

Exploring the Role of Epithelial–Mesenchymal Transcriptional Factors Involved in Hematological Malignancy and Solid Tumors: A Systematic Review
 , , ,
, , ,  ,
,  and
and 
Simple Summary
Abstract
1. Introduction
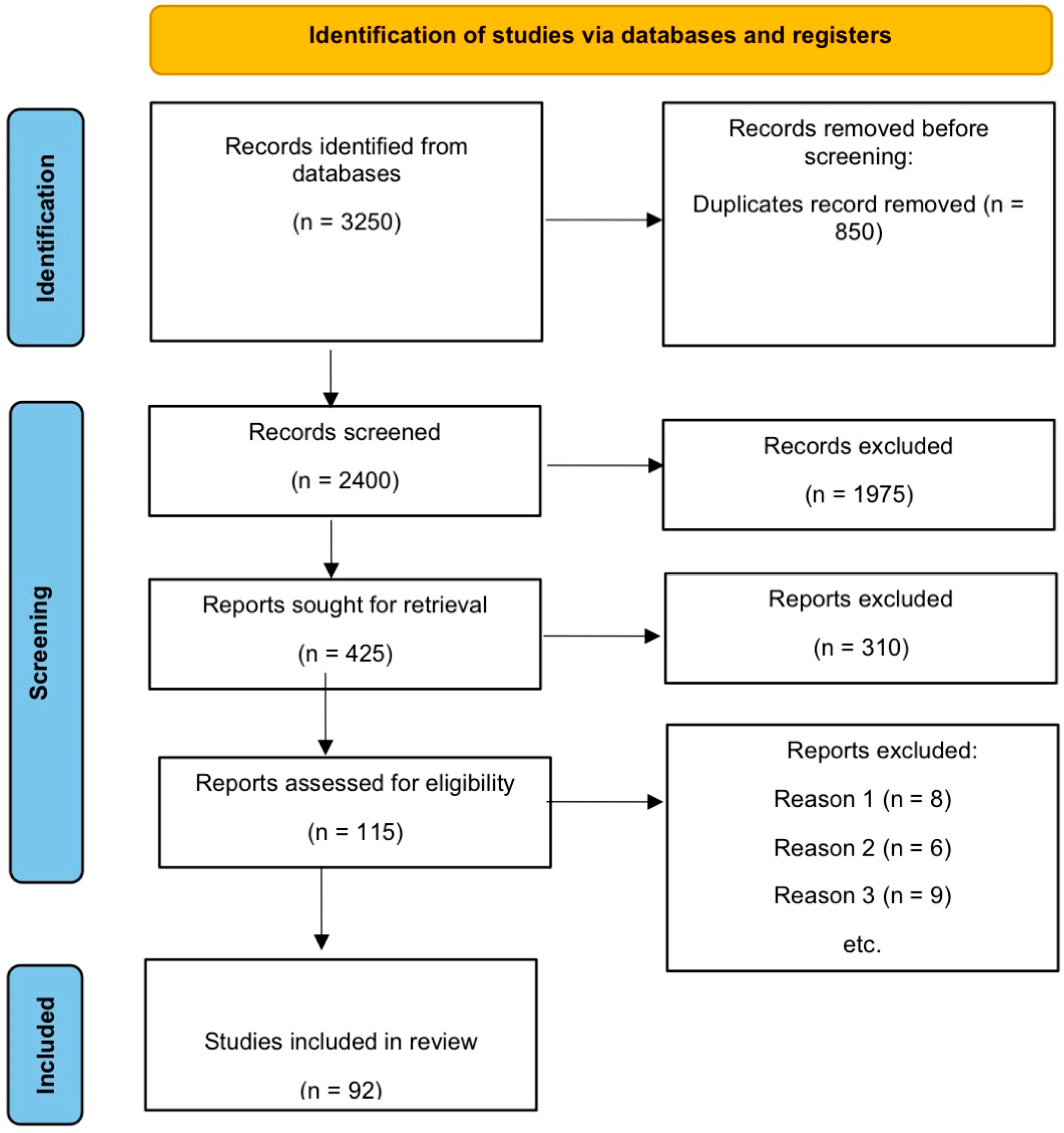
2. Materials and Methods
3. Results
3.1. Hematological Malignancy
3.1.1. ZEB1
3.1.2. ZEB2
3.1.3. TWIST1
3.1.4. TWIST2
3.1.5. SNAI1
3.1.6. SNAI2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptional Factor | Hematological Malignancy | Role/Function | Reference |
|---|---|---|---|
| ZEB1 | Acute Myeloid Leukemia (AML) | Promotes the EMT; increases the stemness and aggressiveness of the disease Regulates the self-renewal ability of leukemic stem cells; enhances the AML malignancy; drug resistance and poor prognosis | [21,29,31] |
| Mantle Cell Lymphoma (MCL) | Reduces apoptosis; enhances proliferation; chemotherapy resistance and worse overall survival | [23,32] | |
| Acute Lymphoblastic Leukemia (ALL) | Promotes stemness and therapy resistance | [33,34] | |
| ZEB2 | Acute Myeloid Leukemia (AML) | Oncogenic activity promotes leukemia stemness, proliferation, and progression | [36,37,39] |
| Acute Lymphoblastic Leukemia (ALL) | Enhances the proliferation and stemness activity of cells; promotes drug resistance | [43] | |
| TWIST1 | Myelodysplastic Syndrome (MDS) | Expression is linked to disease development, reduced apoptosis, enhance cell survival, and therapeutic resistance | [44,45,46] |
| Acute Lymphoblastic Leukemia (ALL) | Resistance to imatinib, mitoxantrone, and daunorubicin; enhances cell proliferation and increases apoptosis, which is associated with worse overall survival | [47] | |
| Chronic Myeloid Leukemia (CML) | Expression level is 100X greater than AM associated with tyrosine kinase inhibitor (TKI) | [27] | |
| Cutaneous T-cell Lymphoma (CTCL) | Expression is associated with disease progression from mycosis fungoides to Sezary syndrome | [48,49,50,51] | |
| Anaplastic Large Cell Lymphoma (ALCL) | Promotes invasiveness and chemoresistance | [26] | |
| Multiple Myeloma (MM) | Enhances invasiveness and metastasis | [28] | |
| TWIST2 | Acute Myeloid Leukemia (AML) | Tumor suppressor; reduces colony formation and cell growth | [52] |
| Acute Lymphoblastic Leukemia (ALL) | Tumor suppressor; enhances chemosensitivity and reduces proliferation | [53] | |
| SNAI1 | Acute Myeloid Leukemia (AML) | Promotes drug resistance, impaired differentiation, and self-renewal | [56] |
| SNAI2 | Acute Myeloid Leukemia (AML) | Enhances proliferation and disease progression; inhibits apoptosis, which is crucial for maintaining the LIC and LSC population | [58] |
| Chronic Myeloid Leukemia (CML) | Promotes chemotherapy resistance and cell survival | [59] |
3.2. Solid Tumors
| Solid Tumors | Role of EMT | Reference |
|---|---|---|
| Breast Cancer | NLRP3 inflammasome promotes the IL-1β level that, in turn, stimulates the EMT; as a result, this increases the tumor progression EMT-TFs increased the drug resistance via enhancing the promoter activity of ABC transporters | [66,90] |
| Ovarian Cancer | The ovaries secrete hormones and growth factors, such as activin A and TGF-β, which induce the EMT and stimulate the migration of cancer cells through the MEK/ERK and P13K/AKT pathways ET-1 activates SNAIL in Taxol- and cisplatin-resistant EOC cell models | [79,92] |
| Pancreatic Cancer | The EGF receptor (EGFR) leads to the upregulation of ZEB1; ZEB1 stimulates the ERK/MAPK signaling pathway, which further enhances the PC invasion Notch-2 activation in pancreatic cancer cells that have undergone the EMT mechanism show resistance to gemcitabine | [80,93] |
| Gastric Cancer | Helicobacter pylori (Hp) infection promotes the EMT by enhancing MMP-7 levels, which, in turn, increase the HB-EGF level. Hp cytotoxin-associated gene A (Cag A) reduces the E-cadherin expression and enhances the vimentin and TWIST1 expression in gastric cancer cell lines The EMT induces resistance against paclitaxel in gastric cancer cell lines | [82,83,95] |
| Lung Cancer | The overexpression of SLUG and SNAIL in lung cancer cell lines with epidermal growth factor receptor (EGFR) mutation induces resistance against gefitinib The PAX6-ZEB2 axis promotes cisplatin resistance and metastasis in NSCLC via the P13K/AKT signaling pathway | [96,97] |
| Hepatocellular Carcinoma | The hypoxic condition induce the EMT in HCC; as a result, invasion, metastasis, and drug resistance increases in HCC. The P13/AKT/HIF-1α pathway plays an active role in the hypoxia-induced EMT, which leads to worse treatment outcomes | [98] |
| Prostate Cancer | SNAIL promotes AR activity and leads to the resistance to AR-targeted therapies like enzalutamide. SNAIL is significantly overexpressed in metastatic tumor cells as compared to benign tumors | [99] |
| Bladder Cancer | The upregulation of TWIST and SLUG, and the downregulation of SNAIL and E-cadherin, are associated with poor survival and more aggressive phenotypes of bladder cancer | [89] |
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pedri, D.; Karras, P.; Landeloos, E.; Marine, J.C.; Rambow, F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 2022, 289, 1352–1368. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 2016, 21–45. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, L.; Liu, S.; Cao, L.L.; Wang, N.; Li, H.C.; Ji, J.F. Interpretation on the report of global cancer statistics 2022. Zhonghua Zhong Liu Za Zhi 2024, 46, 710–721. [Google Scholar] [PubMed]
- Pal, A.; Barrett, T.F.; Paolini, R.; Parikh, A.; Puram, S.V. Partial EMT in head and neck cancer biology: A spectrum instead of a switch. Oncogene 2021, 40, 5049–5065. [Google Scholar] [CrossRef]
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Br. Med. J. (Clin. Res. Ed.) 2021, 372, n71. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, F.; Zhang, P.; Wang, X.; Shen, Y.; Feng, Y.; Jia, Y.; Zhang, R.; Hu, J.; He, A. IGF-1 promotes multiple myeloma progression through PI3K/Akt-mediated epithelial-mesenchymal transition. Life Sci. 2020, 249, 117503. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Mathsyaraja, H.; Ostrowski, M.C. Setting Snail2’s pace during EMT. Nat. Cell Biol. 2012, 14, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Can. Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef]
- Lemma, S.; Karihtala, P.; Haapasaari, K.M.; Jantunen, E.; Soini, Y.; Bloigu, R.; Pasanen, A.K.; Turpeenniemi-Hujanen, T.; Kuittinen, O. Biological roles and prognostic values of the epithelial-mesenchymal transition-mediating transcription factors Twist, ZEB1 and Slug in diffuse large B-cell lymphoma. Histopathology 2013, 62, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Gyorffy, B.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Gibson, H.; Hake, T.; Geyer, S.; Frederickson, J.; Marcucci, G.; Caligiuri, M.A.; Porcu, P.; Mishra, A. Promoter-Specific Hypomethylation Is Associated with Overexpression of PLS3, GATA6, and TWIST1 in the Sezary Syndrome. J. Investig. Dermatol. 2015, 135, 2084–2092. [Google Scholar] [CrossRef]
- Mishra, A.; La Perle, K.; Kwiatkowski, S.; Sullivan, L.A.; Sams, G.H.; Johns, J.; Curphey, D.P.; Wen, J.; McConnell, K.; Qi, J.; et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer Discov. 2016, 6, 986–1005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, P.; Wu, F.; Li, M.; Sharon, D.; Ingham, R.J.; Hitt, M.; McMullen, T.P.; Lai, R. Aberrant expression of the transcriptional factor Twist1 promotes invasiveness in ALK-positive anaplastic large cell lymphoma. Cell Signal 2012, 24, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Cosset, E.; Hamdan, G.; Jeanpierre, S.; Voeltzel, T.; Sagorny, K.; Hayette, S.; Mahon, F.X.; Dumontet, C.; Puisieux, A.; Nicolini, F.E.; et al. Deregulation of TWIST-1 in the CD34+ compartment represents a novel prognostic factor in chronic myeloid leukemia. Blood 2011, 117, 1673–1676. [Google Scholar] [CrossRef]
- Cheong, C.M.; Mrozik, K.M.; Hewett, D.R.; Bell, E.; Panagopoulos, V.; Noll, J.E.; Licht, J.D.; Gronthos, S.; Zannettino, A.C.W.; Vandyke, K. Twist-1 is upregulated by NSD2 and contributes to tumour dissemination and an epithelial-mesenchymal transition-like gene expression signature in t(4;14)-positive multiple myeloma. Cancer Lett. 2020, 475, 99–108. [Google Scholar] [CrossRef]
- Li, L.; Feng, Y.; Hu, S.; Du, Y.; Xu, X.; Zhang, M.; Peng, X.; Chen, F. ZEB1 serves as an oncogene in acute myeloid leukaemia via regufrlating the PTEN/PI3K/AKT signalling pathway by combining with P53. J. Cell Mol. Med. 2021, 25, 5295–5304. [Google Scholar] [CrossRef]
- Almotiri, A.; Alzahrani, H.; Menendez-Gonzalez, J.B.; Abdelfattah, A.; Alotaibi, B.; Saleh, L.; Greene, A.; Georgiou, M.; Gibbs, A.; Alsayari, A.; et al. Zeb1 modulates hematopoietic stem cell fates required for suppressing acute myeloid leukemia. J. Clin. Investig. 2021, 131, e129115. [Google Scholar] [CrossRef]
- Bassani, B.; Simonetti, G.; Cancila, V.; Fiorino, A.; Ciciarello, M.; Piva, A.; Khorasani, A.M.; Chiodoni, C.; Lecis, D.; Gulino, A.; et al. ZEB1 shapes AML immunological niches, suppressing CD8 T cell activity while fostering Th17 cell expansion. Cell Rep. 2024, 43, 113794. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Poohadsuan, J.; Samart, P.; Kiratipaiboon, C.; Rojanasakul, Y.; Issaragrisil, S. Reactive oxygen species mediate cancer stem-like cells and determine bortezomib sensitivity via Mcl-1 and Zeb-1 in mantle cell lymphoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3739–3753. [Google Scholar] [CrossRef]
- Sun, W.; Yang, S.; Shen, W.; Li, H.; Gao, Y.; Zhu, T.H. Identification of DeltaEF1 as a novel target that is negatively regulated by LMO2 in T-cell leukemia. Eur. J. Haematol. 2010, 85, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, J.; Tian, C.; Shi, W.; Jiang, H.; Zhang, Z.; Wang, H.; Zhang, Q.; Sun, W.; Sun, P.; et al. Epigenetic dysregulation of ZEB1 is involved in LMO2-promoted T-cell acute lymphoblastic leukaemia leukaemogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2511–2525. [Google Scholar] [CrossRef] [PubMed]
- Epifanova, E.; Babaev, A.; Newman, A.G.; Tarabykin, V. Role of Zeb2/Sip1 in neuronal development. Brain Res. 2019, 1705, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Saia, M.; Termanini, A.; Rizzi, N.; Mazza, M.; Barbieri, E.; Valli, D.; Ciana, P.; Gruszka, A.M.; Alcalay, M. AML1/ETO accelerates cell migration and impairs cell-to-cell adhesion and homing of hematopoietic stem/progenitor cells. Sci. Rep. 2016, 6, 34957. [Google Scholar] [CrossRef] [PubMed]
- De Conti, G.; Gruszka, A.M.; Valli, D.; Cammarata, A.U.; Righi, M.; Mazza, M.; Pelicci, P.G. A Novel Platform to Test In Vivo Single Gene Dependencies in t(8,21) and t(15,17) AML Confirms Zeb2 as Leukemia Target. Cancers 2020, 12, 3768. [Google Scholar] [CrossRef]
- Shi, X.; Li, J.; Ma, L.; Wen, L.; Wang, Q.; Yao, H.; Ruan, C.; Wu, D.; Zhang, X.; Chen, S. Overexpression of ZEB2-AS1 lncRNA is associated with poor clinical outcomes in acute myeloid leukemia. Oncol. Lett. 2019, 17, 4935–4947. [Google Scholar] [CrossRef]
- Wang, J.; Farkas, C.; Benyoucef, A.; Carmichael, C.; Haigh, K.; Wong, N.; Huylebroeck, D.; Stemmler, M.P.; Brabletz, S.; Brabletz, T.; et al. Interplay between the EMT transcription factors ZEB1 and ZEB2 regulates hematopoietic stem and progenitor cell differentiation and hematopoietic lineage fidelity. PLoS Biol. 2021, 19, e3001394. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mar, B.G.; Zhang, H.; Puram, R.V.; Vazquez, F.; Weir, B.A.; Hahn, W.C.; Ebert, B.; Pellman, D. The EMT regulator ZEB2 is a novel dependency of human and murine acute myeloid leukemia. Blood 2017, 129, 497–508. [Google Scholar] [CrossRef]
- Goossens, S.; Radaelli, E.; Blanchet, O.; Durinck, K.; van der Meulen, J.; Peirs, S.; Taghon, T.; Tremblay, C.S.; Costa, M.; Ghahremani, M.F.; et al. ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat. Commun. 2015, 6, 5794. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Wang, J.; Tremblay, C.S.; De Medts, J.; T’Sas, S.; Nguyen, T.; Saw, J.; Haigh, K.; Curtis, D.J.; Van Vlierberghe, P.; et al. ZEB2 and LMO2 drive immature T-cell lymphoblastic leukemia via distinct oncogenic mechanisms. Haematologica 2019, 104, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Peirs, S.; Van Loocke, W.; Wang, J.; Takawy, M.; Matthijssens, F.; Sonderegger, S.E.; Haigh, K.; Nguyen, T.; Vandamme, N.; et al. Oncogenic ZEB2 activation drives sensitivity toward KDM1A inhibition in T-cell acute lymphoblastic leukemia. Blood 2017, 129, 981–990. [Google Scholar] [CrossRef]
- Li, X.; Marcondes, A.M.; Gooley, T.A.; Deeg, H.J. The helix-loop-helix transcription factor TWIST is dysregulated in myelodysplastic syndromes. Blood 2010, 116, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, F.; Chang, C.; Byon, J.; Papayannopoulou, T.; Deeg, H.J.; Marcondes, A.M. Transcriptional regulation of miR-10a/b by TWIST-1 in myelodysplastic syndromes. Haematologica 2013, 98, 414–419. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Pang, X.; Xie, C.; Deeg, J.H.; Wang, H.; Wan, T.; Wu, J.; Guan, F.; Li, X. Elevated TWIST1 expression in myelodysplastic syndromes/acute myeloid leukemia reduces efficacy of hypomethylating therapy with decitabine. Haematologica 2020, 105, e502. [Google Scholar] [PubMed]
- Wang, N.; Guo, D.; Zhao, Y.Y.; Dong, C.Y.; Liu, X.Y.; Yang, B.X.; Wang, S.; Wang, L.; Liu, Q.; Ren, Q.; et al. TWIST-1 promotes cell growth, drug resistance and progenitor clonogenic capacities in myeloid leukemia and is a novel poor prognostic factor in acute myeloid leukemia. Oncotarget 2015, 6, 20977–20992. [Google Scholar] [CrossRef]
- Goswami, M.; Duvic, M.; Dougherty, A.; Ni, X. Increased Twist expression in advanced stage of mycosis fungoides and Sezary syndrome. J. Cutan. Pathol. 2012, 39, 500–507. [Google Scholar] [CrossRef]
- Chen, S.C.; Liao, T.T.; Yang, M.H. Emerging roles of epithelial-mesenchymal transition in hematological malignancies. J. Biomed. Sci. 2018, 25, 37. [Google Scholar] [CrossRef]
- Chang, T.P.; Vancurova, I. NFκB function and regulation in cutaneous T-cell lymphoma. Am. J. Cancer Res. 2013, 3, 433–445. [Google Scholar] [PubMed]
- Michel, L.; Jean-Louis, F.; Begue, E.; Bensussan, A.; Bagot, M. Use of PLS3, twist, CD158k/KIR3DL2, and NKp46 gene expression combination for reliable Sezary syndrome diagnosis. Blood 2013, 121, 1477–1478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, W.; Cui, J.; Yao, H.; Zhou, H.; Ge, Y.; Xiao, L.; Hu, X.; Liu, B.-H.; Yang, J.; et al. Regulation of p21 by TWIST2 contributes to its tumor-suppressor function in human acute myeloid leukemia. Oncogene 2015, 34, 3000–3010. [Google Scholar] [CrossRef]
- Thathia, S.H.; Ferguson, S.; Gautrey, H.E.; van Otterdijk, S.D.; Hili, M.; Rand, V.; Moorman, A.V.; Meyer, S.; Brown, R.; Strathdee, G. Epigenetic inactivation of TWIST2 in acute lymphoblastic leukemia modulates proliferation, cell survival and chemosensitivity. Haematologica 2012, 97, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Shousha, W.G.; Ramadan, S.S.; El-Saiid, A.S.; Abdelmoneim, A.E.; Abbas, M.A. Expression and clinical significance of SNAI1 and ZEB1 genes in acute myeloid leukemia patients. Mol. Biol. Rep. 2019, 46, 4625–4630. [Google Scholar] [CrossRef]
- Gouda, M.B.Y.; Hassan, N.M.; Kandil, E.I. Bone Marrow Overexpression of SNAI1 is an early indicator of intrinsic drug resistance in patients with De Novo acute myeloid leukemia. J. Gene Med. 2022, 25, e3443. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, C.L.; Wang, J.; Nguyen, T.; Kolawole, O.; Benyoucef, A.; De Maziere, C.; Milne, A.R.; Samuel, S.; Gillinder, K.; Hediyeh-Zadeh, S.; et al. The EMT modulator SNAI1 contributes to AML pathogenesis via its interaction with LSD1. Blood 2020, 136, 957–973. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.M.; Wang, H.S.; Zhang, F.; Zhang, K.S.; Liu, Z.C.; Fang, R.; Wang, H.; Cai, S.-H.; Du, J. Histone deacetylase inhibitor induction of epithelial–mesenchymal transitions via up-regulation of Snail facilitates cancer progression. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, L.; Wu, C.; Yin, G.; Zhu, P.; Zhou, Y.; Hong, Y.; Ni, H.; Qian, Z.; Wu, W.-S. Inhibition of Slug effectively targets leukemia stem cells via the Slc13a3/ROS signaling pathway. Leukemia 2020, 34, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Petta, S.; Iacobucci, I.; Salvestrini, V.; Barbieri, E.; Santucci, M.A. Zinc-finger transcription factor slug contributes to the survival advantage of chronic myeloid leukemia cells. Cell Signal 2010, 22, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes. Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yi, M.; Xiang, B. Novel Insights on Lipid Metabolism Alterations in Drug Resistance in Cancer. Front. Cell Dev. Biol. 2022, 10, 875318. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, Y.; Dai, H.; Han, B. Epithelial-Mesenchymal Transition-Mediated Tumor Therapeutic Resistance. Molecules 2022, 27, 4750. [Google Scholar] [CrossRef] [PubMed]
- Gorgzadeh, A.; Hheidari, A.; Ghanbarikondori, P.; Arastonejad, M.; Goki, T.G.; Aria, M.; Allahyartorkaman, A.; Moazzam, F.; Velisdeh, Z.J.; Saberian, E.; et al. Investigating the properties and cytotoxicity of cisplatin-loaded nano-polybutylcyanoacrylate on breast cancer cells. Asian Pac. J. Cancer Biol. 2023, 8, 345–350. [Google Scholar] [CrossRef]
- Sun, X.; Huang, T.; Zhang, C.; Zhang, S.; Wang, Y.; Zhang, Q.; Liu, Z. Retracted article: Long non-coding RNA LINC00968 reduces cell proliferation and migration and angiogenesis in breast cancer through up-regulation of PROX1 by reducing hsa-miR-423-5p. Cell Cycle 2019, 18, 1908–1924. [Google Scholar] [CrossRef]
- Zhu, L.; Tian, Q.; Gao, H.; Wu, K.; Wang, B.; Ge, G.; Jiang, S.; Wang, K.; Zhou, C.; He, J.; et al. PROX1 promotes breast cancer invasion and metastasis through WNT/β-catenin pathway via interacting with hnRNPK. Int. J. Biol. Sci. 2022, 18, 2032. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Xu, Y.; Peng, T.; Meng, X.; Zou, F. NLRP3 induces the autocrine secretion of IL-1β to promote epithelial–mesenchymal transition and metastasis in breast cancer. Biochem. Biophys. Res. Commun. 2021, 560, 72–79. [Google Scholar] [CrossRef]
- Liu, W.; Xin, M.; Li, Q.; Sun, L.; Han, X.; Wang, J. IL-17A promotes the migration, invasion and the EMT process of lung cancer accompanied by NLRP3 activation. BioMed Res. Int. 2022, 2022, 7841279. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Su, Z.; Zhou, Z.; Wang, S.; Wang, Z.; Tong, X.; Li, C.; Wang, Y.; Chen, X.; Lei, Z.; et al. RNA demethylase ALKBH5 inhibits TGF-β-induced EMT by regulating TGF-β/SMAD signaling in non-small cell lung cancer. FASEB J. 2022, 36, e22283. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, T.W.; Ryan, C.J. Targeting the androgen receptor. Urol. Clin. N. Am. 2012, 39, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.H.; Liu, Z.C.; Zhang, G.; Wei, W.; Wang, X.X.; Wang, H.; Ke, H.P.; Zhang, F.; Wang, H.S.; Cai, S.H.; et al. Tgf-beta and egf induced hla-i downregulation is associated with epithelial-mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol. Immunol. 2015, 65, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhao, J.; Li, Y.; Li, H.; Hu, Z.; Pan, P.; Zhang, Y.; Du, E.; Liu, R.; Xu, Y. ELF5 inhibits TGF-beta-driven epithelial-mesenchymal transition in prostate cancer by repressing SMAD3 activation. Prostate 2015, 75, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Talati, P.; Vogiatzi, P.; Romero-Weaver, A.L.; Abdulghani, J.; Liao, Z.; Leiby, B.; Hoang, D.T.; Mirtti, T.; Alanen, K.; et al. Pharmacologic suppression of JAK1/2 by JAK1/2 inhibitor AZD1480 potently inhibits IL-6-induced experimental prostate cancer metastases formation. Mol. Cancer Ther. 2014, 13, 1246–1258. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and prostate cancer: The role of interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef]
- Wang, H.; Jones, J.; Turner, T.; He, Q.P.; Hardy, S.; Grizzle, W.E.; Welch, D.R.; Yates, C. Clinical and biological significance of kiss1 expression in prostate cancer. Am. J. Pathol. 2012, 180, 1170–1178. [Google Scholar] [CrossRef]
- Hou, L.; Li, Q.; Yu, Y.; Li, M.; Zhang, D. Set8 induces epithelial mesenchymal transition and enhances prostate cancer cell me- tastasis by cooperating with ZEB1. Mol. Med. Rep. 2016, 13, 1681–1688. [Google Scholar] [CrossRef]
- Raatikainen, S.; Aaltomaa, S.; Palvimo, J.J.; Karja, V.; Soini, Y. Twist overexpression predicts biochemical recurrence-free survival in prostate cancer patients treated with radical prostatectomy. Scand. J. Urol. 2015, 49, 51–57. [Google Scholar] [CrossRef]
- Ray, U.; Roy, S.S.; Chowdhury, S.R. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell. Physiol. Biochem. 2017, 41, 805. [Google Scholar] [CrossRef]
- Ha, J.H.; Ward, J.D.; Radhakrishnan, R.; Jayaraman, M.; Song, Y.S.; Dhanasekaran, D.N. Lysophosphatidic acid stimulates epi- thelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Gαi2, Src, and HIF1α signaling nexus. Oncotarget 2016, 7, 37664–37679. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Davis, D.A.; Burdette, J.E. Activin A stimulates migration of the fallopian tube epithelium, an origin of high-grade serous ovarian cancer, through non-canonical signaling. Cancer Lett. 2017, 391, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Shi, X.; Lin, Y.; Tang, J.; Jia, C.; Cao, R.; Sun, J.; Wang, G.; Zhou, L.; Dong, M. Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signalling. J. Exp. Clin. Cancer Res. 2020, 39, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Luo, Y.; Cen, Y.; Qiu, X.; Li, J.; Jie, M.; Yang, S.; Qin, S. MACC1 promotes pancreatic cancer metastasis by interacting with the EMT regulator SNAI1. Cell Death Dis. 2022, 13, 923. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Grabowska, A.M.; Clarke, P.A.; Whelband, E.; Robinson, K.; Argent, R.H.; Tobias, A.; Kumari, R.; Atherton, J.C.; Watson, S.A. Helico- bacter pylori potentiates epithelial: Mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix met- alloproteinase-7. Gut 2010, 59, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zeng, J.; Liang, X.; Wang, W.; Zhou, Y.; Sun, Y.; Liu, S.; Li, W.; Chen, C.; Jia, J. Helicobacter pylori promotes epithelial-mesenchymal transition in gastric cancer by downregulating programmed cell death protein 4 (PDCD4). PLoS ONE 2014, 9, e105306. [Google Scholar]
- Shan, T.; Cui, X.; Li, W.; Lin, W.; Li, Y.; Chen, X.; Wu, T. Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci. 2014, 105, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Li, W.; Wang, Y.; Xie, T.; Cai, Y.; Wang, Z.; Jiang, B. miR-23a inhibits E-cadherin expression and is regulated by AP-1 and NFAT4 complex during Fas-induced EMT in gastrointestinal cancer. Carcinogenesis 2014, 35, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.X.; Cai, Y.D.; Wang, Y.D.; Cui, X.B.; Xie, T.T.; Li, W.J.; Peng, L.; Zhang, Y.; Wang, Z.Q.; Wang, J.; et al. Fas signaling promotes motility and metastasis through epithelial-mesenchymal transition in gastrointestinal cancer. Oncogene 2013, 32, 1183–1192. [Google Scholar] [CrossRef]
- Zhu, M.; Yin, F.; Fan, X.; Jing, W.; Chen, R.; Liu, L.; Zhang, L.; Liu, Y.; Liang, Y.; Bu, F.; et al. Decreased TIP30 promotes Snail-mediated epithelial mesenchymal transition and tumor-initiating properties in hepatocellular carcinoma. Oncogene 2015, 34, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Kobori, L.; Fodor, D.; Jung, I. Epithelial mesenchymal and endothelial mesenchymal transitions in hepato- cellular carcinoma: A review. BioMed Res. Int. 2019, 2019, 2962580. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhang, K.; Wang, X.; Liu, X.; Zhang, Z. Expression of transcription factors snail, slug, and twist in human bladder carcinoma. J. Exp. Clin. Cancer Res. 2010, 29, 119. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Todokoro, I.; Kamioka, H.; Tomono, T.; Ogihara, T. Functional Alterations of Multidrug Resistance-Associated Proteins 2 and 5, and Breast Cancer Resistance Protein upon Snail-Induced Epithelial-Mesenchymal Transition in HCC827 Cells. Biol. Pharm. Bull. 2021, 44, 103–111. [Google Scholar] [CrossRef]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemo- resistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Elaskalani, O.; Razak NB, A.; Falasca, M.; Metharom, P. Epithelial-mesenchymal transition as a therapeutic target for overcoming chemoresistance in pancreatic cancer. World, J. Gastrointest. Oncol. 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial–mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef]
- Dhanuthai, K.; Rojanawatsirivej, S.; Thosaporn, W.; Kintarak, S.; Subarnbhesaj, A.; Darling, M.; Kryshtalskyj, E.; Chiang, C.-P.; Shin, H.-I.; Choi, S.-Y.; et al. Oral cancer: A multicenter study. Med. Oral Patol. Oral Y Cir. Bucal 2018, 23, e23. [Google Scholar] [CrossRef]
- Lee, A.F.; Chen, M.C.; Chen, C.J.; Yang, C.J.; Huang, M.S.; Liu, Y.P. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS ONE 2017, 12, e018038. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Zhang, T.; Liu, Y.B.; Deng, S.H.; Han, R.; Liu, T.; Li, J.; Xu, Y. The PAX6-ZEB2 axis promotes metastasis and cisplatin resistance in non-small cell lung cancer through PI3K/AKT signaling. Cell Death Dis. 2019, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Nan, K.J. Activation of PI3 kinase/Akt/HIF-1α pathway contributes to hypoxia-induced epithelial-mesenchy- mal transition and chemoresistance in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 461–468. [Google Scholar] [PubMed]
- Ware, K.E.; Somarelli, J.A.; Schaeffer, D.; Li, J.; Zhang, T.; Park, S.; Patierno, S.R.; Freedman, J.; Foo, W.C.; Garcia-Blanco, M.A.; et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget 2016, 7, 50507–50521. [Google Scholar] [CrossRef]
- Xie, Q.; Tang, T.; Pang, J.; Xu, J.; Yang, X.; Wang, L.; Huang, Y.; Huang, Z.; Liu, G.; Ting, D.; et al. LSD1 promotes bladder cancer progression by upregu- lating LEF1 and enhancing EMT. Front. Oncol. 2020, 10, 1234. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ang, H.L.; Moghadam, E.R.; Mohammadi, S.; Zarrin, V.; Hushmandi, K.; Samarghandian, S.; Zarrabi, A.; Najafi, M.; Mohammadinejad, R.; et al. MicroRNAs and their influence on the ZEB family: Mechanistic aspects and therapeutic applications in cancer therapy. Biomolecules 2020, 10, 1040. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Ambati, R.; Gundamaraju, R. Exploring the Crosstalk between Inflammation and Epithelial-Mesenchymal Transition in Cancer. Mediat. Inflamm. 2021, 2021, 9918379. [Google Scholar] [CrossRef]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microen- vironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Reuben, J.M.; Lee, B.N.; Li, C.; Gao, H.; Broglio, K.R.; Valero, V.; Jackson, S.A.; Ueno, N.T.; Krishnamurthy, S.; Hortobagyi, G.N.; et al. Circulating tumor cells and biomarkers: Implications for person- alized targeted treatments for metastatic breast cancer. Breast J. 2010, 16, 327–330. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Imani, S.; Hosseini Fard, H.; Cheng, J.; Wei, C.; Fu, J. Prognostic value of EMT-inducing transcription factors (EMT-TFs) in metastatic breast cancer: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 28587. [Google Scholar] [CrossRef]
- Wan, T.; Zhang, T.; Si, X.; Zhou, Y. Overexpression of EMT-inducing transcription factors as a potential poor prognostic factor for hepatocellular carcinoma in Asian populations: A meta-analysis. Oncotarget 2017, 8, 59500–59508. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.J.; Park, S.A.; Lee, S.Y.; Cha, Y.N.; Surh, Y.J. Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of Snail: A potential role of Sox9. Sci. Rep. 2017, 7, 15918. [Google Scholar] [CrossRef]
- Lin, C.W.; Wang, L.K.; Wang, S.P.; Chang, Y.L.; Wu, Y.Y.; Chen, H.Y.; Hsiao, T.H.; Lai, W.Y.; Lu, H.H.; Chang, Y.H.; et al. Daxx inhibits hypoxia-induced lung cancer cell metastasis by suppressing the HIF-1alpha/HDAC1/Slug axis. Nat. Commun. 2016, 7, 13867. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, S.; Zhang, J.; Liu, C.; Li, X.; Guo, W.; Duan, Y.; Chen, X.; Zong, S.; Zheng, J.; et al. Elucidating minimal residual disease of paediatric B-cell acute lymphoblastic leukaemia by single-cell analysis. Nat. Cell Biol. 2022, 24, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Guillaumet-Adkins, A.; Dimitrova, V.; Yun, H.; Drier, Y.; Sotudeh, N.; Rogers, A.J.; Ouseph, M.M.; Nair, M.; Potdar, S.; et al. Single-cell RNA-seq reveals developmental plasticity with coexisting oncogenic states and immune evasion programs in ETP-ALL. Blood 2021, 137, 2463–2480. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Abu-Kaoud, N.; Al Thani, H.; Rafii, A. Epithelial to mesenchymal transition in a clinical perspective. J. Oncol. 2015, 2015, 792182. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K.E.M.T. Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef] [PubMed]



| Database | Keywords |
|---|---|
| PubMed | (“Epithelial Mesenchymal Transition” [tiab] OR “EMT transcriptional factors” [tiab] OR “EMT-TFs” [tiab]) AND (“haematological malignancy” [tiab] OR “haematopoietic tumour” [tiab] OR “solid tumours” [tiab] OR “tumorigenesis” [tiab] OR “carcinogenesis” [tiab]) NOT (“review” [pt] OR “systematic review” [pt]) |
| Google Scholar | Epithelial Mesenchymal transition OR EMT OR EMT transcription factors OR EMT-TFs OR EMT and haematological malignancy OR EMT-TFs and haematological malignancy OR EMT and solid tumors OR EMT-TFs and solid tumors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanwal, R.; Esposito, J.E.; Jawed, B.; Zakir, S.K.; Pulcini, R.; Martinotti, R.; Botteghi, M.; Gaudio, F.; Martinotti, S.; Toniato, E. Exploring the Role of Epithelial–Mesenchymal Transcriptional Factors Involved in Hematological Malignancy and Solid Tumors: A Systematic Review. Cancers 2025, 17, 529. https://doi.org/10.3390/cancers17030529
Kanwal R, Esposito JE, Jawed B, Zakir SK, Pulcini R, Martinotti R, Botteghi M, Gaudio F, Martinotti S, Toniato E. Exploring the Role of Epithelial–Mesenchymal Transcriptional Factors Involved in Hematological Malignancy and Solid Tumors: A Systematic Review. Cancers. 2025; 17(3):529. https://doi.org/10.3390/cancers17030529
Chicago/Turabian StyleKanwal, Rimsha, Jessica Elisabetta Esposito, Bilal Jawed, Syed Khuram Zakir, Riccardo Pulcini, Riccardo Martinotti, Matteo Botteghi, Francesco Gaudio, Stefano Martinotti, and Elena Toniato. 2025. "Exploring the Role of Epithelial–Mesenchymal Transcriptional Factors Involved in Hematological Malignancy and Solid Tumors: A Systematic Review" Cancers 17, no. 3: 529. https://doi.org/10.3390/cancers17030529
APA StyleKanwal, R., Esposito, J. E., Jawed, B., Zakir, S. K., Pulcini, R., Martinotti, R., Botteghi, M., Gaudio, F., Martinotti, S., & Toniato, E. (2025). Exploring the Role of Epithelial–Mesenchymal Transcriptional Factors Involved in Hematological Malignancy and Solid Tumors: A Systematic Review. Cancers, 17(3), 529. https://doi.org/10.3390/cancers17030529