
Immune Resistance in Glioblastoma: Understanding the Barriers to ICI and CAR-T Cell Therapy

 , , , , ,
, , , , ,
Simple Summary
Abstract
1. Introduction
2. PD-1/PD-L1
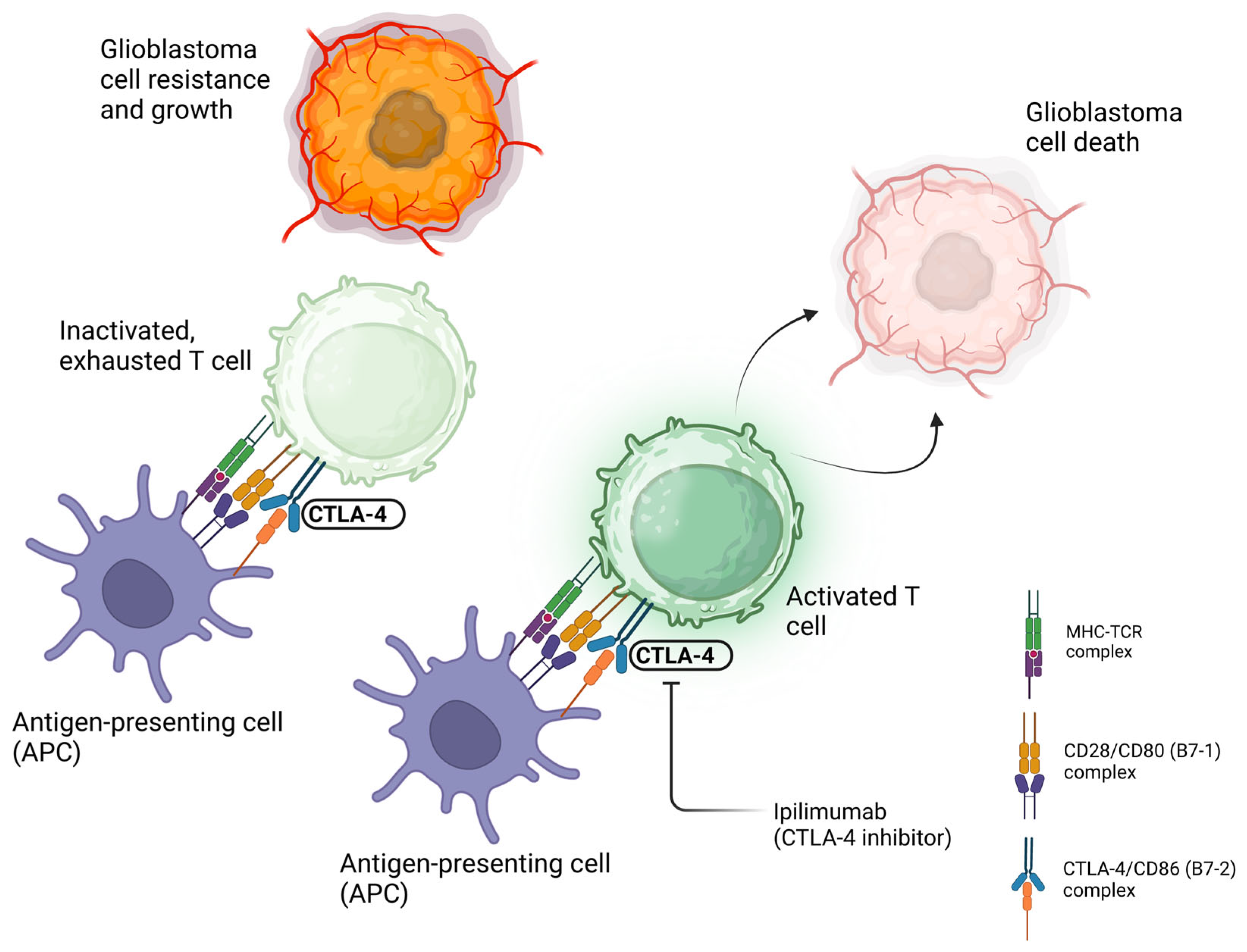
3. CTLA-4
4. Translating ICI Therapy for GBM
4.1. Single ICI Therapy
4.2. ICI as Neoadjuvant Therapy
4.3. Multiple ICI Combination Therapy
4.4. Combination with Vaccine-Based and Viral Therapies
5. CAR T-Cell Therapy in GBM
6. GBM Resistance, Immune Evasion, and Challenges of Assessing Responses
7. Conclusions and Future Directions—The Myeloid-Derived Compartment
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandrasekar, G.; Bansal, V.S.; Panigrahi, M.; Kitambi, S.S. An overview of targets and therapies for glioblastoma multiforme. J. Cancer Res. Ther. 2022, 18, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef]
- Agosti, E.; Zeppieri, M.; De Maria, L.; Tedeschi, C.; Fontanella, M.M.; Panciani, P.P.; Ius, T. Glioblastoma Immunotherapy: A Systematic Review of the Present Strategies and Prospects for Advancements. Int. J. Mol. Sci. 2023, 24, 15037. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Mende, A.L.; Schulte, J.D.; Okada, H.; Clarke, J.L. Current Advances in Immunotherapy for Glioblastoma. Curr. Oncol. Rep. 2021, 23, 21. [Google Scholar] [CrossRef]
- Rocha Pinheiro, S.L.; Lemos, F.F.B.; Marques, H.S.; Silva Luz, M.; de Oliveira Silva, L.G.; Faria Souza Mendes Dos Santos, C.; da Costa Evangelista, K.; Calmon, M.S.; Sande Loureiro, M.; Freire de Melo, F. Immunotherapy in glioblastoma treatment: Current state and future prospects. World J. Clin. Oncol. 2023, 14, 138–159. [Google Scholar] [CrossRef]
- Butler, M.; Prasad, S.; Srivastava, S.K. Targeting Glioblastoma Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1296, 1–9. [Google Scholar] [CrossRef]
- Olivet, M.M.; Brown, M.C.; Reitman, Z.J.; Ashley, D.M.; Grant, G.A.; Yang, Y.; Markert, J.M. Clinical Applications of Immunotherapy for Recurrent Glioblastoma in Adults. Cancers 2023, 15, 3901. [Google Scholar] [CrossRef]
- Ghosh, M.K.; Kumar, S.; Begam, S.; Ghosh, S.; Basu, M. GBM immunotherapy: Exploring molecular and clinical frontiers. Life Sci. 2024, 356, 123018. [Google Scholar] [CrossRef]
- Aghajani, M.; Jalilzadeh, N.; Aghebati-Maleki, A.; Yari, A.; Tabnak, P.; Mardi, A.; Saeedi, H.; Aghebati-Maleki, L.; Baradaran, B. Current approaches in glioblastoma multiforme immunotherapy. Clin. Transl. Oncol. 2024, 26, 1584–1612. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). A Phase II Study of Checkpoint Blockade Immunotherapy in Patients with Somatically Hypermutated Recurrent WHO Grade 4 Glioma. clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04145115 (accessed on 14 October 2024).
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Luksik, A.S.; Yazigi, E.; Shah, P.; Jackson, C.M. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers 2023, 15, 1414. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, F.; Ali, H.; Lathia, J.D.; Chen, P. Immunotherapy for glioblastoma: Current state, challenges, and future perspectives. Cell Mol. Immunol. 2024, 21, 1354–1375. [Google Scholar] [CrossRef]
- Yasinjan, F.; Xing, Y.; Geng, H.; Guo, R.; Yang, L.; Liu, Z.; Wang, H. Immunotherapy: A promising approach for glioma treatment. Front. Immunol 2023, 14, 1255611. [Google Scholar] [CrossRef]
- Waibl Polania, J.; Hoyt-Miggelbrink, A.; Tomaszewski, W.H.; Wachsmuth, L.P.; Lorrey, S.J.; Wilkinson, D.S.; Lerner, E.; Woroniecka, K.; Finlay, J.B.; Ayasoufi, K.; et al. Antigen presentation by tumor-associated macrophages drives T cells from a progenitor exhaustion state to terminal exhaustion. Immunity 2025, 58, 232–246.e6. [Google Scholar] [CrossRef]
- Tang, F.; Wang, Y.; Zeng, Y.; Xiao, A.; Tong, A.; Xu, J. Tumor-associated macrophage-related strategies for glioma immunotherapy. NPJ Precis. Onc. 2023, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signaling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Kuol, N.; Stojanovska, L.; Nurgali, K.; Apostolopoulos, V. PD-1/PD-L1 in Disease. Immunotherapy 2018, 10, 149–160. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef]
- Scheffel, T.B.; Grave, N.; Vargas, P.; Diz, F.M.; Rockenbach, L.; Morrone, F.B. Immunosuppression in Gliomas via PD-1/PD-L1 Axis and Adenosine Pathway. Front. Oncol. 2020, 10, 617385. [Google Scholar] [CrossRef]
- Hu, M.; Li, Y.; Lu, Y.; Wang, M.; Li, Y.; Wang, C.; Li, Q.; Zhao, H. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ 2021, 9, e11306. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.K.A.; et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020, 22, 539–549. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). A Randomized Phase II/III Open-Label Study of Ipilimumab and Nivolumab Versus Temozolomide in Patients with Newly Diagnosed MGMT (Tumor O-6-Methylguanine DNA Methyltransferase) Unmethylated Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04396860 (accessed on 14 October 2024).
- Northwestern University. A Phase, I.I.; Open Label, Clinical Trial of Pre-Surgical and Adjuvant Treatment of Recurrent Malignant Glioma with Tremelimumab and Durvalumab (MEDI4736) Alone and in Combination to Determine Immunologic Changes from Treatment. Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/study/NCT02794883 (accessed on 15 January 2025).
- Alaunos Therapeutics. Protocol ATI001-102 Substudy: Evaluation of Ad-RTS-hIL-12 + Veledimex in Combination with Nivolumab in Subjects with Recurrent or Progressive Glioblastoma. Clinicaltrials.gov. 2021. Available online: https://clinicaltrials.gov/study/NCT03636477 (accessed on 15 January 2025).
- Reardon, D. Phase II Trial of Pembrolizumab and Reirradiation in Bevacizumab Naïve and Bevacizumab Resistant Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT03661723 (accessed on 15 January 2025).
- Reardon, D. Phase II Study of Pembrolizumab (MK-3475) with and Without Bevacizumab for Recurrent Glioblastoma. Clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/study/NCT02337491 (accessed on 15 January 2025).
- NYU Langone Health. A Phase II Open-Label, Single Arm Trial of Nivolumab, Ipilimumab, and Short-Course Radiotherapy in Adults with Newly Diagnosed, MGMT Unmethylated. GlioblastomaClinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/study/NCT03367715 (accessed on 15 January 2025).
- Patil, C.G. Phase Ib/II Trial of Anti-PD-1 Immunotherapy and Stereotactic Radiation in Patients with Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04977375 (accessed on 15 January 2025).
- Istari Oncology, Inc. A Phase 2, Open-Label, Single-Arm Study Evaluating the Efficacy, Safety and Tolerability of Lerapolturev (PVSRIPO) and the Immune Checkpoint Inhibitor Pembrolizumab in the Treatment of Patients with Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04479241 (accessed on 16 January 2025).
- Peereboom, D. Phase II Study of Pembrolizumab Plus SurVaxM for Glioblastoma at First Recurrence. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04013672 (accessed on 16 January 2025).
- Fu, M.; Zhou, Z.; Huang, X.; Chen, Z.; Zhang, L.; Zhang, J.; Hua, W.; Mao, Y. Use of Bevacizumab in recurrent glioblastoma: A scoping review and evidence map. BMC Cancer 2023, 23, 544. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Fisher, J.L.; Ernstoff, M.S.; Fadul, C.E. Vaccine-based immunotherapy for glioblastoma. CNS Oncol. 2013, 2, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Webb, M.J.; Parney, I.F.; Sener, U.T. The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma. Pharmaceutics. 2023, 15, 1134. [Google Scholar] [CrossRef]
- Asija, S.; Chatterjee, A.; Goda, J.S.; Yadav, S.; Chekuri, G.; Purwar, R. Oncolytic immunovirotherapy for high-grade gliomas: A novel and an evolving therapeutic option. Front. Immunol. 2023, 14, 1118246. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Yu, C.; Hsieh, K.; Cherry, D.R.; Nehlsen, A.D.; Resende Salgado, L.; Lazarev, S.; Sindhu, K.K. Immune Escape in Glioblastoma: Mechanisms of Action and Implications for Immune Checkpoint Inhibitors and CAR T-Cell Therapy. Biology 2023, 12, 1528. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Agosti, E.; Garaba, A.; Antonietti, S.; Ius, T.; Fontanella, M.M.; Zeppieri, M.; Panciani, P.P. CAR-T Cells Therapy in Glioblastoma: A Systematic Review on Molecular Targets and Treatment Strategies. Int. J. Mol. Sci. 2024, 25, 7174. [Google Scholar] [CrossRef]
- Shanghai Simnova Biotechnology Co. Ltd. A Phase I Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Antitumor Activity of SNC-109 CAR-T Cell Therapy in Subjects with Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05868083 (accessed on 10 December 2024).
- Chimeric Therapeutics. A Phase 1b Study to Evaluate CHM-1101, a CAR T-Cell Therapy with a Chlorotoxin Tumor-Targeting Domain for Patients with Matrix Metallopeptidase 2 Positive (MMP2+) Recurrent or Progressive Glioblastoma Multiforme. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05627323 (accessed on 10 December 2024).
- Zhang, Y. An Open, Single-Arm, Phase 1 Study to Evaluate the SafetyPreliminary Effectiveness and Determine the Maximal Tolerated Dose of B7-H3-Targeting CAR-T Cell Therapy in Treating Recurrent Glioblastomas. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05241392 (accessed on 10 December 2024).
- Beijing Immunochina Medical Science & Technology Co. Ltd. Clinical Study Evaluating the Safety and Efficacy of WL276 CAR-T Cell Therapy in CD276 Positive Recurrent or Progressive Glioblastoma Patients. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT06691308 (accessed on 10 December 2024).
- Zhang, W. Phase 1 Study of Autologous Tris-CAR-T Cell Locoregional Immunotherapy for Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05577091 (accessed on 10 December 2024).
- University of Florida. Phase I Study—To Assess Safety and Feasibility of IL-8 Receptor Modified Patient-Derived Activated CD70 CAR T Cell Therapy in CD70+ Adult GBM. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05353530 (accessed on 10 December 2024).
- UNC Lineberger Comprehensive Cancer Center. Phase I Study of Intraventricular Infusion of T Cells Expressing B7-H3 Specific Chimeric Antigen Receptors (CAR) in Subjects with Recurrent or Refractory Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05366179 (accessed on 10 December 2024).
- Second Affiliated Hospital, School of Medicine, Zhejiang University. A Pilot Study of Chimeric Antigen Receptor (CAR) T Cells Targeting B7-H3 Antigen in Treating Patients with Recurrent and Refractory Glioblastoma. Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/study/NCT04385173 (accessed on 10 December 2024).
- Shanghai Simnova Biotechnology Co. Ltd. A Phase I Study to Evaluate the Safety, Tolerability and Pharmacokinetics of SNC109 in Patients with Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT06616727 (accessed on 10 December 2024).
- Second Affiliated Hospital, School of Medicine, Zhejiang University. B7-H3-Targeted Chimeric Antigen Receptor (CAR) T Cells in Treating Patients with Recurrent or Refractory Glioblastoma. Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/study/NCT04077866 (accessed on 10 December 2024).
- Chang, L.J. Immunogene-Modified Antigen-Specific T (IgT) Cells for the Treatment of Glioblastoma Multiforme. Clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT03170141 (accessed on 10 December 2024).
- Maus, M.V. INCIPIENT: Intraventricular CARv3-TEAM-E T Cells for Patients with GBM. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT05660369 (accessed on 10 December 2024).
- Okada, H. Phase 1 Study of Autologous Anti-EGFRvIII synNotch Receptor Induced Anti-EphA2/IL-13RAlpha2 CAR (E-SYNC) T Cells in Adult Participants with EGFRvIII+ Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT06186401 (accessed on 10 December 2024).
- City of Hope Medical Center. A Phase 1 Study to Evaluate Chimeric Antigen Receptor (CAR) T Cells with a Chlorotoxin Tumor-Targeting Domain for Patients with MMP2+ Recurrent or Progressive Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04214392 (accessed on 10 December 2024).
- Tcelltech Inc. A Phase, I.; Open-Label, Single/Multiple Dose, Dose-Escalation Study to Evaluate the Safety, Tolerability and Antitumor Activity of TX103 CAR-T Cell Injection (TX103) in Subjects with Recurrent or Progressive Grade 4 Glioma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT06482905 (accessed on 10 December 2024).
- Yang, X. An Open Clinical Study to Evaluate the Safety, Tolerance and Initial Efficacy of Epidermal Growth Factor Receptor Variant III Chimeric Antigen Receptor T(EGFRvIII CAR-T) in the Treatment of Recurrent Glioblastoma. Clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT05802693 (accessed on 10 December 2024).
- Xijing Hospital. A Clinical Study to Investigate the Safety, Tolerance and Efficacy Evaluation of Single-Centre, Open-Label of Local Treatment of CD147-CART in Recurrent Glioblastoma. Clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/study/NCT04045847 (accessed on 10 December 2024).
- City of Hope Medical Center. A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells Combined with Checkpoint Inhibition for Patients with Resectable Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04003649 (accessed on 19 October 2024).
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. New Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef]
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.P.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G.; et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: A phase 1 trial. Nat. Cancer 2024, 5, 517–531. [Google Scholar] [CrossRef]
- Bryan, D.C.; Elizabeth, R.G.; Matthew, J.F.; Mark, B.L.; Christopher, W.M.; Leonora, B.; Sarah, N.; Bob, S.C.; William, T.C.; Kathleen, G.; et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef]
- Yalamarty, S.S.K.; Filipczak, N.; Li, X.; Subhan, M.A.; Parveen, F.; Ataide, J.A.; Rajmalani, B.A.; Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options for Glioblastoma Multiforme (GBM). Cancers 2023, 15, 2116. [Google Scholar] [CrossRef]
- Kang, K.; Xie, F.; Wu, Y.; Wang, Z.; Wang, L.; Long, J.; Lian, X.; Zhang, F. Comprehensive exploration of tumor mutational burden and immune infiltration in diffuse glioma. Int. Immunopharmacol. 2021, 96, 107610. [Google Scholar] [CrossRef]
- Merchant, M.; Ranjan, A.; Pang, Y.; Yu, G.; Kim, O.; Khan, J.; Wu, J. Tumor mutational burden and immunotherapy in gliomas. Trends Cancer 2021, 7, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Chuntova, P.; Chow, F.; Watchmaker, P.B.; Galvez, M.; Heimberger, A.B.; Newell, E.W.; Diaz, A.; DePinho, R.A.; Li, M.O.; Wherry, E.J.; et al. Unique challenges for glioblastoma immunotherapy-discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol. 2021, 23, 356–375. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Okada, H.; Weller, M.; Huang, R.; Finocchiaro, G.; Gilbert, M.R.; Wick, W.; Ellingson, B.M.; Hashimoto, N.; Pollack, I.F.; Brandes, A.A.; et al. Immunotherapy Response Assessment in Neuro-Oncology (iRANO): A Report of the RANO Working Group. Lancet Oncol. 2015, 16, e534–e542. [Google Scholar] [CrossRef]
- Babaei Rikan, S.; Sorayaie Azar, A.; Naemi, A.; Bagherzadeh Mohasefi, J.; Pirnejad, H.; Wiil, U.K. Survival prediction of glioblastoma patients using modern deep learning and machine learning techniques. Sci. Rep. 2024, 14, 2371. [Google Scholar] [CrossRef]
- Moassefi, M.; Faghani, S.; Conte, G.M.; Kowalchuk, R.O.; Vahdati, S.; Crompton, D.J.; Perez-Vega, C.; Cabreja, R.A.D.; Vora, S.A.; Quiñones-Hinojosa, A.; et al. A deep learning model for discriminating true progression from pseudoprogression in glioblastoma patients. J. Neurooncol. 2022, 159, 447–455. [Google Scholar] [CrossRef]
- Larkin, C.J.; Arrieta, V.A.; Najem, H.; Li, G.; Zhang, P.; Miska, J.; Chen, P.; James, C.D.; Sonabend, A.M.; Heimberger, A.B. Myeloid Cell Classification and Therapeutic Opportunities within the Glioblastoma Tumor Microenvironment in the Single Cell-Omics Era. Front. Immunol. 2022, 13, 907605. [Google Scholar] [CrossRef]
- Lin, Y.J.; Wu, C.Y.J.; Wu, J.Y.; Lim, M. The Role of Myeloid Cells in GBM Immunosuppression. Front. Immunol. 2022, 13, 887781. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| ID | Duration | Authors | Phase | Enrollment | Target | Diagnosis | Treatment Arm | Control Arm | Outcomes | Recruitment Status |
|---|---|---|---|---|---|---|---|---|---|---|
| NCT02017717 (CheckMate 143) [32] | 2014–2024 | Reardon et al. | III | 529 | PD-1 | Recurrent GBM (rGBM) | Nivolumab (anti-PD-1) | Bevacizumab (anti-VEGF) | No significant difference in overall survival (OS) | Completed |
| NCT02617589 (CheckMate 498) [33] | 2016–2022 | Omuro et al. | III | 560 | PD-1 | Newly diagnosed GBM (nGBM) | Nivolumab + RT | RT + TMZ | No improvement in median overall survival (mOS) and progression-free survival (PFS) | Completed |
| NCT02667587 (CheckMate 548) [14] | 2016–2024 | Lim et al. | III | 716 | PD-1 | nGBM | Nivolumab + RT + TMZ | RT + TMZ | No improvement in mOS and median PFS (mPFS) | Completed |
| NCT02550249 [34] | 2015–2017 | Schalper et al. | II | 29 | PD-1 | nGBM + rGBM | Neoadjuvant nivolumab | Single arm | No improvement in mOS and mPFS | Completed |
| NCT02337686 [35] | 2015- | de Groot et al. | II | 18 | PD-1 | rGBM | Neoadjuvant pembrolizumab | Single arm | No improvement in mOS and mPFS | Active, not recruiting |
| NCT04396860 [36] | 2020- | Lassman et al. | II/III | 159 | PD-1 + CTLA-4 | nGBM + unmethylated MGMT | Nivolumab + ipilimumab + RT | RT + TMZ | No improvement in PFS | Active, not recruiting |
| NCT04145115 [15] | 2020- | Dunn et al. | II | 37 | PD-1 + CTLA-4 | rGBM + high mutational burden (TMB) | Nivolumab + ipilimumab | Single arm | No results posted | Suspended |
| NCT02794883 [37] | 2016–2020 | Raizer et al. | II | 36 | PD-L1 + CTLA-4 | rGBM | Durvalumab + tremelimumab | Parallel assignment | No improvement in mOS and mPFS | Completed |
| NCT03636477 [38] | 2018–2021 | Gelb et al. | I | 21 | PD-1 | rGBM | Nivolumab + Ad-RTS-hIL-12 + Veledimex | Single arm | Established safety; no improvement in OS and PFS | Completed |
| NCT03661723 [39] | 2018–2024 | Reardon et al. | II | 60 | PD-1 | rGBM | Pembrolizumab + Bevacizumab + Reirradiation | Parallel assignment | No improvement in mPFS and mOS | Completed |
| NCT02337491 [40] | 2015–2020 | Reardon et al. | II | 80 | PD-1 | rGBM | Pembrolizumab +/− Bevacizumab | Parallel assignment | No improvement in mOS and PFS at 6 months | Completed |
| NCT03367715 [41] | 2018–2022 | NYU Langone | II | 10 | PD-1 + CTLA-4 | MGMT unmethylated nGBM | Nivolumab + ipilimumab + short-course RT | Single arm | No improvement in PFS and OS | Completed |
| NCT04977375 [42] | 2021- | Patil et al. | I/II | 10 | PD-1 | rGBM | Pembrolizumab + stereotactic radiosurgery + surgical resection | Single arm | No results posted | Recruiting |
| NCT04479241 [43] | 2020–2024 | Franklin et al. | II | 25 | PD-1 | rGBM | Lerapolturev (PVSRIPO) + pembrolizumab | Single arm | No results posted | Completed |
| NCT04013672 [44] | 2020–2022 | Peereboom et al. | II | 41 | PD-1 | rGBM | Pembrolizumab + SurVaxM | Single arm | No improvement of PFS at 6 months | Completed |
| ID | Duration | Authors/Sponsor | Phase | Enrollment | Target/Therapy | Diagnosis | Outcomes | Status |
|---|---|---|---|---|---|---|---|---|
| NCT05868083 [64] | 2022–2024 | Shanghai Simnova Biotechnology | I | 16 | SNC-109 CAR-T cells | rGBM | No results posted | Recruiting |
| NCT05627323 [65] | 2023– | Litten et al. Chimeric Therapeutics | I | 42 | CHM-1101 CAR-T cells | MMP2+ rGBM or progressive GBM (pGBM) | No results posted | Active, not recruiting |
| NCT05241392 [66] | 2022– | Zhang et al. Beijing Tiantan Hospital | I | 30 | Anti-B7-H3 CAR-T cells | rGBM | No results posted | Active |
| NCT06691308 [67] | 2024– | Cheng et al. Beijing Immunochina Medical Science & Technology | I | 6 | WL276 CAR-T cells | rGBM or pGBM | No results posted | Not yet recruiting |
| NCT05577091 [68] | 2023– | Zhang et al. Beijing Tiantan Hospital | I | 10 | Tris-CAR-T cells | rGBM | No results posted | Recruiting |
| NCT05353530 [69] | 2023 | Ghiaseddin et al. University of Florida | I | 18 | IL-8 CD70 CAR-T cells | CD70+ newly diagnosed GBM (ndGBM) | No results posted | Recruiting |
| NCT05366179 [70] | 2022– | Rauf et al. UNC Lineberger Comprehensive Cancer Center | I | 36 | B7-H3 CAR-T cells | rGBM | No results posted | Recruiting |
| NCT04385173 [71] | 2022– | Zhejiang University | I | 12 | B7-H3 CAR-T cells | rGBM | No results posted | Unknown |
| NCT06616727 [72] | 2023– | Shanghai Simnova Biotechnology | I | 50 | SNC-109 CAR-T cells | rGBM | No results posted | Enrolling by invitation |
| NCT04077866 [73] | 2023– | Zhejiang University | I/II | 40 | B7-H3 CAR-T cells | rGBM | No results posted | Recruiting |
| NCT03170141 [74] | 2020– | Chang et al. Shenzhen Geno-Immune Medical Institute | I | 20 | Autologous IgT cells | rGBM | No results posted | Enrolling by invitation |
| NCT05660369 [75] | 2023– | Maus et al. | I | 26 | CARv3-TEAM-E T cells | rGBM or ndGBM | No results posted | Recruiting |
| NCT06186401 [76] | 2024– | Okada et al. University of California San Francisco | I | 20 | Anti-EphA2/IL-13alpha2 CAR (E-SYNC) T cells | EGFRvIII+ ndGBM or rGBM | No results posted | Recruiting |
| NCT04214392 [77] | 2020– | Badie et al. City of Hope Medical Center | I | 36 | CLTX-CAR T cells | MPP2+ rGBM or pGBM | No results posted | Recruiting |
| NCT06482905 [78] | 2024– | Ji et al. Tcelltech inc. | I | 52 | B7-H3 CAR-T cells | rGBM or pGBM | No results posted | Not yet recruiting |
| NCT05802693 [79] | 2023– | Yang et al. Beijing Tsinghua Chang Gung Hospital | I | 22 | EGFRvIII CAR-T cells | rGBM | No results posted | Not yet recruiting |
| NCT04045847 [80] | 2019– | Xijing Hospital | I | 31 | Anti-CD147 CAR-T cells | rGBM | No results posted | Unknown |
| NCT04003649 [81] | 2019– | Badie et al. City of Hope Medical Center | I | 60 | IL13Rα2-CAR T cells + ICIs (nivolumab and ipilimumab) | rGBM | No results posted | Recruiting |
| NCT01454596 [82] | 2012–2019 | Rosenberg et al. National Cancer Institute (NCI) | I/II | 18 | EGFRvIII CAR-T cells | rGBM | mOS 6.9 months, PFS 1.3 months | Completed |
| ID | Duration | Authors | Phase | Enrollment | Target/Therapy | Diagnosis | Outcomes | Status |
|---|---|---|---|---|---|---|---|---|
| NCT03726515 [85] | 2021–2023 | Bagley et al. | I | 7 | EGFRvIII CAR-T cells + pembrolizumab (anti-PD-1) | ndGBM | mOS 11.8 months, mPFS 5.2 months | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckert, T.; Zobaer, M.S.; Boulos, J.; Alexander-Bryant, A.; Baker, T.G.; Rivers, C.; Das, A.; Vandergrift, W.A.; Martinez, J.; Zukas, A.; et al. Immune Resistance in Glioblastoma: Understanding the Barriers to ICI and CAR-T Cell Therapy. Cancers 2025, 17, 462. https://doi.org/10.3390/cancers17030462
Eckert T, Zobaer MS, Boulos J, Alexander-Bryant A, Baker TG, Rivers C, Das A, Vandergrift WA, Martinez J, Zukas A, et al. Immune Resistance in Glioblastoma: Understanding the Barriers to ICI and CAR-T Cell Therapy. Cancers. 2025; 17(3):462. https://doi.org/10.3390/cancers17030462
Chicago/Turabian StyleEckert, Thomas, M. S. Zobaer, Jessie Boulos, Angela Alexander-Bryant, Tiffany G. Baker, Charlotte Rivers, Arabinda Das, William A. Vandergrift, Jaime Martinez, Alicia Zukas, and et al. 2025. "Immune Resistance in Glioblastoma: Understanding the Barriers to ICI and CAR-T Cell Therapy" Cancers 17, no. 3: 462. https://doi.org/10.3390/cancers17030462
APA StyleEckert, T., Zobaer, M. S., Boulos, J., Alexander-Bryant, A., Baker, T. G., Rivers, C., Das, A., Vandergrift, W. A., Martinez, J., Zukas, A., Lindhorst, S. M., Patel, S., Strickland, B., & Rowland, N. C. (2025). Immune Resistance in Glioblastoma: Understanding the Barriers to ICI and CAR-T Cell Therapy. Cancers, 17(3), 462. https://doi.org/10.3390/cancers17030462