. 2009, 41, 531–538. [Google Scholar] [CrossRef]
- Waser, M.; Mesaeli, N.; Spencer, C.; Michalak, M. Regulation of Calreticulin Gene Expression by Calcium. J. Cell Biol. 1997, 138, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.O.; Capra, J.D.; Sontheimer, R.D. Calreticulin Is Transcriptionally Upregulated by Heat Shock, Calcium and Heavy Metals. Mol. Immunol. 1996, 33, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, D.H.; Kendall, J.M.; Sheikh, F.N.; Campbell, A.K. Induction of Calreticulin Expression in HeLa Cells by Depletion of the Endoplasmic Reticulum Ca2+ Store and Inhibition of N-Linked Glycosylation. Biochem. J. 1996, 318, 555–560. [Google Scholar] [CrossRef]
- Ní Fhlathartaigh, M.; McMahon, J.; Reynolds, R.; Connolly, D.; Higgins, E.; Counihan, T.; FitzGerald, U. Calreticulin and Other Components of Endoplasmic Reticulum Stress in Rat and Human Inflammatory Demyelination. Acta Neuropathol. Commun. 2013, 1, 37. [Google Scholar] [CrossRef]
- Khanna, N.C.; Waisman, D.M. Development of a Radioimmunoassay for Quantitation of Calregulin in Bovine Tissues. Biochemistry 1986, 25, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.; Michalak, M. Expression of Calreticulin in Escherichia Coli and Identification of Its Ca2+ Binding Domains. J. Biol. Chem. 1991, 266, 21458–21465. [Google Scholar] [CrossRef]
- Schrag, J.D.; Bergeron, J.J.; Li, Y.; Borisova, S.; Hahn, M.; Thomas, D.Y.; Cygler, M. The Structure of Calnexin, an ER Chaperone Involved in Quality Control of Protein Folding. Mol. Cell 2001, 8, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Chouquet, A.; Païdassi, H.; Ling, W.L.; Frachet, P.; Houen, G.; Arlaud, G.J.; Gaboriaud, C. X-Ray Structure of the Human Calreticulin Globular Domain Reveals a Peptide-Binding Area and Suggests a Multi-Molecular Mechanism. PLoS ONE 2011, 6, e17886. [Google Scholar] [CrossRef]
- Kozlov, G.; Pocanschi, C.L.; Rosenauer, A.; Bastos-Aristizabal, S.; Gorelik, A.; Williams, D.B.; Gehring, K. Structural Basis of Carbohydrate Recognition by Calreticulin. J. Biol. Chem. 2010, 285, 38612–38620. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.; Spamer, C.; Heilmann, C.; Michalak, M. Identification of the Zn2+ Binding Region in Calreticulin. FEBS Lett. 1995, 376, 53–57. [Google Scholar] [CrossRef]
- Saito, Y.; Ihara, Y.; Leach, M.R.; Cohen-Doyle, M.F.; Williams, D.B. Calreticulin Functions in Vitro as a Molecular Chaperone for Both Glycosylated and Non-Glycosylated Proteins. EMBO J. 1999, 18, 6718–6729. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.; Bydoun, M.; Holloway, R.; Waisman, D. Annexin A2 Heterotetramer: Structure and Function. Int. J. Mol. Sci. 2013, 14, 6259–6305. [Google Scholar] [CrossRef]
- Maffey, K.G.; Keil, L.B.; DeBari, V.A. The Influence of Lipid Composition and Divalent Cations on Annexin V Binding to Phospholipid Mixtures. Ann. Clin. Lab. Sci. 2001, 31, 85–90. [Google Scholar]
- Leach, M.R.; Cohen-Doyle, M.F.; Thomas, D.Y.; Williams, D.B. Localization of the Lectin, ERp57 Binding, and Polypeptide Binding Sites of Calnexin and Calreticulin. J. Biol. Chem. 2002, 277, 29686–29697. [Google Scholar] [CrossRef]
- Kapoor, M.; Ellgaard, L.; Gopalakrishnapai, J.; Schirra, C.; Gemma, E.; Oscarson, S.; Helenius, A.; Surolia, A. Mutational Analysis Provides Molecular Insight into the Carbohydrate-Binding Region of Calreticulin: Pivotal Roles of Tyrosine-109 and Aspartate-135 in Carbohydrate Recognition. Biochemistry 2004, 43, 97–106. [Google Scholar] [CrossRef]
- Peterson, J.R.; Ora, A.; Van, P.N.; Helenius, A. Transient, Lectin-like Association of Calreticulin with Folding Intermediates of Cellular and Viral Glycoproteins. Mol. Biol. Cell 1995, 6, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Wearsch, P.A.; Mitchell, D.A.; Wassenberg, J.J.; Gilboa, E.; Nicchitta, C.V. Calreticulin Displays in Vivo Peptide-Binding Activity and Can Elicit CTL Responses against Bound Peptides. J. Immunol. 1999, 162, 6426–6432. [Google Scholar] [CrossRef] [PubMed]
- Vassilakos, A.; Michalak, M.; Lehrman, M.A.; Williams, D.B. Oligosaccharide Binding Characteristics of the Molecular Chaperones Calnexin and Calreticulin. Biochemistry 1998, 37, 3480–3490. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.M.; Mancino, L.; Thammavongsa, V.; Cantley, R.L.; Raghavan, M. A Polypeptide Binding Conformation of Calreticulin Is Induced by Heat Shock, Calcium Depletion, or by Deletion of the C-Terminal Acidic Region. Mol. Cell 2004, 15, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Stafford, W.F.; Bouvier, M. The Metal Ion Binding Properties of Calreticulin Modulate Its Conformational Flexibility and Thermal Stability. Biochemistry 2001, 40, 11193–11201. [Google Scholar] [CrossRef] [PubMed]
- Lum, R.; Ahmad, S.; Hong, S.J.; Chapman, D.C.; Kozlov, G.; Williams, D.B. Contributions of the Lectin and Polypeptide Binding Sites of Calreticulin to Its Chaperone Functions in Vitro and in Cells. J. Biol. Chem. 2016, 291, 19631–19641. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Goldberg, A.L.; Ho, S.; Rohde, M.F.; Bush, K.T.; Sherman, M. A Set of Endoplasmic Reticulum Proteins Possessing Properties of Molecular Chaperones Includes Ca(2+)-Binding Proteins and Members of the Thioredoxin Superfamily. J. Biol. Chem. 1994, 269, 1744–1749. [Google Scholar] [CrossRef]
- Burns, K.; Duggan, B.; Atkinson, E.A.; Famulski, K.S.; Nemer, M.; Bleackley, R.C.; Michalak, M. Modulation of Gene Expression by Calreticulin Binding to the Glucocorticoid Receptor. Nature 1994, 367, 476–480. [Google Scholar] [CrossRef]
- Michalak, M.; Burns, K.; Andrin, C.; Mesaeli, N.; Jass, G.H.; Busaan, J.L.; Opas, M. Endoplasmic Reticulum Form of Calreticulin Modulates Glucocorticoid-Sensitive Gene Expression. J. Biol. Chem. 1996, 271, 29436–29445. [Google Scholar] [CrossRef] [PubMed]
- Walther, R.F.; Lamprecht, C.; Ridsdale, A.; Groulx, I.; Lee, S.; Lefebvre, Y.A.; Haché, R.J.G. Nuclear Export of the Glucocorticoid Receptor Is Accelerated by Cell Fusion-Dependent Release of Calreticulin. J. Biol. Chem. 2003, 278, 37858–37864. [Google Scholar] [CrossRef]
- Ellgaard, L.; Bettendorff, P.; Braun, D.; Herrmann, T.; Fiorito, F.; Jelesarov, I.; Güntert, P.; Helenius, A.; Wüthrich, K. NMR Structures of 36 and 73-Residue Fragments of the Calreticulin P-Domain. J. Mol. Biol. 2002, 322, 773–784. [Google Scholar] [CrossRef]
- Sakono, M.; Seko, A.; Takeda, Y.; Ito, Y. PDI Family Protein ERp29 Forms 1:1 Complex with Lectin Chaperone Calreticulin. Biochem. Biophys. Res. Commun. 2014, 452, 27–31. [Google Scholar] [CrossRef]
- Kozlov, G.; Bastos-Aristizabal, S.; Määttänen, P.; Rosenauer, A.; Zheng, F.; Killikelly, A.; Trempe, J.-F.; Thomas, D.Y.; Gehring, K. Structural Basis of Cyclophilin B Binding by the Calnexin/Calreticulin P-Domain. J. Biol. Chem. 2010, 285, 35551–35557. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.; Burns, K.; Andrin, C.; Michalak, M. Interaction of Calreticulin with Protein Disulfide Isomerase. J. Biol. Chem. 1995, 270, 31338–31344. [Google Scholar] [CrossRef]
- Corbett, E.F.; Oikawa, K.; Francois, P.; Tessier, D.C.; Kay, C.; Bergeron, J.J.; Thomas, D.Y.; Krause, K.H.; Michalak, M. Ca2+ Regulation of Interactions between Endoplasmic Reticulum Chaperones. J. Biol. Chem. 1999, 274, 6203–6211. [Google Scholar] [CrossRef] [PubMed]
- Avezov, E.; Konno, T.; Zyryanova, A.; Chen, W.; Laine, R.; Crespillo-Casado, A.; Melo, E.P.; Ushioda, R.; Nagata, K.; Kaminski, C.F.; et al. Retarded PDI Diffusion and a Reductive Shift in Poise of the Calcium Depleted Endoplasmic Reticulum. BMC Biol. 2015, 13, 2. [Google Scholar] [CrossRef]
- Ellgaard, L.; Riek, R.; Herrmann, T.; Güntert, P.; Braun, D.; Helenius, A.; Wüthrich, K. NMR Structure of the Calreticulin P-Domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3133–3138. [Google Scholar] [CrossRef] [PubMed]
- Frickel, E.-M.; Riek, R.; Jelesarov, I.; Helenius, A.; Wuthrich, K.; Ellgaard, L. TROSY-NMR Reveals Interaction between ERp57 and the Tip of the Calreticulin P-Domain. Proc. Natl. Acad. Sci. USA 2002, 99, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Wijeyesakere, S.J.; Gafni, A.A.; Raghavan, M. Calreticulin Is a Thermostable Protein with Distinct Structural Responses to Different Divalent Cation Environments. J. Biol. Chem. 2011, 286, 8771–8785. [Google Scholar] [CrossRef]
- Damiani, E.; Heilmann, C.; Salvatori, S.; Margreth, A. Characterization of High-Capacity Low-Affinity Calcium Binding Protein of Liver Endoplasmic Reticulum: Calsequestrin-like and Divergent Properties. Biochem. Biophys. Res. Commun. 1989, 165, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Villamil Giraldo, A.M.; Lopez Medus, M.; Gonzalez Lebrero, M.; Pagano, R.S.; Labriola, C.A.; Landolfo, L.; Delfino, J.M.; Parodi, A.J.; Caramelo, J.J. The Structure of Calreticulin C-Terminal Domain Is Modulated by Physiological Variations of Calcium Concentration. J. Biol. Chem. 2010, 285, 4544–4553. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R.B. A C-Terminal Signal Prevents Secretion of Luminal ER Proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Sönnichsen, B.; Füllekrug, J.; Nguyen Van, P.; Diekmann, W.; Robinson, D.G.; Mieskes, G. Retention and Retrieval: Both Mechanisms Cooperate to Maintain Calreticulin in the Endoplasmic Reticulum. J. Cell Sci. 1994, 107, 2705–2717. [Google Scholar] [CrossRef] [PubMed]
- Conte, I.L.; Keith, N.; Gutiérrez-Gonzalez, C.; Parodi, A.J.; Caramelo, J.J. The Interplay between Calcium and the in Vitro Lectin and Chaperone Activities of Calreticulin. Biochemistry 2007, 46, 4671–4680. [Google Scholar] [CrossRef]
- Wijeyesakere, S.J.; Bedi, S.K.; Huynh, D.; Raghavan, M. The C-Terminal Acidic Region of Calreticulin Mediates Phosphatidylserine Binding and Apoptotic Cell Phagocytosis. J. Immunol. 2016, 196, 3896–3909. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.R.; Lynch, N.J.; Day, A.J.; Schwaeble, W.J.; Sim, R.B. The C1q and Collectin Binding Site within C1q Receptor (Cell Surface Calreticulin). Immunopharmacology 1997, 38, 73–80. [Google Scholar] [CrossRef]
- Stuart, G.R.; Lynch, N.J.; Lu, J.; Geick, A.; Moffatt, B.E.; Sim, R.B.; Schwaeble, W.J. Localisation of the C1q Binding Site within C1q Receptor/Calreticulin. FEBS Lett. 1996, 397, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.T.; Nguyen, T.Q.; Yang, Y.S.; Capra, J.D.; Sontheimer, R.D. Calreticulin Binds hYRNA and the 52-kDa Polypeptide Component of the Ro/SS-A Ribonucleoprotein Autoantigen. J. Immunol. 1996, 156, 4484–4491. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Imaishi, K.; Hagiwara, Y.; Horibe, T.; Hayano, T.; Takahashi, N.; Urade, R.; Kato, K.; Kikuchi, M. ERp57 Binds Competitively to Protein Disulfide Isomerase and Calreticulin. Biochem. Biophys. Res. Commun. 2005, 331, 224–230. [Google Scholar] [CrossRef]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.-U.; et al. The Co-Translocation of ERp57 and Calreticulin Determines the Immunogenicity of Cell Death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef]
- Fraser, S.A.; Karimi, R.; Michalak, M.; Hudig, D. Perforin Lytic Activity Is Controlled by Calreticulin. J. Immunol. 2000, 164, 4150–4155. [Google Scholar] [CrossRef] [PubMed]
- Andrin, C.; Pinkoski, M.J.; Burns, K.; Atkinson, E.A.; Krahenbuhl, O.; Hudig, D.; Fraser, S.A.; Winkler, U.; Tschopp, J.; Opas, M.; et al. Interaction between a Ca2+-Binding Protein Calreticulin and Perforin, a Component of the Cytotoxic T-Cell Granules. Biochemistry 1998, 37, 10386–10394. [Google Scholar] [CrossRef]
- Fraser, S.A.; Michalak, M.; Welch, W.H.; Hudig, D. Calreticulin, a Component of the Endoplasmic Reticulum and of Cytotoxic Lymphocyte Granules, Regulates Perforin-Mediated Lysis in the Hemolytic Model System. Biochem. Cell Biol. Biochim. Biol. Cell. 1998, 76, 881–887. [Google Scholar] [CrossRef]
- Rojiani, M.V.; Finlay, B.B.; Gray, V.; Dedhar, S. In Vitro Interaction of a Polypeptide Homologous to Human Ro/SS-A Antigen (Calreticulin) with a Highly Conserved Amino Acid Sequence in the Cytoplasmic Domain of Integrin Alpha Subunits. Biochemistry 1991, 30, 9859–9866. [Google Scholar] [CrossRef] [PubMed]
- Ohkuro, M.; Kim, J.-D.; Kuboi, Y.; Hayashi, Y.; Mizukami, H.; Kobayashi-Kuramochi, H.; Muramoto, K.; Shirato, M.; Michikawa-Tanaka, F.; Moriya, J.; et al. Calreticulin and Integrin Alpha Dissociation Induces Anti-Inflammatory Programming in Animal Models of Inflammatory Bowel Disease. Nat. Commun. 2018, 9, 1982. [Google Scholar] [CrossRef] [PubMed]
- Vinaik, R.; Kozlov, G.; Gehring, K. Structure of the Non-Catalytic Domain of the Protein Disulfide Isomerase-Related Protein (PDIR) Reveals Function in Protein Binding. PLoS ONE 2013, 8, e62021. [Google Scholar] [CrossRef]
- McDonnell, J.M.; Jones, G.E.; White, T.K.; Tanzer, M.L. Calreticulin Binding Affinity for Glycosylated Laminin. J. Biol. Chem. 1996, 271, 7891–7894. [Google Scholar] [CrossRef] [PubMed]
- Thielmann, Y.; Weiergräber, O.H.; Mohrlüder, J.; Willbold, D. Structural Framework of the GABARAP-Calreticulin Interface--Implications for Substrate Binding to Endoplasmic Reticulum Chaperones. FEBS J. 2009, 276, 1140–1152. [Google Scholar] [CrossRef]
- Mohrlüder, J.; Stangler, T.; Hoffmann, Y.; Wiesehan, K.; Mataruga, A.; Willbold, D. Identification of Calreticulin as a Ligand of GABARAP by Phage Display Screening of a Peptide Library. FEBS J. 2007, 274, 5543–5555. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Devi, M.S.; Dhople, V.M.; Jesudasan, R.A. A Mouse Protein That Localizes to Acrosome and Sperm Tail Is Regulated by Y-Chromosome. BMC Cell Biol. 2013, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiao, X.; D’Atri, I.; Ono, F.; Nelson, R.; Chan, C.-C.; Nakaya, N.; Ma, Z.; Ma, Y.; Cai, X.; et al. Mutation in the Intracellular Chloride Channel CLCC1 Associated with Autosomal Recessive Retinitis Pigmentosa. PLoS Genet. 2018, 14, e1007504. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Willis, A.C.; Jensenius, J.C.; Jackson, J.; Sim, R.B. Structure and Homology of Human C1q Receptor (Collectin Receptor). Immunology 1993, 78, 341–348. [Google Scholar]
- Eggleton, P.; Lieu, T.S.; Zappi, E.G.; Sastry, K.; Coburn, J.; Zaner, K.S.; Sontheimer, R.D.; Capra, J.D.; Ghebrehiwet, B.; Tauber, A.I. Calreticulin Is Released from Activated Neutrophils and Binds to C1q and Mannan-Binding Protein. Clin. Immunol. Immunopathol. 1994, 72, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Wicker-Planquart, C.; Dufour, S.; Tacnet-Delorme, P.; Bally, I.; Delneste, Y.; Frachet, P.; Housset, D.; Thielens, N.M. Molecular and Cellular Interactions of Scavenger Receptor SR-F1 With Complement C1q Provide Insights Into Its Role in the Clearance of Apoptotic Cells. Front. Immunol. 2020, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.A.; deCathelineau, A.; Hoffmann, P.R.; Bratton, D.; Ghebrehiwet, B.; Fadok, V.A.; Henson, P.M. C1q and Mannose Binding Lectin Engagement of Cell Surface Calreticulin and CD91 Initiates Macropinocytosis and Uptake of Apoptotic Cells. J. Exp. Med. 2001, 194, 781–795. [Google Scholar] [CrossRef]
- Duus, K.; Pagh, R.T.; Holmskov, U.; Højrup, P.; Skov, S.; Houen, G. Interaction of Calreticulin with CD40 Ligand, TRAIL and Fas Ligand. Scand. J. Immunol. 2007, 66, 501–507. [Google Scholar] [CrossRef]
- Orr, A.W.; Pedraza, C.E.; Pallero, M.A.; Elzie, C.A.; Goicoechea, S.; Strickland, D.K.; Murphy-Ullrich, J.E. Low Density Lipoprotein Receptor–Related Protein Is a Calreticulin Coreceptor That Signals Focal Adhesion Disassembly. J. Cell Biol. 2003, 161, 1179–1189. [Google Scholar] [CrossRef]
- Goicoechea, S.; Pallero, M.A.; Eggleton, P.; Michalak, M.; Murphy-Ullrich, J.E. The Anti-Adhesive Activity of Thrombospondin Is Mediated by the N-Terminal Domain of Cell Surface Calreticulin. J. Biol. Chem. 2002, 277, 37219–37228. [Google Scholar] [CrossRef]
- Goicoechea, S.; Orr, A.W.; Pallero, M.A.; Eggleton, P.; Murphy-Ullrich, J.E. Thrombospondin Mediates Focal Adhesion Disassembly through Interactions with Cell Surface Calreticulin. J. Biol. Chem. 2000, 275, 36358–36368. [Google Scholar] [CrossRef]
- Kretsinger, R.H.; Nockolds, C.E. Carp Muscle Calcium-Binding Protein. II. Structure Determination and General Description. J. Biol. Chem. 1973, 248, 3313–3326. [Google Scholar] [CrossRef]
- Filipenko, N.R.; Kang, H.M.; Waisman, D.M. Characterization of the Ca2+-Binding Sites of Annexin II Tetramer. J. Biol. Chem. 2001, 276, 38877–38884. [Google Scholar] [CrossRef]
- Madureira, P.A.; O’Connell, P.A.; Surette, A.P.; Miller, V.A.; Waisman, D.M. The Biochemistry and Regulation of S100A10: A Multifunctional Plasminogen Receptor Involved in Oncogenesis. J. Biomed. Biotechnol. 2012, 2012, 353687. [Google Scholar] [CrossRef]
- Williams, D.B. Beyond Lectins: The Calnexin/Calreticulin Chaperone System of the Endoplasmic Reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef]
- Samtleben, S.; Jaepel, J.; Fecher, C.; Andreska, T.; Rehberg, M.; Blum, R. Direct Imaging of ER Calcium with Targeted-Esterase Induced Dye Loading (TED). JoVE J. Vis. Exp. 2013, 75, e50317. [Google Scholar] [CrossRef]
- Romani, A.; Scarpa, A. Regulation of Cell Magnesium. Arch. Biochem. Biophys. 1992, 298, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.M.P. Magnesium in Eukaryotes. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov, E.A., Eds.; Springer: New York, NY, USA, 2013; pp. 1255–1264. ISBN 978-1-4614-1533-6. [Google Scholar]
- Chabosseau, P.; Tuncay, E.; Meur, G.; Bellomo, E.A.; Hessels, A.; Hughes, S.; Johnson, P.R.V.; Bugliani, M.; Marchetti, P.; Turan, B.; et al. Mitochondrial and ER-Targeted eCALWY Probes Reveal High Levels of Free Zn2+. ACS Chem. Biol. 2014, 9, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.D.; Wang, F.; MacDiarmid, C.W.; Clark, S.; Lyons, T.; Eide, D.J. Zinc and the Msc2 Zinc Transporter Protein Are Required for Endoplasmic Reticulum Function. J. Cell Biol. 2004, 166, 325–335. [Google Scholar] [CrossRef]
- Grabarek, Z. Insights into Modulation of Calcium Signaling by Magnesium in Calmodulin, Troponin C and Related EF-Hand Proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Nara, M.; Morii, H.; Tanokura, M. Coordination to Divalent Cations by Calcium-Binding Proteins Studied by FTIR Spectroscopy. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 2319–2327. [Google Scholar] [CrossRef]
- Strynadka, N.C.; James, M.N. Crystal Structures of the Helix-Loop-Helix Calcium-Binding Proteins. Annu. Rev. Biochem. 1989, 58, 951–998. [Google Scholar] [CrossRef] [PubMed]
- van de Put, F.H.M.M.; Elliott, A.C. The Endoplasmic Reticulum Can Act as a Functional Ca2+ Store in All Subcellular Regions of the Pancreatic Acinar Cell. J. Biol. Chem. 1997, 272, 27764–27770. [Google Scholar] [CrossRef]
- Waisman, D.M. Annexin II Tetramer: Structure and Function. MolCell Biochem 1995, 149/150, 301–322. [Google Scholar]
- Bedard, K.; Szabo, E.; Michalak, M.; Opas, M. Cellular Functions of Endoplasmic Reticulum Chaperones Calreticulin, Calnexin, and ERp57. Int. Rev. Cytol. 2005, 245, 91–121. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and Cancer. Cell Res. 2021, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Camacho, P.; Lechleiter, J.D. Calreticulin Inhibits Repetitive Intracellular Ca2+ Waves. Cell 1995, 82, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Zuppini, A.; Arnaudeau, S.; Lynch, J.; Ahsan, I.; Krause, R.; Papp, S.; De Smedt, H.; Parys, J.B.; Muller-Esterl, W.; et al. Functional Specialization of Calreticulin Domains. J. Cell Biol. 2001, 154, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Arnaudeau, S.; Frieden, M.; Nakamura, K.; Castelbou, C.; Michalak, M.; Demaurex, N. Calreticulin Differentially Modulates Calcium Uptake and Release in the Endoplasmic Reticulum and Mitochondria. J. Biol. Chem. 2002, 277, 46696–46705. [Google Scholar] [CrossRef]
- Sadasivan, B.; Lehner, P.J.; Ortmann, B.; Spies, T.; Cresswell, P. Roles for Calreticulin and a Novel Glycoprotein, Tapasin, in the Interaction of MHC Class I Molecules with TAP. Immunity 1996, 5, 103–114. [Google Scholar] [CrossRef]
- Gao, B.; Adhikari, R.; Howarth, M.; Nakamura, K.; Gold, M.C.; Hill, A.B.; Knee, R.; Michalak, M.; Elliott, T. Assembly and Antigen-Presenting Function of MHC Class I Molecules in Cells Lacking the ER Chaperone Calreticulin. Immunity 2002, 16, 99–109. [Google Scholar] [CrossRef]
- Hebert, D.N.; Foellmer, B.; Helenius, A. Calnexin and Calreticulin Promote Folding, Delay Oligomerization and Suppress Degradation of Influenza Hemagglutinin in Microsomes. EMBO J. 1996, 15, 2961–2968. [Google Scholar] [CrossRef]
- Dedhar, S.; Rennie, P.S.; Shago, M.; Hagesteijn, C.-Y.L.; Yang, H.; Filmus, J.; Hawley, R.G.; Bruchovsky, N.; Cheng, H.; Matusik, R.J.; et al. Inhibition of Nuclear Hormone Receptor Activity by Calreticulin. Nature 1994, 367, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Black, B.E.; Love, D.C.; Hanover, J.A.; Leszyk, J.; Paschal, B.M. Calreticulin Is a Receptor for Nuclear Export. J. Cell Biol. 2001, 152, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.-C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of Pre-Apoptotic Calreticulin Exposure in Immunogenic Cell Death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin Is the Dominant Pro-Phagocytic Signal on Multiple Human Cancers and Is Counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of LRP on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Ye, Z.-H.; Huang, M.-Y.; Lu, J.-J. Regulation of CD47 Expression in Cancer Cells. Transl. Oncol. 2020, 13. [Google Scholar] [CrossRef]
- Liu, N.; Fine, R.E.; Simons, E.; Johnson, R.J. Decreasing Calreticulin Expression Lowers the Ca2+ Response to Bradykinin and Increases Sensitivity to Ionomycin in NG-108-15 Cells. J. Biol. Chem. 1994, 269, 28635–28639. [Google Scholar] [CrossRef]
- Bastianutto, C.; Clementi, E.; Codazzi, F.; Podini, P.; De Giorgi, F.; Rizzuto, R.; Meldolesi, J.; Pozzan, T. Overexpression of Calreticulin Increases the Ca2+ Capacity of Rapidly Exchanging Ca2+ Stores and Reveals Aspects of Their Lumenal Microenvironment and Function. J. Cell Biol. 1995, 130, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.S.; Park, C.S.; Choi, K.; Ahnn, J.; Kim, J.I.; Eom, S.H.; Kaufman, S.J.; Song, W.K. Calreticulin Couples Calcium Release and Calcium Influx in Integrin-Mediated Calcium Signaling. Mol. Biol. Cell 2000, 11, 1433–1443. [Google Scholar] [CrossRef]
- Fadel, M.P.; Dziak, E.; Lo, C.M.; Ferrier, J.; Mesaeli, N.; Michalak, M.; Opas, M. Calreticulin Affects Focal Contact-Dependent but Not Close Contact-Dependent Cell-Substratum Adhesion. J. Biol. Chem. 1999, 274, 15085–15094. [Google Scholar] [CrossRef]
- Coppolino, M.G.; Dedhar, S. Ligand-Specific, Transient Interaction between Integrins and Calreticulin during Cell Adhesion to Extracellular Matrix Proteins Is Dependent upon Phosphorylation/Dephosphorylation Events. Biochem. J. 1999, 340, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, M.G.; Woodside, M.J.; Demaurex, N.; Grinstein, S.; St-Arnaud, R.; Dedhar, S. Calreticulin Is Essential for Integrin-Mediated Calcium Signalling and Cell Adhesion. Nature 1997, 386, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.I.; Rahman, M.; Michaels, J.; Callaghan, M.; Deveau-Rosen, J.; Michalak, M.; Gurtner, G.C.; Nanney, L.B. Calreticulin Enhances Wound Healing Via Multiple Biological Effects. Wound Repair Regen. 2005, 13, A4–A27. [Google Scholar] [CrossRef]
- Gold, L.I.; Rahman, M.; Blechman, K.M.; Greives, M.R.; Churgin, S.; Michaels, J.; Callaghan, M.J.; Cardwell, N.L.; Pollins, A.C.; Michalak, M.; et al. Overview of the Role for Calreticulin in the Enhancement of Wound Healing through Multiple Biological Effects. J. Investig. Dermatol. Symp. Proc. 2006, 11, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Donnelly, S.; Roake, W.; Brown, S.; Young, P.; Naik, H.; Wordsworth, P.; Isenberg, D.A.; Reid, K.B.M.; Eggleton, P. Impaired Recognition of Apoptotic Neutrophils by the C1q/Calreticulin and CD91 Pathway in Systemic Lupus Erythematosus. Arthritis Rheum. 2006, 54, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Panaretakis, T.; Tesniere, A.; Joza, N.; Tufi, R.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Leveraging the Immune System during Chemotherapy: Moving Calreticulin to the Cell Surface Converts Apoptotic Death from “Silent” to Immunogenic. Cancer Res. 2007, 67, 7941–7944. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-Calreticulin in Immunogenic Chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef]
- Basu, S.; Srivastava, P.K. Calreticulin, a Peptide-Binding Chaperone of the Endoplasmic Reticulum, Elicits Tumor- and Peptide-Specific Immunity. J. Exp. Med. 1999, 189, 797–802. [Google Scholar] [CrossRef]
- Totary-Jain, H.; Naveh-Many, T.; Riahi, Y.; Kaiser, N.; Eckel, J.; Sasson, S. Calreticulin Destabilizes Glucose Transporter-1 mRNA in Vascular Endothelial and Smooth Muscle Cells under High-Glucose Conditions. Circ. Res. 2005, 97, 1001–1008. [Google Scholar] [CrossRef]
- Nickenig, G.; Michaelsen, F.; Müller, C.; Berger, A.; Vogel, T.; Sachinidis, A.; Vetter, H.; Böhm, M. Destabilization of AT(1) Receptor mRNA by Calreticulin. Circ. Res. 2002, 90, 53–58. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Okura, G.C.; Woods, J.W.; Allen, E.A.; Miller, V.A.; Kempster, E.; Hancock, M.A.; Gujar, S.; Slibinskas, R.; Waisman, D.M. Identification and Characterization of Calreticulin as a Novel Plasminogen Receptor. J. Biol. Chem. 2023, 300, 105465. [Google Scholar] [CrossRef]
- Mesaeli, N.; Nakamura, K.; Zvaritch, E.; Dickie, P.; Dziak, E.; Krause, K.H.; Opas, M.; MacLennan, D.H.; Michalak, M. Calreticulin Is Essential for Cardiac Development. J. Cell Biol. 1999, 144, 857–868. [Google Scholar] [CrossRef]
- Rauch, F.; Prud’homme, J.; Arabian, A.; Dedhar, S.; St-Arnaud, R. Heart, Brain, and Body Wall Defects in Mice Lacking Calreticulin. Exp. Cell Res. 2000, 256, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, M.G.; Dedhar, S. Calreticulin. Int. J. Biochem. Cell Biol. 1998, 30, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Robertson, M.; Liu, G.; Dickie, P.; Nakamura, K.; Guo, J.Q.; Duff, H.J.; Opas, M.; Kavanagh, K.; Michalak, M. Complete Heart Block and Sudden Death in Mice Overexpressing Calreticulin. J. Clin. Investig. 2001, 107, 1245. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Neuronal Calcium Signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Orkand, R.K.; Kettenmann, H. Glial Calcium: Homeostasis and Signaling Function. Physiol. Rev. 1998, 78, 99–141. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, J.A.; Parekh, A.B. Respiring Mitochondria Determine the Pattern of Activation and Inactivation of the Store-Operated Ca(2+) Current I(CRAC). EMBO J. 2000, 19, 6401–6407. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling Endoplasmic Reticulum Stress to the Cell Death Program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium Homeostasis and Cancer: Insights from Endoplasmic Reticulum-Centered Organelle Communications. Trends Cell Biol. 2023, 33, 312–323. [Google Scholar] [CrossRef]
- Gangola, P.; Rosen, B.P. Maintenance of Intracellular Calcium in Escherichia Coli. J. Biol. Chem. 1987, 262, 12570–12574. [Google Scholar] [CrossRef] [PubMed]
- Watkins, N.J.; Knight, M.R.; Trewavas, A.J.; Campbell, A.K. Free Calcium Transients in Chemotactic and Non-Chemotactic Strains of Escherichia Coli Determined by Using Recombinant Aequorin. Biochem. J. 1995, 306, 865–869. [Google Scholar] [CrossRef]
- Takenaka, H.; Adler, P.N.; Katz, A.M. Calcium Fluxes across the Membrane of Sarcoplasmic Reticulum Vesicles. J. Biol. Chem. 1982, 257, 12649–12656. [Google Scholar] [CrossRef]
- Mignery, G.A.; Südhof, T.C.; Takei, K.; De Camilli, P. Putative Receptor for Inositol 1,4,5-Trisphosphate Similar to Ryanodine Receptor. Nature 1989, 342, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; Knudson, C.M.; Imagawa, T.; Leung, A.T.; Sutko, J.L.; Kahl, S.D.; Raab, C.R.; Madson, L. Identification and Characterization of the High Affinity [3H]Ryanodine Receptor of the Junctional Sarcoplasmic Reticulum Ca2+ Release Channel. J. Biol. Chem. 1987, 262, 6460–6463. [Google Scholar] [CrossRef]
- Inui, M.; Saito, A.; Fleischer, S. Purification of the Ryanodine Receptor and Identity with Feet Structures of Junctional Terminal Cisternae of Sarcoplasmic Reticulum from Fast Skeletal Muscle. J. Biol. Chem. 1987, 262, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Erdmann, F.; Jung, M.; Wagner, R.; Cavalie, A.; Zimmermann, R. Sec61 Complexes Form Ubiquitous ER Ca2+ Leak Channels. Channels 2011, 5, 228–235. [Google Scholar] [CrossRef]
- Lièvremont, J.P.; Rizzuto, R.; Hendershot, L.; Meldolesi, J. BiP, a Major Chaperone Protein of the Endoplasmic Reticulum Lumen, Plays a Direct and Important Role in the Storage of the Rapidly Exchanging Pool of Ca2+. J. Biol. Chem. 1997, 272, 30873–30879. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.K.; Mee, C.; Xu, W.; Liu, L.; Blond, S.; Cooper, A.; Charles, I.G.; Hawkins, A.R. The Affinity of a Major Ca2+ Binding Site on GRP78 Is Differentially Enhanced by ADP and ATP. J. Biol. Chem. 2006, 281, 8796–8805. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an Essential and Conserved Component of Store-Operated Ca2+ Channel Function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A Mutation in Orai1 Causes Immune Deficiency by Abrogating CRAC Channel Function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 Is a Plasma Membrane Protein Essential for Store-Operated Ca2+ Entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-Wide RNAi Screen of Ca(2+) Influx Identifies Genes That Regulate Ca(2+) Release-Activated Ca(2+) Channel Activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed]
- Shim, A.H.-R.; Tirado-Lee, L.; Prakriya, M. Structural and Functional Mechanisms of CRAC Channel Regulation. J. Mol. Biol. 2015, 427, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Ho-Schleyer, S.C.; Travers, K.J.; Weissman, J.S. Biochemical Basis of Oxidative Protein Folding in the Endoplasmic Reticulum. Science 2000, 290, 1571–1574. [Google Scholar] [CrossRef]
- Bader, M.; Muse, W.; Ballou, D.P.; Gassner, C.; Bardwell, J.C. Oxidative Protein Folding Is Driven by the Electron Transport System. Cell 1999, 98, 217–227. [Google Scholar] [CrossRef]
- Ziegler, D.M.; Poulsen, L.L. Protein Disulfide Bond Synthesis: A Possible Intracellular Mechanism. Trends Biochem. Sci. 1977, 2, 79–81. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized Redox State of Glutathione in the Endoplasmic Reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Flatmark, T. Binding of the Viral Immunogenic Octapeptide VSV8 to Native Glucose-Regulated Protein Grp94 (Gp96) and Its Inhibition by the Physiological Ligands ATP and Ca2+. FEBS J. 2006, 273, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Mery, L.; Mesaeli, N.; Michalak, M.; Opas, M.; Lew, D.P.; Krause, K.H. Overexpression of Calreticulin Increases Intracellular Ca2+ Storage and Decreases Store-Operated Ca2+ Influx. J. Biol. Chem. 1996, 271, 9332–9339. [Google Scholar] [CrossRef] [PubMed]
- John, L.M.; Lechleiter, J.D.; Camacho, P. Differential Modulation of SERCA2 Isoforms by Calreticulin. J. Cell Biol. 1998, 142, 963–973. [Google Scholar] [CrossRef]
- Llewelyn Roderick, H.; Llewellyn, D.H.; Campbell, A.K.; Kendall, J.M. Role of Calreticulin in Regulating Intracellular Ca2+ Storage and Capacitative Ca2+ Entry in HeLa Cells. Cell Calcium 1998, 24, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Longo, F.J.; Wintermantel, M.R.; Jiang, X.; Clark, R.A.; DeLisle, S. Calreticulin Modulates Capacitative Ca2+ Influx by Controlling the Extent of Inositol 1,4,5-Trisphosphate-Induced Ca2+ Store Depletion. J. Biol. Chem. 2000, 275, 36676–36682. [Google Scholar] [CrossRef] [PubMed]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of Calcium Homeostasis and Flux between the Endoplasmic Reticulum and the Cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.Y.; Milankov, K.; Michalak, M.; Wilkins, J.; Dedhar, S. Cell Attachment to Extracellular Matrix Substrates Is Inhibited upon Downregulation of Expression of Calreticulin, an Intracellular Integrin Alpha-Subunit-Binding Protein. J. Cell Sci. 1994, 107, 589–600. [Google Scholar] [CrossRef]
- Helenius, A. How N-Linked Oligosaccharides Affect Glycoprotein Folding in the Endoplasmic Reticulum. Mol. Biol. Cell 1994, 5, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Lamriben, L.; Graham, J.B.; Adams, B.M.; Hebert, D.N. N-Glycan Based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic Cph. Den. 2016, 17, 308. [Google Scholar] [CrossRef]
- Wijeyesakere, S.J.; Gagnon, J.K.; Arora, K.; Brooks, C.L.; Raghavan, M. Regulation of Calreticulin-Major Histocompatibility Complex (MHC) Class I Interactions by ATP. Proc. Natl. Acad. Sci. USA 2015, 112, E5608–E5617. [Google Scholar] [CrossRef]
- Kozlov, G.; Muñoz-Escobar, J.; Castro, K.; Gehring, K. Mapping the ER Interactome: The P Domains of Calnexin and Calreticulin as Plurivalent Adapters for Foldases and Chaperones. Structure 2017, 25, 1415–1422.e3. [Google Scholar] [CrossRef] [PubMed]
- Corbett, E.F.; Michalak, K.M.; Oikawa, K.; Johnson, S.; Campbell, I.D.; Eggleton, P.; Kay, C.; Michalak, M. The Conformation of Calreticulin Is Influenced by the Endoplasmic Reticulum Luminal Environment. J. Biol. Chem. 2000, 275, 27177–27185. [Google Scholar] [CrossRef]
- Olson, E.; Raghavan, M. Major Histocompatibility Complex Class I Assembly within Endolysosomal Pathways. Curr. Opin. Immunol. 2023, 84, 102356. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, G.; Bangia, N.; Pan, M.; Cresswell, P. A Role for Calnexin in the Assembly of the MHC Class I Loading Complex in the Endoplasmic Reticulum1. J. Immunol. 2001, 166, 1703–1709. [Google Scholar] [CrossRef]
- Kanaseki, T.; Blanchard, N.; Hammer, G.E.; Gonzalez, F.; Shastri, N. ERAAP Synergizes with MHC Class I Molecules to Make the Final Cut in the Antigenic Peptide Precursors in the Endoplasmic Reticulum. Immunity 2006, 25, 795–806. [Google Scholar] [CrossRef]
- Lan, B.H.; Becker, M.; Freund, C. The Mode of Action of Tapasin on Major Histocompatibility Class I (MHC-I) Molecules. J. Biol. Chem. 2023, 299, 102987. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Osorio, M.; Zúñiga, M.C.; Edidin, M. Selective Export of MHC Class I Molecules from the ER after Their Dissociation from TAP. Immunity 2000, 13, 841–851. [Google Scholar] [CrossRef]
- Arshad, N.; Cresswell, P. Tumor-Associated Calreticulin Variants Functionally Compromise the Peptide Loading Complex and Impair Its Recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fu, H.; Flutter, B.; Powis, S.J.; Gao, B. Suppression of MHC Class I Surface Expression by Calreticulin’s P-Domain in a Calreticulin Deficient Cell Line. Biochim. Biophys. Acta 2010, 1803, 544–552. [Google Scholar] [CrossRef]
- Wearsch, P.A.; Peaper, D.R.; Cresswell, P. Essential Glycan-Dependent Interactions Optimize MHC Class I Peptide Loading. Proc. Natl. Acad. Sci. USA 2011, 108, 4950–4955. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Wearsch, P.A.; Peaper, D.R.; Cresswell, P.; Reinisch, K.M. Insights into MHC Class I Peptide Loading from the Structure of the Tapasin-ERp57 Thiol Oxidoreductase Heterodimer. Immunity 2009, 30, 21–32. [Google Scholar] [CrossRef]
- Del Cid, N.; Jeffery, E.; Rizvi, S.M.; Stamper, E.; Peters, L.R.; Brown, W.C.; Provoda, C.; Raghavan, M. Modes of Calreticulin Recruitment to the Major Histocompatibility Complex Class I Assembly Pathway. J. Biol. Chem. 2010, 285, 4520–4535. [Google Scholar] [CrossRef]
- Howe, C.; Garstka, M.; Al-Balushi, M.; Ghanem, E.; Antoniou, A.N.; Fritzsche, S.; Jankevicius, G.; Kontouli, N.; Schneeweiss, C.; Williams, A.; et al. Calreticulin-Dependent Recycling in the Early Secretory Pathway Mediates Optimal Peptide Loading of MHC Class I Molecules. EMBO J. 2009, 28, 3730–3744. [Google Scholar] [CrossRef] [PubMed]
- Païdassi, H.; Tacnet-Delorme, P.; Verneret, M.; Gaboriaud, C.; Houen, G.; Duus, K.; Ling, W.L.; Arlaud, G.J.; Frachet, P. Investigations on the C1q-Calreticulin-Phosphatidylserine Interactions Yield New Insights into Apoptotic Cell Recognition. J. Mol. Biol. 2011, 408, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Verneret, M.; Tacnet-Delorme, P.; Osman, R.; Awad, R.; Grichine, A.; Kleman, J.-P.; Frachet, P. Relative Contribution of C1q and Apoptotic Cell-Surface Calreticulin to Macrophage Phagocytosis. J. Innate Immun. 2014, 6, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Cockram, T.O.J.; Puigdellívol, M.; Brown, G.C. Calreticulin and Galectin-3 Opsonise Bacteria for Phagocytosis by Microglia. Front. Immunol. 2019, 10, 2647. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M. ERP57 Membrane Translocation Dictates the Immunogenicity of Tumor Cell Death by Controlling the Membrane Translocation of Calreticulin. J. Immunol. 2008, 181, 2533–2543. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Leclair, P.; Pedari, F.; Vieira, H.; Monajemi, M.; Sly, L.M.; Reid, G.S.; Lim, C.J. Integrins and ERp57 Coordinate to Regulate Cell Surface Calreticulin in Immunogenic Cell Death. Front. Oncol. 2019, 9, 411. [Google Scholar] [CrossRef]
- Tanaka, M.; Kataoka, H.; Yano, S.; Sawada, T.; Akashi, H.; Inoue, M.; Suzuki, S.; Inagaki, Y.; Hayashi, N.; Nishie, H.; et al. Immunogenic Cell Death Due to a New Photodynamic Therapy (PDT) with Glycoconjugated Chlorin (G-Chlorin). Oncotarget 2016, 7, 47242–47251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Yang, M.; Zhang, J.; Yin, Y.; Fan, X.; Zhang, Y.; Qin, S.; Zhang, H.; Yu, F. Immunogenic Cell Death Induction by Ionizing Radiation. Front. Immunol. 2021, 12, 705361. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Lin, K. Immunogenic Cell Death-Based Oncolytic Virus Therapy: A Sharp Sword of Tumor Immunotherapy. Eur. J. Pharmacol. 2024, 981, 176913. [Google Scholar] [CrossRef]
- Chen, L.; Wen, Y.; Xiong, J.; Chen, Y.; Chen, C. An Immunogenic Cell Death-Related Gene Signature Reflects Immune Landscape and Predicts Prognosis in Melanoma Independently of BRAF V600E Status. BioMed Res. Int. 2023, 2023, 1189022. [Google Scholar] [CrossRef]
- Cifric, S.; Turi, M.; Folino, P.; Clericuzio, C.; Barello, F.; Maciel, T.; Anderson, K.C.; Gulla, A. DAMPening Tumor Immune Escape: The Role of Endoplasmic Reticulum Chaperones in Immunogenic Chemotherapy. Antioxid. Redox Signal. 2024, 41, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, H.; Inoue, H.; Shiraishi, Y.; Hirayama, A.; Nakanishi, T.; Ando, H.; Nakajima, M.; Shinozaki, S.; Ogata, H.; Okamura, K.; et al. Impact of Increased Plasma Levels of Calreticulin on Prognosis of Patients with Advanced Lung Cancer Undergoing Combination Treatment of Chemotherapy and Immune Checkpoint Inhibitors. Lung Cancer 2023, 181, 107264. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-Y.; Pyo, A.; Yun, M.; Thangam, R.; You, S.-H.; Zhang, Y.; Jung, Y.; Nguyen, D.-H.; Venu, A.; Kim, H.S.; et al. Imaging Calreticulin for Early Detection of Immunogenic Cell Death During Anticancer Treatment. J. Nucl. Med. 2021, 62, 956–960. [Google Scholar] [CrossRef]
- Feng, M.; Marjon, K.D.; Zhu, F.; Weissman-Tsukamoto, R.; Levett, A.; Sullivan, K.; Kao, K.S.; Markovic, M.; Bump, P.A.; Jackson, H.M.; et al. Programmed Cell Removal by Calreticulin in Tissue Homeostasis and Cancer. Nat. Commun. 2018, 9, 3194. [Google Scholar] [CrossRef] [PubMed]
- Andriani, A.; Latagliata, R.; Anaclerico, B.; Spadea, A.; Rago, A.; Di Veroli, A.; Spirito, F.; Porrini, R.; De Muro, M.; Crescenzi Leonetti, S.; et al. Spleen Enlargement Is a Risk Factor for Thrombosis in Essential Thrombocythemia: Evaluation on 1,297 Patients. Am. J. Hematol. 2016, 91, 318–321. [Google Scholar] [CrossRef]
- Mancuso, S.; Accurso, V.; Santoro, M.; Raso, S.; Contrino, A.D.; Perez, A.; Di Piazza, F.; Florena, A.M.; Russo, A.; Siragusa, S. The Essential Thrombocythemia, Thrombotic Risk Stratification, and Cardiovascular Risk Factors. Adv. Hematol. 2020, 2020, 9124821. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, F.; Colombi, M.; Calori, R.; Zilioli, V.R.; Bramanti, S.; Iurlo, A.; Zanella, A. Analysis of Risk Factors Predicting Thrombotic and/or Haemorrhagic Complications in 306 Patients with Essential Thrombocythemia. Hematol. Oncol. 2007, 25, 115–120. [Google Scholar] [CrossRef]
- Regev, A.; Stark, P.; Blickstein, D.; Lahav, M. Thrombotic Complications in Essential Thrombocythemia with Relatively Low Platelet Counts. Am. J. Hematol. 1997, 56, 168–172. [Google Scholar] [CrossRef]
- Małecki, R.; Gacka, M.; Kuliszkiewicz-Janus, M.; Jakobsche-Policht, U.; Kwiatkowski, J.; Adamiec, R.; Undas, A. Altered Plasma Fibrin Clot Properties in Essential Thrombocythemia. Platelets 2016, 27, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.L.; Keragala, C.B. The Fibrinolytic System: Mysteries and Opportunities. HemaSphere 2021, 5, e570. [Google Scholar] [CrossRef] [PubMed]
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An Enigmatic Zymogen. Blood 2021, 137, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Aisina, R.B.; Mukhametova, L.I. Structure and Function of Plasminogen/Plasmin System. Russ. J. Bioorganic Chem. 2014, 40, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y. Hypofibrinolytic State and High Thrombin Generation May Play a Major Role in SARS-COV-2 Associated Thrombosis. J. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Contreras, O.A.; Martinez de Lizarrondo, S.; Bardou, I.; Orset, C.; Pruvost, M.; Anfray, A.; Frigout, Y.; Hommet, Y.; Lebouvier, L.; Montaner, J.; et al. Hyperfibrinolysis Increases Blood–Brain Barrier Permeability by a Plasmin- and Bradykinin-Dependent Mechanism. Blood 2016, 128, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Plow, E.F.; Waisman, D.M.; Parmer, R.J. Plasminogen Receptors. J. Biomed. Biotechnol. 2012, 2012, 130735. [Google Scholar] [CrossRef]
- Plow, E.F.; Doeuvre, L.; Das, R. So Many Plasminogen Receptors: Why? J. Biomed. Biotechnol. 2012, 2012, 141806. [Google Scholar] [CrossRef]
- Miles, L.A.; Parmer, R.J. Plasminogen Receptors: The First Quarter Century. Semin. Thromb. Hemost. 2013, 39, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Godier, A.; Hunt, B.J. Plasminogen Receptors and Their Role in the Pathogenesis of Inflammatory, Autoimmune and Malignant Disease. J. Thromb. Haemost. JTH 2013, 11, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.L. Fibrinolysis: From Blood to the Brain. J. Thromb. Haemost. JTH 2017, 15, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Guglielmelli, P.; Bordoni, R.; Casetti, I.; Milanesi, C.; Sant’Antonio, E.; Ferretti, V.; Pancrazzi, A.; Rotunno, G.; et al. Acquired Copy-Neutral Loss of Heterozygosity of Chromosome 1p as a Molecular Event Associated with Marrow Fibrosis in MPL-Mutated Myeloproliferative Neoplasms. Blood 2013, 121, 4388–4395. [Google Scholar] [CrossRef]
- Pérez Encinas, M.M.; Sobas, M.; Gómez-Casares, M.T.; Abuin Blanco, A.; Noya Pereira, M.S.; Raya, J.M.; Andrade-Campos, M.M.; Álvarez Larrán, A.; Lewandowski, K.; Łukasz, S.; et al. The Risk of Thrombosis in Essential Thrombocythemia Is Associated with the Type of CALR Mutation: A Multicentre Collaborative Study. Eur. J. Haematol. 2021, 106, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Rotunno, G.; Mannarelli, C.; Guglielmelli, P.; Pacilli, A.; Pancrazzi, A.; Pieri, L.; Fanelli, T.; Bosi, A.; Vannucchi, A.M.; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. Impact of Calreticulin Mutations on Clinical and Hematological Phenotype and Outcome in Essential Thrombocythemia. Blood 2014, 123, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the Thrombopoietin Receptor by Mutant Calreticulin in CALR-Mutant Myeloproliferative Neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-Terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the Requirements for the Pathogenic Interaction between Mutant Calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.-I.; Defour, J.-P.; Nivarthi, H.; Hug, E.; et al. Calreticulin Mutants as Oncogenic Rogue Chaperones for TpoR and Traffic-Defective Pathogenic TpoR Mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef]
- Kuwabara, K.; Pinsky, D.J.; Schmidt, A.M.; Benedict, B.; Brett, B.; Ogawa, S.; Broekman, M.J.; Marcus, A.J.; Sciacca, R.R.; Michalak, M.; et al. Calreticulin, an Antithrombotic Agent Which Binds to Vitamin K-Dependent Coagulation Factors, Stimulates Endothelial Nitric Oxide Production, and Limits Thrombosis in Canine Coronary Arteries (∗). J. Biol. Chem. 1995, 270, 8179–8187. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Stewart, M.; Ritchie, B.; Mesaeli, N.; Raha, S.; Kolodziejczyk, D.; Hobman, M.L.; Liu, L.Y.; Etches, W.; Nation, N.; et al. Calreticulin, a Potential Vascular Regulatory Protein, Reduces Intimal Hyperplasia after Arterial Injury. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2019, 77, 748–760.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Kroemer, G.; Kepp, O. Secreted Calreticulin Mutants Subvert Anticancer Immunosurveillance. OncoImmunology 2020, 9, 1708126. [Google Scholar] [CrossRef]
- Gigoux, M.; Holmström, M.O.; Zappasodi, R.; Park, J.J.; Pourpe, S.; Bozkus, C.C.; Mangarin, L.M.B.; Redmond, D.; Verma, S.; Schad, S.; et al. Calreticulin Mutant Myeloproliferative Neoplasms Induce MHC-I Skewing, Which Can Be Overcome by an Optimized Peptide Cancer Vaccine. Sci. Transl. Med. 2022, 14, eaba4380. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, J.; Elbanna, Y.A.; Kurylowicz, K.; Ciboddo, M.; Greenbaum, H.S.; Arellano, N.S.; Rodriguez, D.; Evers, M.; Bock-Hughes, A.; Liu, C.; et al. Type I but Not Type II Calreticulin Mutations Activate the IRE1α/XBP1 Pathway of the Unfolded Protein Response to Drive Myeloproliferative Neoplasms. Blood Cancer Discov. 2022, 3, 298–315. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okura, G.C.; Bharadwaj, A.G.; Waisman, D.M. Calreticulin—From the Endoplasmic Reticulum to the Plasma Membrane—Adventures of a Wandering Protein. Cancers 2025, 17, 288. https://doi.org/10.3390/cancers17020288
Okura GC, Bharadwaj AG, Waisman DM. Calreticulin—From the Endoplasmic Reticulum to the Plasma Membrane—Adventures of a Wandering Protein. Cancers. 2025; 17(2):288. https://doi.org/10.3390/cancers17020288
Chicago/Turabian StyleOkura, Gillian C., Alamelu G. Bharadwaj, and David M. Waisman. 2025. "Calreticulin—From the Endoplasmic Reticulum to the Plasma Membrane—Adventures of a Wandering Protein" Cancers 17, no. 2: 288. https://doi.org/10.3390/cancers17020288
APA StyleOkura, G. C., Bharadwaj, A. G., & Waisman, D. M. (2025). Calreticulin—From the Endoplasmic Reticulum to the Plasma Membrane—Adventures of a Wandering Protein. Cancers, 17(2), 288. https://doi.org/10.3390/cancers17020288