
Adoptive T Cell Therapy Targeting MAGE-A4



Simple Summary
Abstract
1. Introduction
2. MAGE A4 Functionality and the Beginnings of T Cell Therapy Development
3. Current MAGE A4 T Cell Therapy Trials
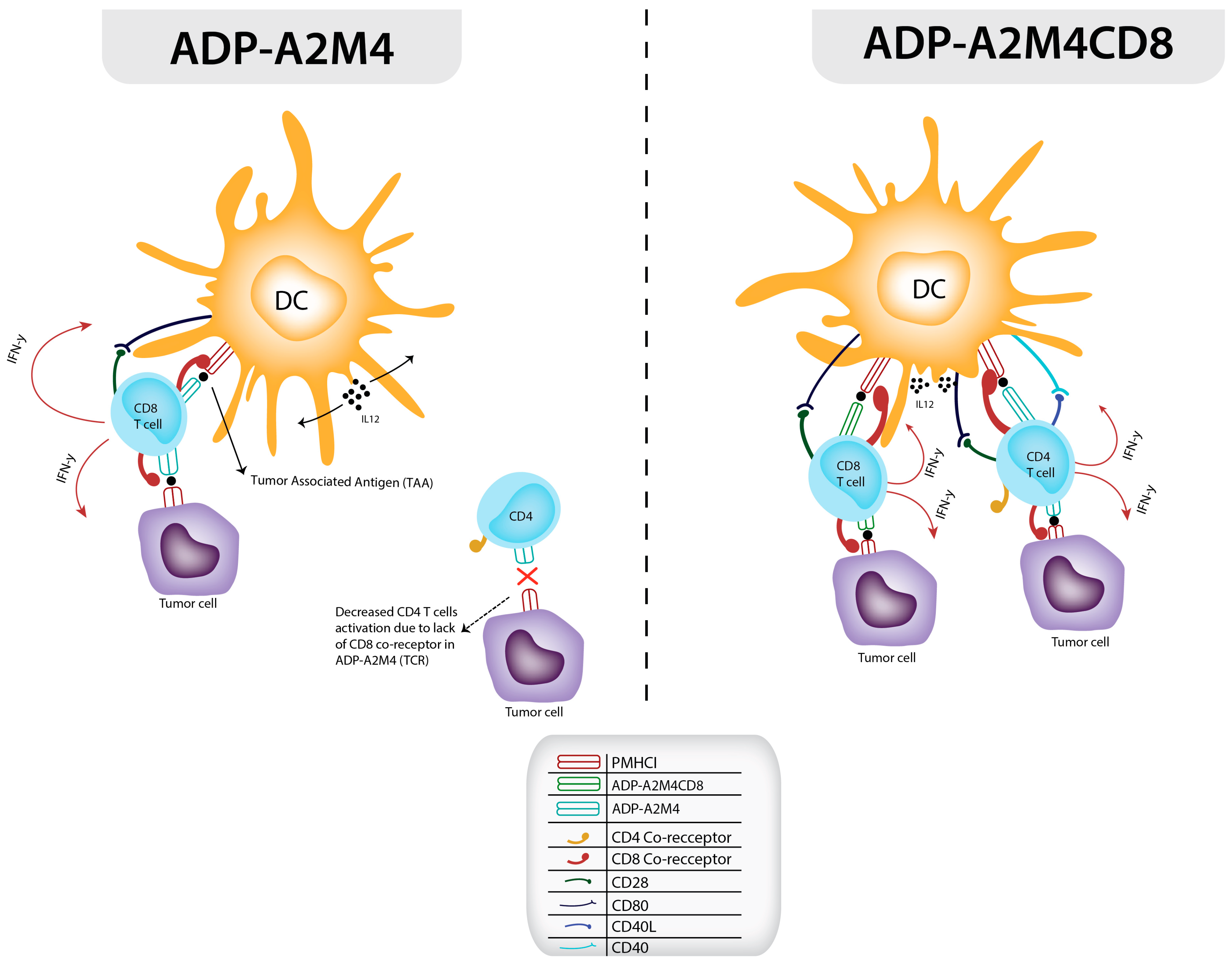
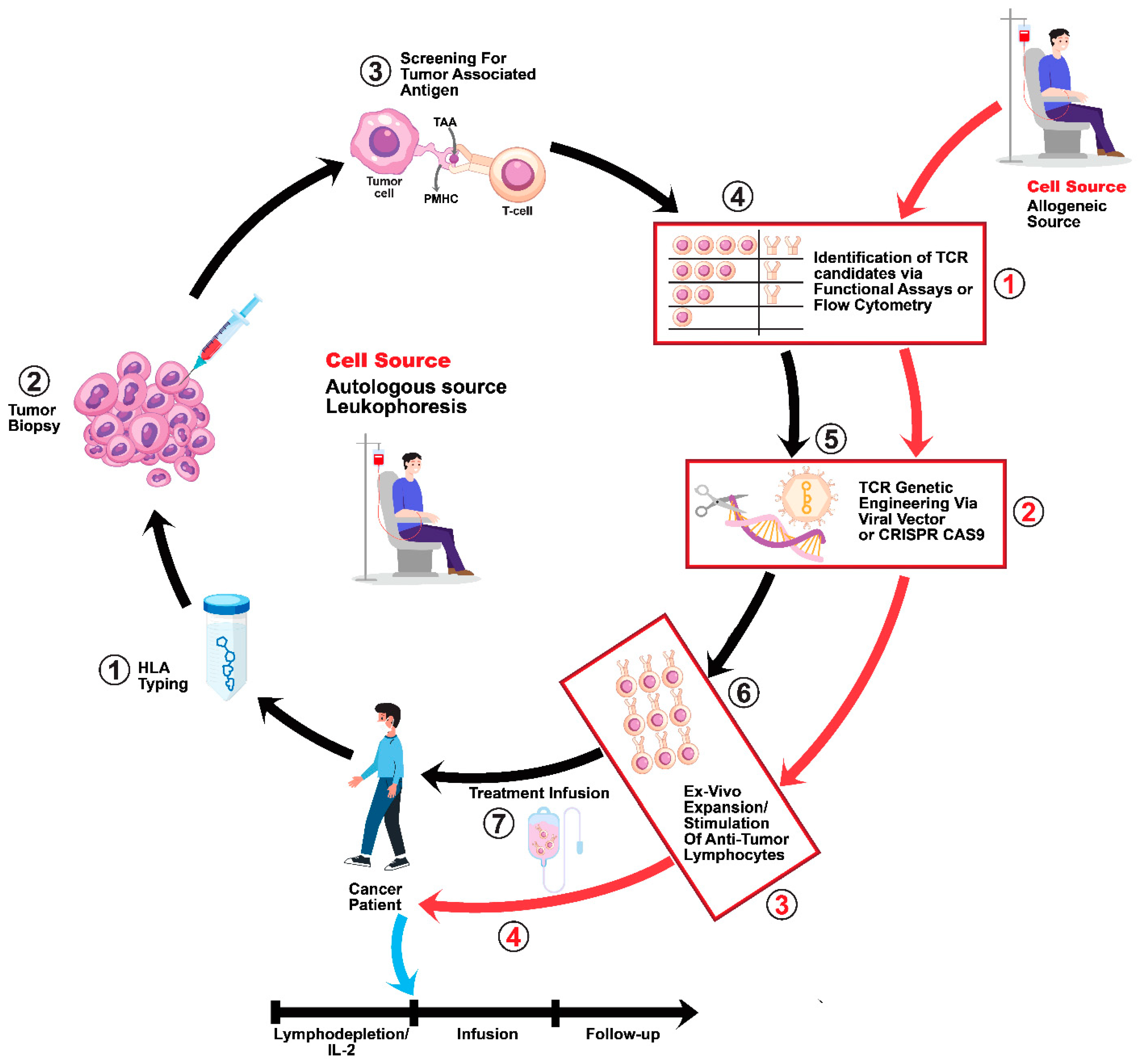
4. Development and Application of Affinity-Enhanced Autologous MAGE A4 TCR Therapy via the CD8 α Co-Receptor
5. Allogeneic Bispecific MAGE A4 BiTE Therapy
6. Current Challenges and the Future Outlook for Autologous TCR Immunotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Magalhaes, I.; Carvalho-Queiroz, C.; Hartana, C.A.; Kaiser, A.; Lukic, A.; Mints, M.; Nilsson, O.; Grönlund, H.; Mattsson, J.; Berglund, S. Facing the Future: Challenges and Opportunities in Adoptive T Cell Therapy in Cancer. Expert Opin. Biol. Ther. 2019, 19, 811–827. [Google Scholar] [CrossRef]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-Cell-Transfer Therapy for the Treatment of Patients with Cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Zhao, Y.; Bennett, A.D.; Zheng, Z.; Wang, Q.J.; Robbins, P.F.; Yu, L.Y.L.; Li, Y.; Molloy, P.E.; Dunn, S.M.; Jakobsen, B.K.; et al. High-Affinity TCRs Generated by Phage Display Provide CD4+ T Cells with the Ability to Recognize and Kill Tumor Cell Lines. J. Immunol. 2007, 179, 5845–5854. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients after Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.A.; Krueger, J. Chimeric Antigen Receptor T Cell Therapy Comes to Clinical Practice. Curr. Oncol. 2020, 27 (Suppl. 2), S115–S123. [Google Scholar] [CrossRef]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the Uneven Road of Antigen Heterogeneity in Solid Tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Llopis, P.M.; Bouffard, A.A.; Scarfó, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T Cell Killing Requires the IFNγR Pathway in Solid but Not Liquid Tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Thibaut, R.; Bost, P.; Milo, I.; Cazaux, M.; Lemaître, F.; Garcia, Z.; Amit, I.; Breart, B.; Cornuot, C.; Schwikowski, B.; et al. Bystander IFN-γ Activity Promotes Widespread and Sustained Cytokine Signaling Altering the Tumor Microenvironment. Nat. Cancer 2020, 1, 302–314. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-Engineered T Cells for Cancer Therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, O.; Chen, Y.T.; Seitz, G.; Villena-Heinsen, C.; Old, L.J.; Pfreundschuh, M. Expression of Multiple Cancer/Testis (CT) Antigens in Breast Cancer and Melanoma: Basis for Polyvalent CT Vaccine Strategies. Int. J. Cancer 1998, 78, 387–389. [Google Scholar] [CrossRef]
- Peled, N.; Oton, A.B.; Hirsch, F.R.; Bunn, P. MAGE A3 Antigen-Specific Cancer Immunotherapeutic. Immunotherapy 2009, 1, 19–25. [Google Scholar] [CrossRef]
- Sigalotti, L.; Covre, A.; Zabierowski, S.; Himes, B.; Colizzi, F.; Natali, P.G.; Herlyn, M.; Maio, M. Cancer Testis Antigens in Human Melanoma Stem Cells: Expression, Distribution, and Methylation Status. J. Cell. Physiol. 2008, 215, 287–291. [Google Scholar] [CrossRef]
- Otte, M.; Zafrakas, M.; Riethdorf, L.; Pichlmeier, U.; Löning, T.; Jänicke, F.; Pantel, K. MAGE-A Gene Expression Pattern in Primary Breast Cancer. Cancer Res. 2001, 61, 6682–6687. [Google Scholar]
- Gure, A.O.; Chua, R.; Williamson, B.; Gonen, M.; Ferrera, C.A.; Gnjatic, S.; Ritter, G.; Simpson, A.J.G.; Chen, Y.-T.; Old, L.J.; et al. Cancer-Testis Genes Are Coordinately Expressed and Are Markers of Poor Outcome in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2005, 11, 8055–8062. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-D.; Park, H.-R.; Song, M.-H.; Shin, D.-H.; Lee, C.-H.; Lee, M.-K.; Lee, S.-Y. Pattern of Cancer/Testis Antigen Expression in Lung Cancer Patients. Int. J. Mol. Med. 2012, 29, 656–662. [Google Scholar] [CrossRef]
- Ayyoub, M.; Scarlata, C.-M.; Hamaï, A.; Pignon, P.; Valmori, D. Expression of MAGE-A3/6 in Primary Breast Cancer Is Associated with Hormone Receptor Negative Status, High Histologic Grade, and Poor Survival. J. Immunother. 2014, 37, 73–76. [Google Scholar] [CrossRef]
- Simpson, A.J.G.; Caballero, O.L.; Jungbluth, A.; Chen, Y.-T.; Old, L.J. Cancer/Testis Antigens, Gametogenesis and Cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Hou, Z.; Liang, X.; Wang, X.; Zhou, Z.; Shi, G. Myeloid-derived Suppressor Cells Infiltration in Non-small-cell Lung Cancer Tumor and MAGE-A4 and NY-ESO-1 Expression. Oncol. Lett. 2020, 19, 3982–3992. [Google Scholar] [CrossRef]
- Freiberger, S.N.; Holzmann, D.; Morand, G.B.; Hüllner, M.; Levesque, M.P.; Dummer, R.; Koelzer, V.H.; Rupp, N.J. Combinational Expression of Tumor Testis Antigens NY-ESO-1, MAGE-A3, and MAGE-A4 Predicts Response to Immunotherapy in Mucosal Melanoma Patients. J. Cancer Res. Clin. Oncol. 2023, 149, 5645–5653. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor Regression in Patients with Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Wermke, M.; Tsimberidou, A.-M.; Mohamed, A.; Mayer-Mokler, A.; Satelli, A.; Reinhardt, C.; Araujo, D.; Maurer, D.; Blumenschein, G.J.; Singh, H.; et al. 959 Safety and Anti-Tumor Activity of TCR-Engineered Autologous, PRAME-Directed T Cells across Multiple Advanced Solid Cancers at Low Doses—Clinical Update on the ACTengine® IMA203 Trial. J. Immunother. Cancer 2021, 9 (Suppl. 2), A1–A1054. [Google Scholar] [CrossRef]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A Gene Encoding an Antigen Recognized by Cytolytic T Lymphocytes on a Human Melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Cabezón, T.; Gromova, I.; Gromov, P.; Serizawa, R.; Timmermans Wielenga, V.; Kroman, N.; Celis, J.E.; Moreira, J.M.A. Proteomic Profiling of Triple-Negative Breast Carcinomas in Combination with a Three-Tier Orthogonal Technology Approach Identifies Mage-A4 as Potential Therapeutic Target in Estrogen Receptor Negative Breast Cancer. Mol. Cell. Proteom. MCP 2013, 12, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Valmori, D.; Dutoit, V.; Rubio-Godoy, V.; Chambaz, C.; Liénard, D.; Guillaume, P.; Romero, P.; Cerottini, J.C.; Rimoldi, D. Frequent Cytolytic T-Cell Responses to Peptide MAGE-A10(254-262) in Melanoma. Cancer Res. 2001, 61, 509–512. [Google Scholar]
- Alves, P.M.S.; Lévy, N.; Bouzourene, H.; Viatte, S.; Bricard, G.; Ayyoub, M.; Vuilleumier, H.; Givel, J.-C.R.; Halkic, N.; Speiser, D.E.; et al. Molecular and Immunological Evaluation of the Expression of Cancer/Testis Gene Products in Human Colorectal Cancer. Cancer Immunol. Immunother. 2007, 56, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Sasajima, K.; Sato, Y.; Watanabe, H.; Matsutani, T.; Iida, S.; Hosone, M.; Tsukui, T.; Maeda, S.; Shimizu, K.; et al. MAGE-A Protein and MAGE-A10 Gene Expressions in Liver Metastasis in Patients with Stomach Cancer. Br. J. Cancer 2008, 99, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Mengus, C.; Schultz-Thater, E.; Coulot, J.; Kastelan, Z.; Goluza, E.; Coric, M.; Spagnoli, G.C.; Hudolin, T. MAGE-A10 Cancer/Testis Antigen Is Highly Expressed in High-Grade Non-Muscle-Invasive Bladder Carcinomas. Int. J. Cancer 2013, 132, 2459–2463. [Google Scholar] [CrossRef]
- Marcar, L.; MacLaine, N.J.; Hupp, T.R.; Meek, D.W. Mage-A Cancer/Testis Antigens Inhibit P53 Function by Blocking Its Interaction with Chromatin. Cancer Res. 2010, 70, 10362–10370. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Mutter-Rottmayer, E.; Greenwalt, A.M.; Goldfarb, D.; Yan, F.; Yang, Y.; Martinez-Chacin, R.C.; Pearce, K.H.; Tateishi, S.; Major, M.B.; et al. A Neomorphic Cancer Cell-Specific Role of MAGE-A4 in Trans-Lesion Synthesis. Nat. Commun. 2016, 7, 12105. [Google Scholar] [CrossRef]
- Higashitsuji, H.; Itoh, K.; Nagao, T.; Dawson, S.; Nonoguchi, K.; Kido, T.; Mayer, R.J.; Arii, S.; Fujita, J. Reduced Stability of Retinoblastoma Protein by Gankyrin, an Oncogenic Ankyrin-Repeat Protein Overexpressed in Hepatomas. Nat. Med. 2000, 6, 96–99. [Google Scholar] [CrossRef]
- Li, J.; Guo, Y. Gankyrin Oncoprotein: Structure, Function, and Involvement in Cancer. Curr. Chem. Biol. 2010, 4, 13–19. [Google Scholar] [CrossRef]
- Chambost, H.; Van Baren, N.; Brasseur, F.; Godelaine, D.; Xerri, L.; Landi, S.J.; Theate, I.; Plumas, J.; Spagnoli, G.C.; Michel, G.; et al. Expression of Gene MAGE-A4 in Reed-Sternberg Cells. Blood 2000, 95, 3530–3533. [Google Scholar]
- Yakirevich, E.; Sabo, E.; Lavie, O.; Mazareb, S.; Spagnoli, G.C.; Resnick, M.B. Expression of the MAGE-A4 and NY-ESO-1 Cancer-Testis Antigens in Serous Ovarian Neoplasms. Clin. Cancer Res. 2003, 9, 6453–6460. [Google Scholar]
- Iura, K.; Kohashi, K.; Ishii, T.; Maekawa, A.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Matsumoto, Y.; Iwamoto, Y.; et al. MAGEA4 expression in bone and soft tissue tumors: Its utility as a target for immunotherapy and diagnostic marker combined with NY-ESO-1. Virchows Arch. 2017, 471, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Caballero, O.L.; Chen, Y.-T. Cancer/Testis (CT) Antigens: Potential Targets for Immunotherapy. Cancer Sci. 2009, 100, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primer 2020, 6, 92. [Google Scholar] [CrossRef]
- Sharma, P.; Shen, Y.; Wen, S.; Bajorin, D.F.; Reuter, V.E.; Old, L.J.; Jungbluth, A.A. Cancer-Testis Antigens: Expression and Correlation with Survival in Human Urothelial Carcinoma. Clin. Cancer Res. 2006, 12, 5442–5447. [Google Scholar] [CrossRef]
- Kakimoto, T.; Matsumine, A.; Kageyama, S.; Asanuma, K.; Matsubara, T.; Nakamura, T.; Iino, T.; Ikeda, H.; Shiku, H.; Sudo, A. Immunohistochemical Expression and Clinicopathological Assessment of the Cancer Testis Antigens NY-ESO-1 and MAGE-A4 in High-Grade Soft-Tissue Sarcoma. Oncol. Lett. 2019, 17, 3937–3943. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, O.; Pfreundschuh, M. Serological Identification of Human Tumor Antigens. Curr. Opin. Immunol. 1997, 9, 709–716. [Google Scholar] [CrossRef]
- Iura, K.; Kohashi, K.; Hotokebuchi, Y.; Ishii, T.; Maekawa, A.; Yamada, Y.; Yamamoto, H.; Iwamoto, Y.; Oda, Y. Cancer-Testis Antigens PRAME and NY-ESO-1 Correlate with Tumour Grade and Poor Prognosis in Myxoid Liposarcoma. J. Pathol. Clin. Res. 2015, 1, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Shen, J.; Tang, Q.; Yang, Z.; Yin, J.; Li, Z.; Xie, X.; Huang, G.; Lev, D.; Wang, J. Cancer-Testis Antigens Expressed in Osteosarcoma Identified by Gene Microarray Correlate with a Poor Patient Prognosis. Cancer 2012, 118, 1845–1855. [Google Scholar] [CrossRef]
- Sugita, M.; Geraci, M.; Gao, B.; Powell, R.L.; Hirsch, F.R.; Johnson, G.; Lapadat, R.; Gabrielson, E.; Bremnes, R.; Bunn, P.A.; et al. Combined Use of Oligonucleotide and Tissue Microarrays Identifies Cancer/Testis Antigens as Biomarkers in Lung Carcinoma1. Cancer Res. 2002, 62, 3971–3979. [Google Scholar] [PubMed]
- Duffour, M.-T.; Chaux, P.; Lurquin, C.; Cornelis, G.; Boon, T.; Van der Bruggen, P. A MAGE-A4 Peptide Presented by HLA-A2 Is Recognized by Cytolytic T Lymphocytes. Eur. J. Immunol. 1999, 29, 3329–3337. [Google Scholar] [CrossRef]
- Celis, E.; Tsai, V.; Crimi, C.; DeMars, R.; Wentworth, P.A.; Chesnut, R.W.; Grey, H.M.; Sette, A.; Serra, H.M. Induction of Anti-Tumor Cytotoxic T Lymphocytes in Normal Humans Using Primary Cultures and Synthetic Peptide Epitopes. Proc. Natl. Acad. Sci. USA 1994, 91, 2105–2109. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Schluesener, H.J. Human T Lymphocytes Recognize a Peptide of Single Point-Mutated, Oncogenic Ras Proteins. J. Exp. Med. 1991, 173, 273–276. [Google Scholar] [CrossRef]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical Evaluation of an Affinity-Enhanced MAGE-A4-Specific T-Cell Receptor for Adoptive T-Cell Therapy. OncoImmunology 2020, 9, 1682381. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Ikeda, H.; Miyahara, Y.; Imai, N.; Ishihara, M.; Saito, K.; Sugino, S.; Ueda, S.; Ishikawa, T.; Kokura, S.; et al. Adoptive Transfer of MAGE-A4 T-Cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clin. Cancer Res. 2015, 21, 2268–2277. [Google Scholar] [CrossRef]
- Redmond, D.; Poran, A.; Elemento, O. Single-Cell TCRseq: Paired Recovery of Entire T-Cell Alpha and Beta Chain Transcripts in T-Cell Receptors from Single-Cell RNAseq. Genome Med. 2016, 8, 80. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Joo, J.; Riley, J.P.; Yu, Z.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Characterization of Genetically Modified T-Cell Receptors That Recognize the CEA:691-699 Peptide in the Context of HLA-A2.1 on Human Colorectal Cancer Cells. Clin. Cancer Res. 2009, 15, 169–180. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and Persistence of Adoptively Transferred Autologous CD19-Targeted T Cells in Patients with Relapsed or Chemotherapy Refractory B-Cell Leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Chernysheva, A.D.; Kirou, K.A.; Crow, M.K. T Cell Proliferation Induced by Autologous Non-T Cells Is a Response to Apoptotic Cells Processed by Dendritic Cells. J. Immunol. 2002, 169, 1241–1250. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Targeting Treg Cells in Cancer Immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef]
- Brocker, T. Survival of Mature CD4 T Lymphocytes Is Dependent on Major Histocompatibility Complex Class II–Expressing Dendritic Cells. J. Exp. Med. 1997, 186, 1223–1232. [Google Scholar] [CrossRef]
- Colf, L.A.; Bankovich, A.J.; Hanick, N.A.; Bowerman, N.A.; Jones, L.L.; Kranz, D.M.; Garcia, K.C. How a Single T Cell Receptor Recognizes Both Self and Foreign MHC. Cell 2007, 129, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.V.; Kulkarni-Kale, U. T-Cell Epitope Prediction Methods: An Overview. In Immunoinformatics; De, R.K., Tomar, N., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 333–364. [Google Scholar] [CrossRef]
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking Predictions of MHC Class I Restricted T Cell Epitopes in a Comprehensively Studied Model System. PLoS Comput. Biol. 2020, 16, e1007757. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Wada, H.; Yamasaki, M.; Miyata, H.; Nishikawa, H.; Sato, E.; Kageyama, S.; Shiku, H.; Mori, M.; Doki, Y. High Expression of MAGE-A4 and MHC Class I Antigens in Tumor Cells and Induction of MAGE-A4 Immune Responses Are Prognostic Markers of CHP-MAGE-A4 Cancer Vaccine. Vaccine 2014, 32, 5901–5907. [Google Scholar] [CrossRef]
- Hong, D.; Van Tine, B.; Biswas, S.; McAlpine, C.; Johnson, M.; Olszanski, A.; Clarke, J.; Araujo, D.; Blumenschein, G.; Kebriaei, P.; et al. Autologous T Cell Therapy for MAGE-A4 Solid Cancers in HLA-A*02 Patients: A Phase 1 Trial. Nat. Med. 2023, 29, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Van Tine, B.A.; Olszanski, A.J.; Johnson, M.L.; Liebner, D.A.; Trivedi, T.; Lin, Q.; Elefant, E.; Dryer-Minnerly, R.; Navenot, J.-M.; et al. Phase I Dose Escalation and Expansion Trial to Assess the Safety and Efficacy of ADP-A2M4 SPEAR T Cells in Advanced Solid Tumors. J. Clin. Oncol. 2020, 38 (Suppl. 15), 102. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Van Tine, B.A.; Attia, S.; Blay, J.-Y.; Strauss, S.J.; Valverde Morales, C.M.; Abdul Razak, A.R.; Van Winkle, E.; Trivedi, T.; Biswas, S.; et al. SPEARHEAD-1: A Phase 2 Trial of Afamitresgene Autoleucel (Formerly ADP-A2M4) in Patients with Advanced Synovial Sarcoma or Myxoid/Round Cell Liposarcoma. J. Clin. Oncol. 2021, 39 (Suppl. 15), 11504. [Google Scholar] [CrossRef]
- Roche, H.L. An Open-Label, Multicenter, Phase I Study to Evaluate Safety, Pharmacokinetics, and Preliminary Anti-Tumor Activity of RO7444973 in Participants with Unresectable and/or Metastatic MAGE-A4-Positive Solid Tumors; Clinical Trial Registration NCT05129280; 2023. Available online: https://clinicaltrials.gov/study/NCT05129280 (accessed on 31 December 2023).
- Rossetti, R.; Brand, H.; Lima, S.C.G.; Furtado, I.P.; Silveira, R.M.; Fantacini, D.M.C.; Covas, D.T.; de Souza, L.E.B. Combination of Genetically Engineered T Cells and Immune Checkpoint Blockade for the Treatment of Cancer. Immunother. Adv. 2022, 2, ltac005. [Google Scholar] [CrossRef]
- Douglass, J.; Hsiue, E.H.-C.; Mog, B.J.; Hwang, M.S.; DiNapoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific Antibodies Targeting Mutant RAS Neoantigens. Sci. Immunol. 2021, 6, eabd5515. [Google Scholar] [CrossRef]
- Hong, D.; Clarke, J.; Johanns, T.; Kebriaei, P.; Heymach, J.; Galal, A.; Saibil, S.; Sacher, A.; Brophy, F.; Betts, G.; et al. 379 Initial Safety, Efficacy, and Product Attributes from the SURPASS Trial with ADP-A2M4CD8, a SPEAR T-Cell Therapy Incorporating an Affinity Optimized TCR Targeting MAGE-A4 and a CD8α Co-Receptor. J. Immunother. Cancer 2020, 8 (Suppl. 3), A1–A559. [Google Scholar] [CrossRef]
- Hong, D.S.; Jalal, S.I.; Elimova, E.; Ajani, J.A.; Blum Murphy, M.A.; Cervantes, A.; Evans, T.R.J.; Park, H.; Lin, Q.; Noto, P.; et al. SURPASS-2 Trial Design: A Phase 2, Open-Label Study of ADP-A2M4CD8 SPEAR T Cells in Advanced Esophageal or Esophagogastric Junction Cancers. J. Clin. Oncol. 2022, 40 (Suppl. 4), TPS363. [Google Scholar] [CrossRef]
- Cohen, E.E.; Dunn, L.; Neupane, P.; Gibson, M.; Leidner, R.; Savvides, P.; Hyland, N.; Trivedi, T.; Dudley, M.; Biswas, S.; et al. 976TiP SPEARHEAD-2 Trial Design: A Phase II Pilot Trial of ADP-A2M4 in Combination with Pembrolizumab in Patients with Recurrent or Metastatic Head and Neck Cancer. Ann. Oncol. 2020, 31, S685. [Google Scholar] [CrossRef]
- Blumenschein, J.; Davar, D.; Gutierrez, R.; Segal, N.; Johnson, M.; Dar, M.; Marshall, S. A Phase I/II First-in-Human Study of a Novel Anti-MAGE-A4 TCR/Anti-CD3 Bispecific (IMC-C103C) as Monotherapy and in Combination with Atezolizumab in HLA-A*02:01-Positive Patients with MAGE-A4-Positive Advanced Solid Tumors (IMC-C103C-101). J. Clin. Oncol. 2020, 38, TPS3165. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive Cell Therapy for Patients with Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The Third Dimension Bridges the Gap between Cell Culture and Live Tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Araujo, D.M.; Razak, A.R.A.; Agulnik, M.; Attia, S.; Blay, J.-Y.; Garcia, I.C.; Charlson, J.A.; Choy, E.; Demetri, G.D.; et al. Afamitresgene Autoleucel for Advanced Synovial Sarcoma and Myxoid Round Cell Liposarcoma (SPEARHEAD-1): An International, Open-Label, Phase 2 Trial. Lancet 2024, 403, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Afamitresgene Autoleucel: First Approval. Mol. Diagn. Ther. 2024, 28, 861–866. [Google Scholar] [CrossRef]
- Davari, K.; Holland, T.; Prassmayer, L.; Longinotti, G.; Ganley, K.P.; Pechilis, L.J.; Diaconu, I.; Nambiar, P.R.; Magee, M.S.; Schendel, D.J.; et al. Development of a CD8 Co-Receptor Independent T-Cell Receptor Specific for Tumor-Associated Antigen MAGE-A4 for next Generation T-Cell-Based Immunotherapy. J. Immunother. Cancer 2021, 9, e002035. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Brilha, S.S.; Weber, A.M.; Pachnio, A.; Wiedermann, G.E.; Dauleh, S.; Ahmed, T.; Pope, G.R.; Quinn, L.L.; Docta, R.Y.; et al. Enhancing Efficacy of TCR-Engineered CD4+ T Cells Via Coexpression of CD8α. J. Immunother. 2023, 46, 132–144. [Google Scholar] [CrossRef]
- Blum Murphy, M.A.; Ajani, J.A.; Van Tine, B.A.; Clarke, J.M.; Butler, M.O.; Lawrence, D.P.; Johnson, M.L.; Cervantes, A.; Moreno, V.; Hong, D.S.; et al. Safety and Efficacy from the Phase 1 SURPASS Trial of ADP-A2M4CD8, a next-Generation T-Cell Receptor T-Cell Therapy, in Patients with Advanced Esophageal, Esophagogastric Junction, or Gastric Cancer. J. Clin. Oncol. 2023, 41 (Suppl. 4), 349. [Google Scholar] [CrossRef]
- Van Tine, B.A.; Ganjoo, K.N.; Blay, J.-Y.; Valverde, C.; Araujo, D.M.; Abdul Razak, A.R.; Le Cesne, A.; Schuetze, S.; Wagner, M.J.; Attia, S.; et al. The SPEARHEAD-1 Trial of Afamitresgene Autoleucel (Afami-Cel [Formerly ADP-A2M4]): Analysis of Overall Survival in Advanced Synovial Sarcoma. J. Clin. Oncol. 2023, 41 (Suppl. 16), 11563. [Google Scholar] [CrossRef]
- Wyer, J.R.; Willcox, B.E.; Gao, G.F.; Gerth, U.C.; Davis, S.J.; Bell, J.I.; van der Merwe, P.A.; Jakobsen, B.K. T Cell Receptor and Coreceptor CD8αα Bind Peptide-MHC Independently and with Distinct Kinetics. Immunity 1999, 10, 219–225. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—New Insights into Old Paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Sillito, F.; Holler, A.; Stauss, H.J. Engineering CD4+ T Cells to Enhance Cancer Immunity. Cells 2020, 9, 1721. [Google Scholar] [CrossRef] [PubMed]
- Ahrends, T.; Borst, J. The Opposing Roles of CD4+ T Cells in Anti-tumour Immunity. Immunology 2018, 154, 582–592. [Google Scholar] [CrossRef]
- Sun, J.C.; Bevan, M.J. Defective CD8 T Cell Memory Following Acute Infection without CD4 T Cell Help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef]
- Garber, K. Driving T-Cell Immunotherapy to Solid Tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Purbhoo, M.A.; Boulter, J.M.; Price, D.A.; Vuidepot, A.L.; Hourigan, C.S.; Dunbar, P.R.; Olson, K.; Dawson, S.J.; Phillips, R.E.; Jakobsen, B.K.; et al. The Human CD8 Coreceptor Effects Cytotoxic T Cell Activation and Antigen Sensitivity Primarily by Mediating Complete Phosphorylation of the T Cell Receptor Zeta Chain. J. Biol. Chem. 2001, 276, 32786–32792. [Google Scholar] [CrossRef] [PubMed]
- Hunder, N.N.; Wallen, H.; Cao, J.; Hendricks, D.W.; Reilly, J.Z.; Rodmyre, R.; Jungbluth, A.; Gnjatic, S.; Thompson, J.A.; Yee, C. Treatment of Metastatic Melanoma with Autologous CD4+ T Cells against NY-ESO-1. N. Engl. J. Med. 2008, 358, 2698–2703. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Roberts, I.M.; Thompson, J.A.; Margolin, K.A.; Bhatia, S.; Lee, S.M.; Sloan, H.L.; Lai, I.P.; Farrar, E.A.; Wagener, F.; et al. T-Cell Therapy Using Interleukin-21-Primed Cytotoxic T-Cell Lymphocytes Combined with Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression. J. Clin. Oncol. 2016, 34, 3787–3795. [Google Scholar] [CrossRef]
- Wooldridge, L.; van den Berg, H.A.; Glick, M.; Gostick, E.; Laugel, B.; Hutchinson, S.L.; Milicic, A.; Brenchley, J.M.; Douek, D.C.; Price, D.A.; et al. Interaction between the CD8 Coreceptor and Major Histocompatibility Complex Class I Stabilizes T Cell Receptor-Antigen Complexes at the Cell Surface. J. Biol. Chem. 2005, 280, 27491–27501. [Google Scholar] [CrossRef]
- Willemsen, R.A.; Sebestyén, Z.; Ronteltap, C.; Berrevoets, C.; Drexhage, J.; Debets, R. CD8 Alpha Coreceptor to Improve TCR Gene Transfer to Treat Melanoma: Down-Regulation of Tumor-Specific Production of IL-4, IL-5, and IL-10. J. Immunol. 2006, 177, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Ronteltap, C.; Heuveling, M.; Debets, R.; Bolhuis, R. Redirecting Human CD4+ T Lymphocytes to the MHC Class I-Restricted Melanoma Antigen MAGE-A1 by TCR Alphabeta Gene Transfer Requires CD8alpha. Gene Ther. 2005, 12, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.M.; Calvo, E.; Asch, A.; Butler, M.; Zugazagoitia, J.; Charlson, J.; Cervantes, A.; Tine, B.V.; Aggen, D.; Clarke, J.; et al. 1019O Clinical and Translational Data from the Phase I SURPASS Trial of ADP-A2M4CD8 T Cell Receptor (TCR) T Cell Therapy Alone or Combined with Nivolumab in Solid Tumors. Ann. Oncol. 2023, 34, S620–S621. [Google Scholar] [CrossRef]
- Adaptimmune. A Phase 2, Open-Label, Randomized, Non-Comparative Clinical Trial of ADP-A2M4CD8 Monotherapy and in Combination with Nivolumab in Subjects with Recurrent Ovarian Cancers; Clinical Trial Registration NCT05601752; 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05601752 (accessed on 12 June 2023).
- Goebeler, M.-E.; Bargou, R.C. T Cell-Engaging Therapies—BiTEs and Beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.-F.; Rivas, J.M.; Andersson, B.S. T-Cell Receptor-Based Therapy: An Innovative Therapeutic Approach for Solid Tumors. J. Hematol. Oncol. 2021, 14, 102. [Google Scholar] [CrossRef]
- Arnaud, M.; Bobisse, S.; Chiffelle, J.; Harari, A. The Promise of Personalized TCR-Based Cellular Immunotherapy for Cancer Patients. Front. Immunol. 2021, 12, 701636. [Google Scholar] [CrossRef]
- Hsiue, E.H.C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; Dinapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a Neoantigen Derived from a Common TP53 Mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of Regular Cytolytic T Cell Synapses by Bispecific Single-Chain Antibody Constructs on MHC Class I-Negative Tumor Cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martínez-Lopez, J.; Crump, M.; Thomas, D.N.; et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J. Clin. Oncol. 2021, 39, 1959–1970. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-Cell Maturation Antigen × CD3 Bispecific Antibody, in Patients with Relapsed or Refractory Multiple Myeloma (MajesTEC-1): A Multicentre, Open-Label, Single-Arm, Phase 1 Study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Mau-Sørensen, M.; Dittrich, C.; Dienstmann, R.; Lassen, U.; Büchler, W.; Martinius, H.; Tabernero, J. A Phase I Trial of Intravenous Catumaxomab: A Bispecific Monoclonal Antibody Targeting EpCAM and the T Cell Coreceptor CD3. Cancer Chemother. Pharmacol. 2015, 75, 1065–1073. [Google Scholar] [CrossRef]
- Immunocore Ltd. A Phase II Randomized, Open-Label, Multi-Center Study of the Safety and Efficacy of IMCgp100 Compared with Investigator Choice in HLA-A*0201 Positive Patients with Previously Untreated Advanced Uveal Melanoma; Clinical Trial Registration NCT03070392; 2023. Available online: https://clinicaltrials.gov/study/NCT03070392 (accessed on 31 December 2023).
- Immunocore Ltd. Phase 1/2 Study of IMC-F106C in Advance PRAME-Positive Cancers; Clinical Trial Registration NCT04262466; 2023. Available online: https://clinicaltrials.gov/study/NCT04262466 (accessed on 31 December 2023).
- Immunocore Ltd. A Phase I/II Study of IMCnyeso, HLA-A*0201-Restricted, NY-ESO-1- and LAGE-1A-Specific Soluble T Cell Receptor and Anti-CD3 Bispecific Molecule, in HLA-A*0201 Positive Patients with Advanced NY-ESO-1 and/or LAGE—1A Positive Cancer; Clinical Trial Registration NCT03515551; 2022. Available online: https://clinicaltrials.gov/study/NCT03515551 (accessed on 31 December 2023).
- Dolgin, E. First Soluble TCR Therapy Opens ‘New Universe’ of Cancer Targets. Nat. Biotechnol. 2022, 40, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Nathan, P.; Sacco, J.J.; Orloff, M.; Hernandez-Aya, L.F.; Yang, J.; Luke, J.J.; Butler, M.O.; Stanhope, S.; Collins, L.; et al. Phase I Study of Safety, Tolerability, and Efficacy of Tebentafusp Using a Step-Up Dosing Regimen and Expansion in Patients with Metastatic Uveal Melanoma. J. Clin. Oncol. 2022, 40, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-Redirected Tumor Cell Killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.; Jakobsen, B.K. ImmTACs. Oncoimmunology 2013, 2, e22891. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Vrohlings, M.; Jungmichel, S.; Tosevski, I.; Vantellini, A.; Knobel, P.; Sanchez, N.; Ross, E.; Kerckhoven, M.V.; Giacomazzi, G.; Liivrand, M.; et al. 1397 CDR404, an Antibody-Based Bispecific & Bivalent T-Cell Engager Targeted against MAGE-A4, for Squamous Non-Small Cell Lung Cancer (SQ-NSCLC). J. Immunother. Cancer 2023, 11 (Suppl. 1), 1552. [Google Scholar] [CrossRef]
- Immunocore Ltd. A Phase 1/2 First-in-Human Study of the Safety and Efficacy of IMC-C103C as Single Agent and in Combination with Atezolizumab in HLA-A*0201-Positive Patients with Advanced MAGE-A4-Positive Cancer; Clinical Trial Registration NCT03973333; 2023. Available online: https://clinicaltrials.gov/study/NCT03973333 (accessed on 31 December 2023).
- Davar, D.; Sweis, R.F.; Blumenschein, G., Jr.; Gutierrez, R.; Melero, I.; Chen, H.A.; Thistlethwaite, F.; Moore, K.N.; Segal, N.H.; Garralda, E.; et al. 91P Phase I Dose Escalation of IMC-C103C, a CD3×MAGE-A4 T-Cell Receptor (TCR) Bispecific Protein. Ann. Oncol. 2021, 32, S1411–S1413. [Google Scholar] [CrossRef]
- Sweis, R.; Garralda, E.; Saavedra Santa Gadea, O.; Moore, K.N.; Davar, D.; Hamid, O.; Segal, N.H.; Evans, T.R.J.; Dar, M.; Yuan, Y.; et al. 157P Phase I Expansion of IMC-C103C, a MAGE-A4×CD3 ImmTAC Bispecific Protein, in Ovarian Carcinoma. Immuno-Oncol. Technol. 2022, 16, 100269. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M. Chimeric Antigen Receptor-Modified T Cells for the Treatment of Solid Tumors: Defining the Challenges and next Steps. Pharmacol. Ther. 2016, 166, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Lum, L.G. In Situ Immunization by Bispecific Antibody Targeted T Cell Therapy in Breast Cancer. Oncoimmunology 2015, 5, e1055061. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Norkina, O.; Lum, L.G. In Vitro Synthesis of Primary Specific Anti-Breast Cancer Antibodies by Normal Human Peripheral Blood Mononuclear Cells. Cancer Immunol. Immunother. 2011, 60, 1707–1720. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Montemurro, A.; Schuster, V.; Povlsen, H.R.; Bentzen, A.K.; Jurtz, V.; Chronister, W.D.; Crinklaw, A.; Hadrup, S.R.; Winther, O.; Peters, B.; et al. NetTCR-2.0 Enables Accurate Prediction of TCR-Peptide Binding by Using Paired TCRα and β Sequence Data. Commun. Biol. 2021, 4, 1060. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, E.; Huuhtanen, J.; Mustjoki, S.; Heinonen, M.; Lähdesmäki, H. Predicting Recognition between T Cell Receptors and Epitopes with TCRGP. PLoS Comput. Biol. 2021, 17, e1008814. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Hollenbach, J.; Shi, X.; Shi, W.; Chopek, M.; Fernández-Viña, M.A. Analysis of the Frequencies of HLA-A, B, and C Alleles and Haplotypes in the Five Major Ethnic Groups of the United States Reveals High Levels of Diversity in These Loci and Contrasting Distribution Patterns in These Populations. Hum. Immunol. 2001, 62, 1009–1030. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Kamata, T.; Tanaka, K.; Ihara, F.; Takami, M.; Suzuki, H.; Nakajima, T.; Ikeuchi, T.; Kawasaki, Y.; Hanaoka, H.; et al. Phase II Study of α-Galactosylceramide-Pulsed Antigen-Presenting Cells in Patients with Advanced or Recurrent Non-Small Cell Lung Cancer. J. Immunother. Cancer 2020, 8, e000316. [Google Scholar] [CrossRef]
- Fujii, S.; Shimizu, K. Immunotherapy with Artificial Adjuvant Vector Cells. Oncoimmunology 2013, 2, e23432. [Google Scholar] [CrossRef]
- The Immunological Basis of Endotoxin-Induced Tumor Regression. Requirement for a Pre-Existing State of Concomitant Anti-Tumor Immunity. J. Exp. Med. 1978, 148, 1560–1569. [CrossRef]
- Mechanisms of Anti-Tumor Action of Corynebacterium Parvum. I. Potentiated Tumor-Specific Immunity and Its Therapeutic Limitations. J. Exp. Med. 1981, 154, 609–620. [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and Local Immunity Following Adoptive Transfer of NY-ESO-1 SPEAR T Cells in Synovial Sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome after CD19 Chimeric Antigen Receptor–Modified T-Cell Therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.; Ghaly, R.; Shohdy, K.S.; Thistlethwaite, F. Lymphodepleting Chemotherapy Practices and Effect on Safety and Efficacy Outcomes in Patients with Solid Tumours Undergoing T Cell Receptor-Engineered T Cell (TCR-T) Therapy: A Systematic Review and Meta-Analysis. Cancer Immunol. Immunother. 2023, 72, 805–814. [Google Scholar] [CrossRef]
- Bobisse, S.; Zanovello, P.; Rosato, A. T-Cell Receptor Gene Transfer by Lentiviral Vectors in Adoptive Cell Therapy. Expert Opin. Biol. Ther. 2007, 7, 893–906. [Google Scholar] [CrossRef]
- Motozono, C.; Bridgeman, J.S.; Price, D.A.; Sewell, A.K.; Ueno, T. Clonotypically Similar Hybrid Aβ T Cell Receptors Can Exhibit Markedly Different Surface Expression, Antigen Specificity and Cross-Reactivity. Clin. Exp. Immunol. 2015, 180, 560–570. [Google Scholar] [CrossRef]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-Mediated TCR Replacement Generates Superior Anticancer Transgenic T Cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef]
- Spear, T.T.; Foley, K.C.; Garrett-Mayer, E.; Nishimura, M.I. TCR Modifications That Enhance Chain Pairing in Gene-Modified T Cells Can Augment Cross-Reactivity and Alleviate CD8 Dependence. J. Leukoc. Biol. 2018, 103, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Petrozziello, E.; Sturmheit, T.; Mondino, A. Exploiting Cytokines in Adoptive T-Cell Therapy of Cancer. Immunotherapy 2015, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive Transfer of Effector CD8+ T Cells Derived from Central Memory Cells Establishes Persistent T Cell Memory in Primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Powell, D.J.; Rosenberg, S.A.; Restifo, N.P. Adoptive Immunotherapy for Cancer: Building on Success. Nat. Rev. Immunol. 2006, 6, 383–393. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely Related T-Memory Stem Cells Correlate with In Vivo Expansion of CAR.CD19-T Cells and Are Preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical Responses with T Lymphocytes Targeting Malignancy-Associated κ Light Chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Mei, Y.; Cai, D.; Han, W. Standardizing CAR-T Therapy: Getting It Scaled Up. Biotechnol. Adv. 2019, 37, 239–245. [Google Scholar] [CrossRef]
- Foley, C.R.; Swan, S.L.; Swartz, M.A. Engineering Challenges and Opportunities in Autologous Cellular Cancer Immunotherapy. J. Immunol. 2024, 212, 188–198. [Google Scholar] [CrossRef]
- Vasileiou, S.; Lulla, P.D.; Tzannou, I.; Watanabe, A.; Kuvalekar, M.; Callejas, W.L.; Bilgi, M.; Wang, T.; Wu, M.J.; Kamble, R.; et al. T-Cell Therapy for Lymphoma Using Nonengineered Multiantigen-Targeted T Cells Is Safe and Produces Durable Clinical Effects. J. Clin. Oncol. 2021, 39, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2016, 22, 436–447. [Google Scholar] [CrossRef]
- Perez, C.; Jukica, A.; Listopad, J.J.; Anders, K.; Kühl, A.A.; Loddenkemper, C.; Blankenstein, T.; Charo, J. Permissive Expansion and Homing of Adoptively Transferred T Cells in Tumor-Bearing Hosts. Int. J. Cancer 2015, 137, 359–371. [Google Scholar] [CrossRef]
- Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunostimulatory Cytokines. Oncoimmunology 2013, 2, e24850. [Google Scholar] [CrossRef]
- Hong, D.S.; Butler, M.O.; Pachynski, R.K.; Sullivan, R.; Kebriaei, P.; Boross-Harmer, S.; Ghobadi, A.; Frigault, M.J.; Dumbrava, E.E.; Sauer, A.; et al. Phase 1 Clinical Trial Evaluating the Safety and Anti-Tumor Activity of ADP-A2M10 SPEAR T-Cells in Patients with MAGE-A10+ Head and Neck, Melanoma, or Urothelial Tumors. Front. Oncol. 2022, 12, 818679. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| NCT Number | Sponsor | Status and Study Dates | Phase | Tumor Type | Intervention | Therapy Type | Outcomes |
| NCT04044768 | Adaptimmune | Start: 8/13/19 Stop: 10/10/2021 | Phase 2 | Synovial sarcoma and myxoid/round cell liposarcoma | Genetic: afamitresgene autoleucel (previously, ADP-A2M4) | Autologous | ORR: 34% (95% CI, 20.86–49.31%), SD: 51.1% ORR in SS: 35.9% ORR in MRCLS: 25.0% PFS: 8.7 months |
| NCT03132922 | Adaptimmune | Start: 5/15/17 Stop: 12/27/2022 | Phase 1 | Urinary bladder cancer, melanoma, head and neck cancer, ovarian cancer, non-small cell lung cancer, esophageal cancer, gastric cancer, synovial sarcoma, myxoid round cell liposarcoma, and gastroesophageal junction cancer | A4c1032-T cells combined with low-dose radiation | Autologous | (No CR, all PRs in SS) PR: 25% SD: 39% PD: 17.8% NE: 17.8% |
| NCT05129280 | Hoffmann-La Roche | Terminated due to program discontinuation | Phase 1 | Solid tumors | RO7444973 | Allogeneic, bispecific T cell engager of MAGE-A4 and CD3 | Not available |
| NCT03973333 | Immunocore Ltd. | Ongoing Start: 5/17/19 | Phases 1 and 2 | Ovarian carcinoma | IMC-C103C in combination with Atezolizumab | Allogeneic, bispecific T cell engager of MAGE-A4 and CD3 | 15 pts MAGE A4 + PR: 1/15 SD: 5/15 Pretreated OC w/low MAGE-A4 |
| NCT05601752 | Adaptimmune | Ongoing Start: 06/26/23 | Phase 2 | Ovarian cancer | ADP-A2M4CD8 cells with Nivolumab | Autologous | Not available |
| NCT04752358 | Adaptimmune | Ongoing Start: 2/12/23 | Phase 2 | Esophageal cancer and esophagogastric junction cancer | ADP-A2M4CD8 | Autologous | Not available |
| NCT05642455 | Adaptimmune | Ongoing Start: 6/30/23 | Phases 1 and 2 | Synovial sarcoma, malignant peripheral nerve sheath tumor (MPNST), neuroblastoma and osteosarcoma | Afamitresgene autoleucel | Autologous | Not available |
| NCT02096614 | Mie University | Start: 04/2014 Stop: 03/2021 | Phase 1 | Solid tumors | TBI-1201 Drug: Cyclophosphamide Drug: Fludarabine | Autologous | Not available |
| NCT01694472 | Tianjin Medical University Cancer Institute and Hospital | Start: 07/2012 Stop: 12/2016 | Phase 1 | Solid tumors | MAGE-A4 TCR gene-modified T cells | Autologous | Not available |
| NCT04408898 | Adaptimmune | Withdrawn Start: 07/02/2020 Stop: 12/2021 | Phase 2 | Head and neck cancer | ADP-A2M4 in combination with pembrolizumab | Autologous | Not available |
| NCT04199559 | Henan Cancer Hospital | Start: 12/01/2019 Stop: 01/01/2022 | Phases 1 and 2 | Non-small cell lung cancer | Autologous dendritic cells pulsed with antigen peptides and nivolumab | Autologous | Not available |
| NCT01333046 | Baylor College of Medicine | Ongoing Start: 01/2012 | Phase 1 | Hodgkin and non-Hodgkin lymphoma | MultiTAA T cells and azacytidine | Autologous Expanded polyclonal T cells reactive to five TAAs: PRAME, SSX2, MAGEA4, SURVIVIN and NY-ESO-1 | Not available |
| NCT03247309 | Immatics US, Inc. | Ongoing Start: 12/19/2018 | Phase 1 | Head and neck cancers and lung cancer | IMA201 | Autologous | Not available |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandora, K.; Chandora, A.; Saeed, A.; Cavalcante, L. Adoptive T Cell Therapy Targeting MAGE-A4. Cancers 2025, 17, 413. https://doi.org/10.3390/cancers17030413
Chandora K, Chandora A, Saeed A, Cavalcante L. Adoptive T Cell Therapy Targeting MAGE-A4. Cancers. 2025; 17(3):413. https://doi.org/10.3390/cancers17030413
Chicago/Turabian StyleChandora, Kapil, Akshay Chandora, Anwaar Saeed, and Ludimila Cavalcante. 2025. "Adoptive T Cell Therapy Targeting MAGE-A4" Cancers 17, no. 3: 413. https://doi.org/10.3390/cancers17030413
APA StyleChandora, K., Chandora, A., Saeed, A., & Cavalcante, L. (2025). Adoptive T Cell Therapy Targeting MAGE-A4. Cancers, 17(3), 413. https://doi.org/10.3390/cancers17030413