
The Complexity of Malignant Glioma Treatment

 , ,
, ,  , , , , ,
, , , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. The Impactful Tumor Microenvironment
2. The Influence of the GBM TME on Prognosis
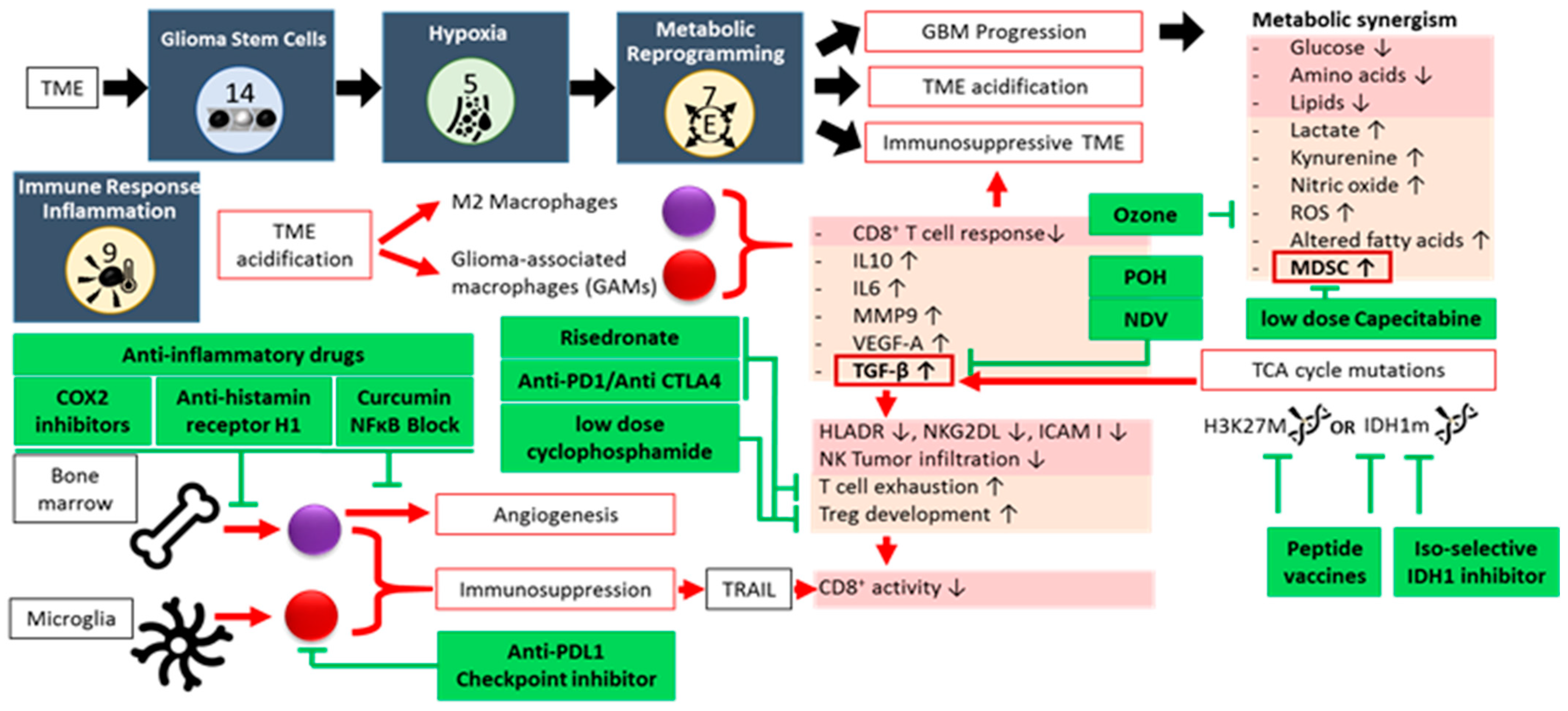
2.1. Glioma Stem-like Cells (GSCs)
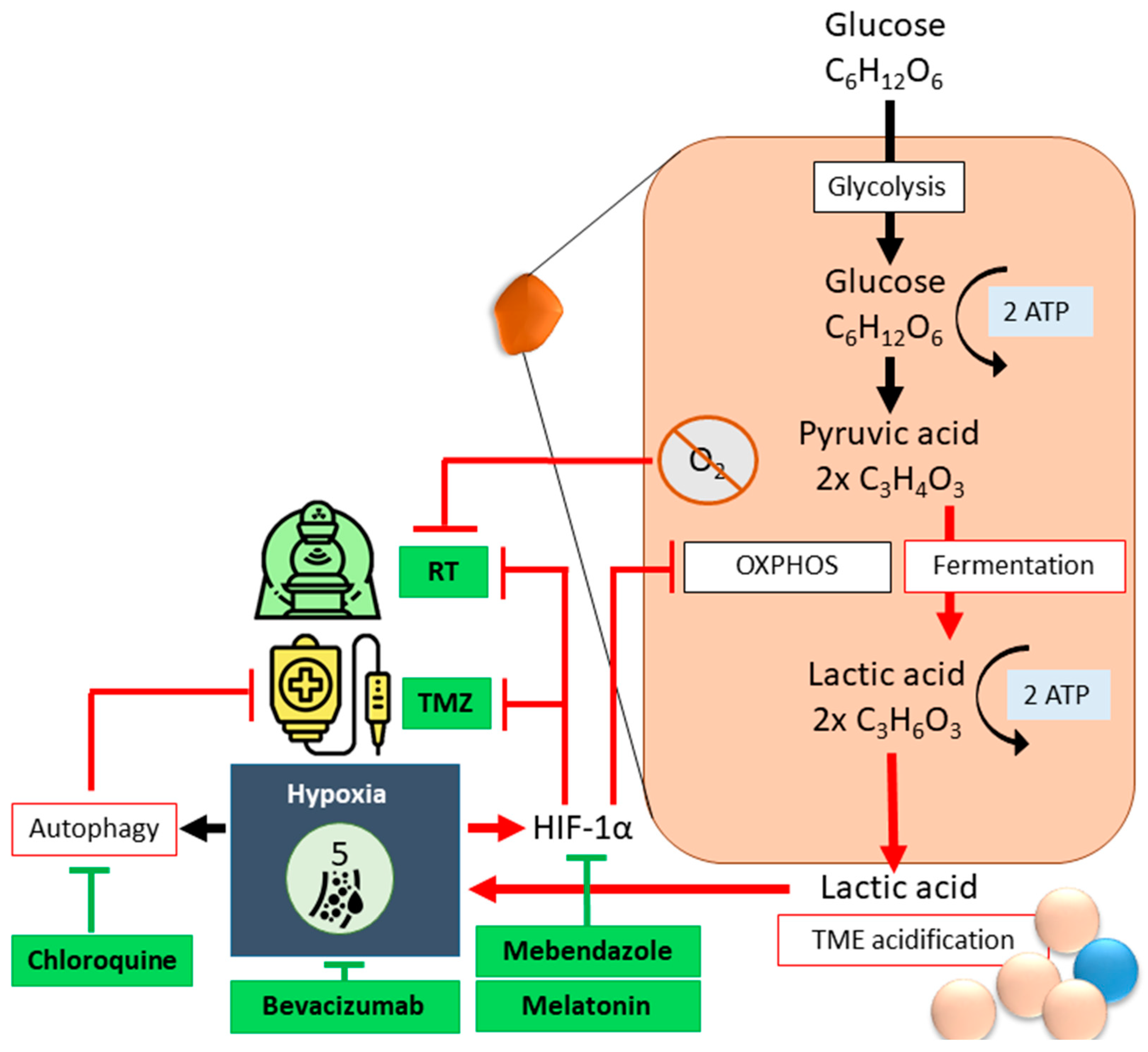
2.2. Hypoxia
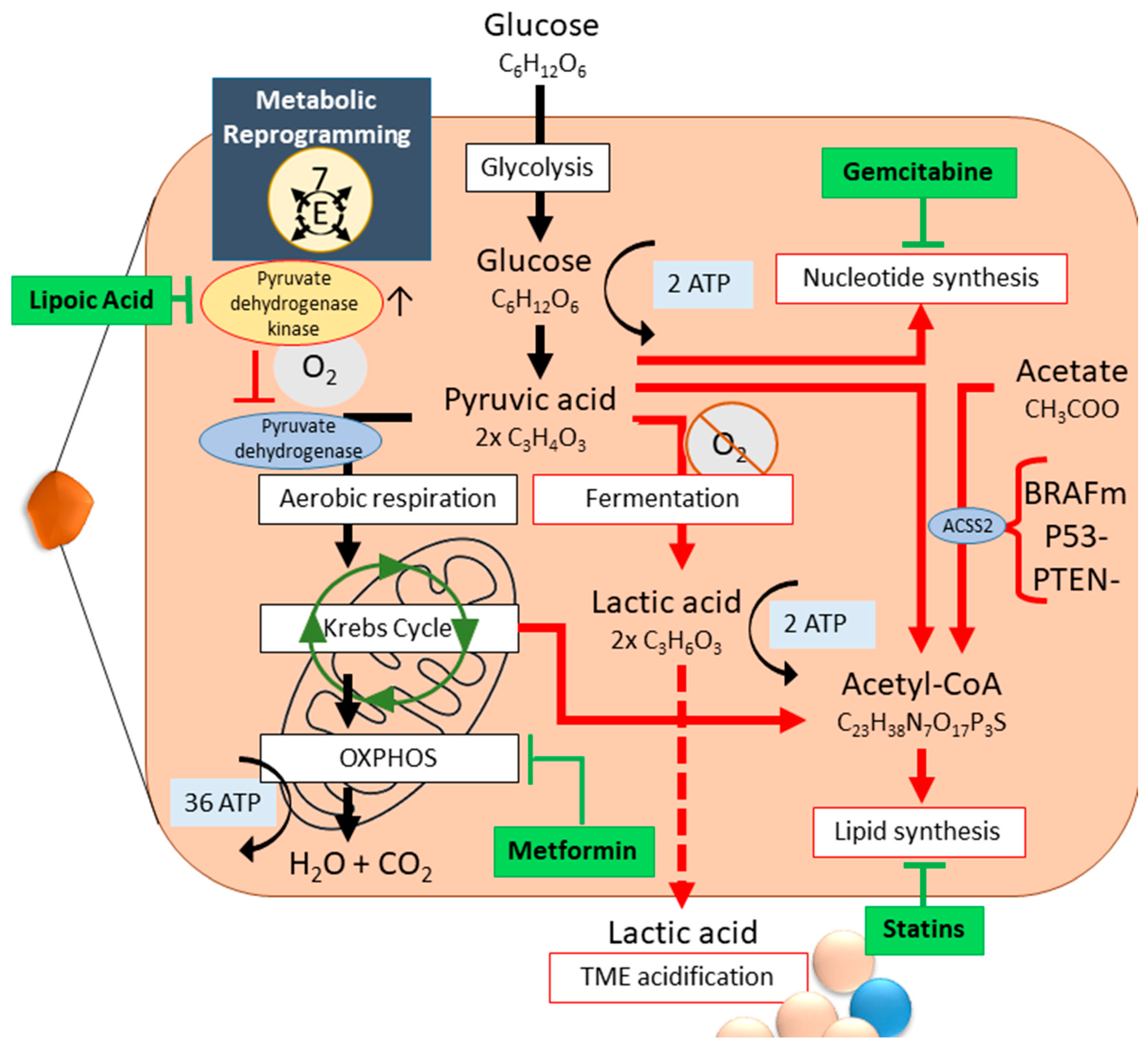
2.3. Metabolic Reprogramming
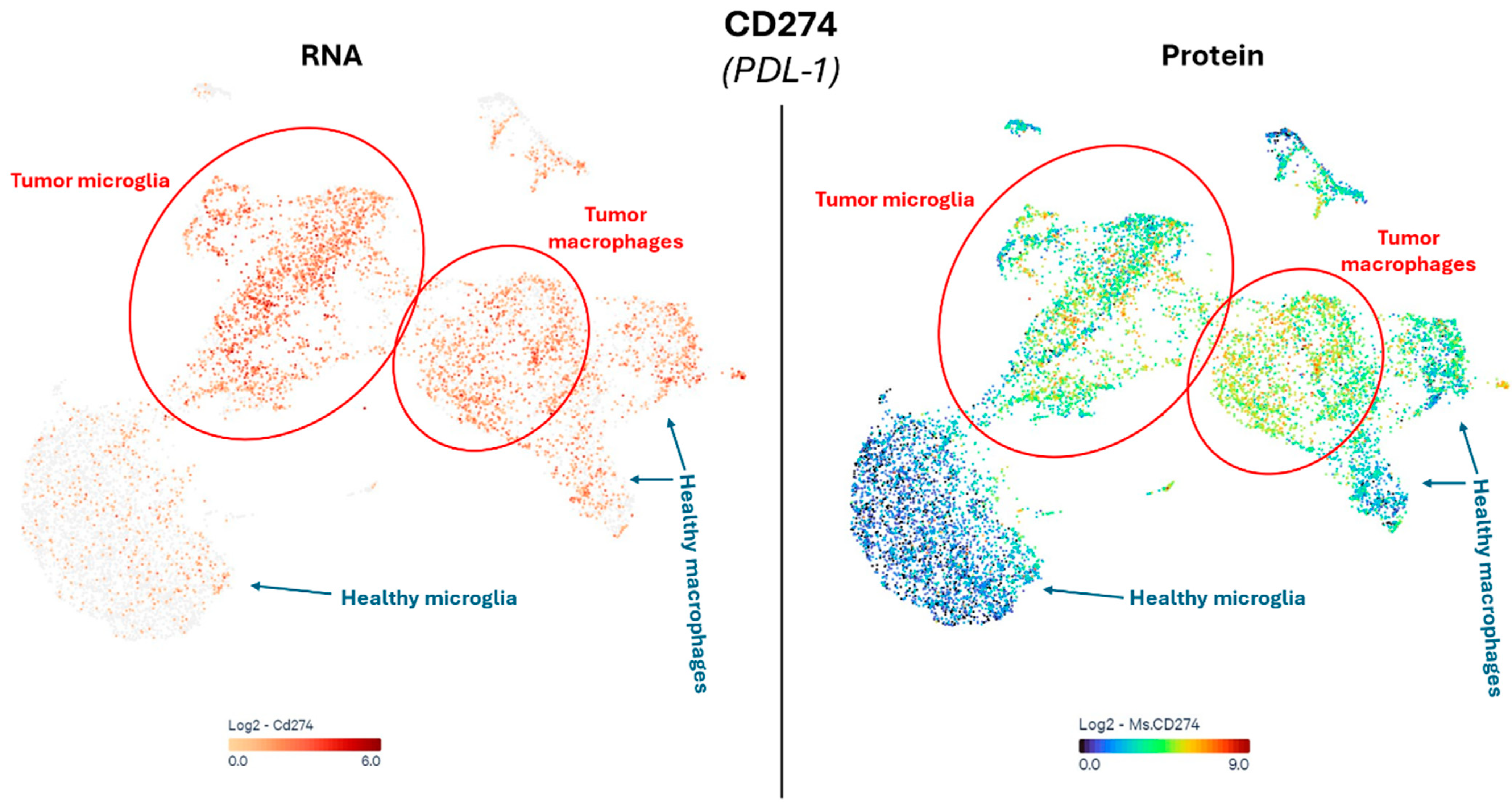
2.4. Immune Suppression and Inflammation
2.5. Neuron-Glioma Interaction
3. GBM Versus Pediatric-Type Diffuse High-Grade Gliomas
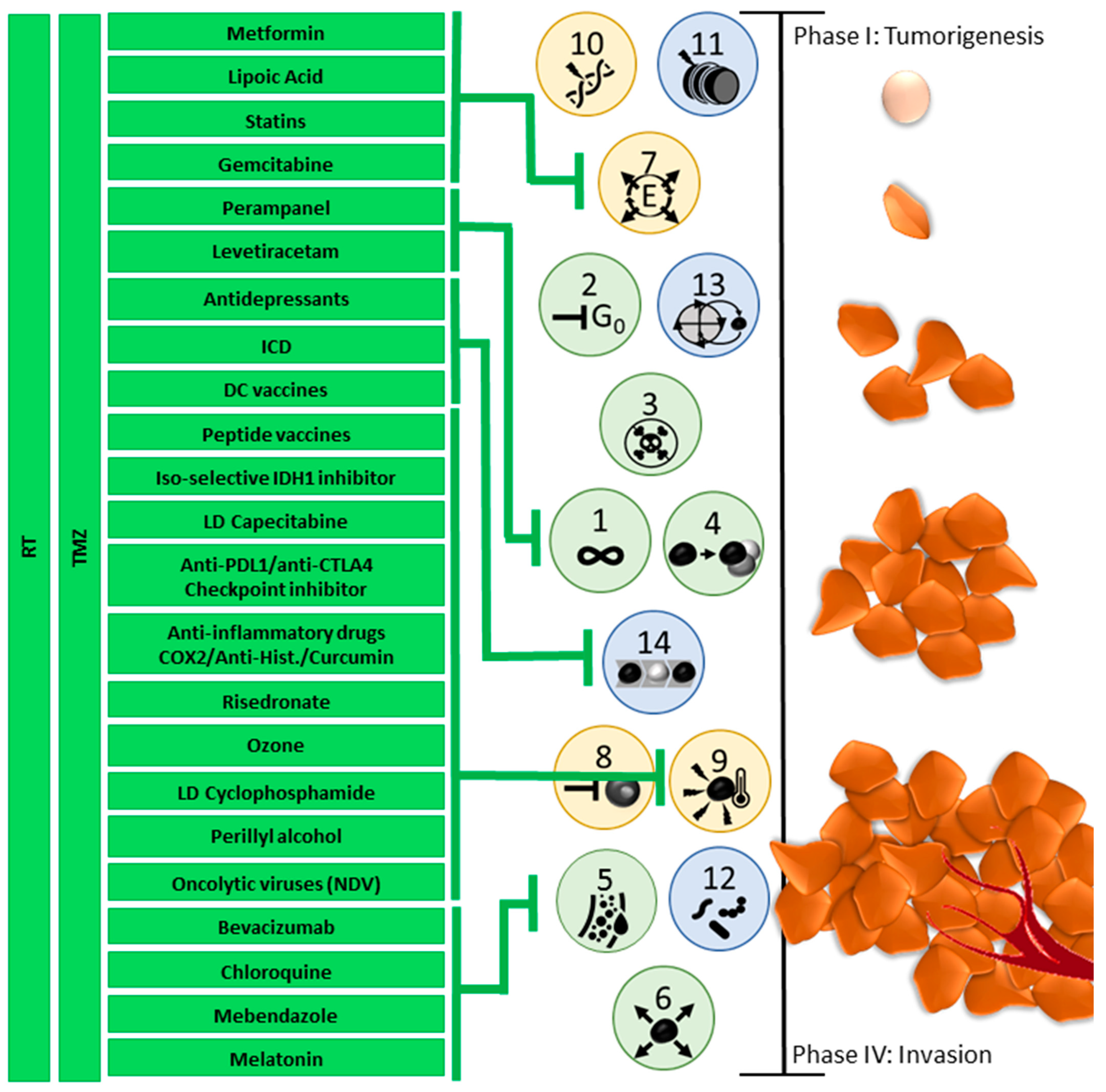
4. Need for Innovative Multiphase Individualized Combination Treatment
5. Optimizing Therapy Requires Careful Consideration and Constant Monitoring
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TME | Tumor microenvironment |
| GSCs | Glioma stem-like cells |
| CNS | Central Nervous System |
| GBM | Glioblastoma |
| Tregs | T regulatory cells |
| NK | Natural Killer cells |
| GAMs | Glioma- or Tumor-associated microglia and macrophages, also known as TAMs |
| MDSC | Myeloid-derived suppressor cells |
| TAN | Tumor-associated neutrophils |
| BBB | Blood–brain barrier |
| SoC | Standard-of-Care |
| RCT | Radiochemotherapy |
| RT | Radiotherapy |
| CTx | Chemotherapy |
| Sc-seq | Single cell sequencing |
| miRNA | Micro-RNA |
| GTMEI | GBM-associated TIME immune cell infiltration |
| WHO | World Health Organization |
| Tcm | Central memory T cells |
| Tfh | T follicular helper cells |
| TMZ | Temozolomide |
| EMT | Epithelial-to-mesenchymal transition |
| VEGF | Vascular endothelial growth factor |
| EPCs | Endothelial progenitor cells |
| HIF | Hypoxia-inducible factor |
| CSF | Cerebrospinal fluid |
| LLMs | Lipid-laden macrophages |
| HMGB | high mobility growth factor |
| PDK | pyruvate dehydrogenase kinase |
| IL | Interleukin |
| ROS | Reactive oxygen species |
| OXPHOS | Oxidative phosphorylation |
| ACSS2 | acetyl-CoA synthase |
| TGF | transforming growth factor |
| NO | Nitric oxide |
| FU | Fluorouracil |
| MMP | matrix metalloproteinases |
| CITE-seq | cellular indexing of transcriptomes and epitomes by sequencing |
| CyTOF | cytometry by time of flight |
| ER | endoplasmic reticulum |
| CPM | Cyclophosphamide |
| NDV | Newcastle Disease Virus |
| DMG | Diffuse midline gliomas |
| DIPG | diffuse intrinsic pontine glioma |
| pDHGG | diffuse pediatric-type high-grade glioma |
| H3 G34-mutant DHG | diffuse hemispheric glioma H3G34-mutant |
| H3 K27-altered DMG | diffuse midline glioma H3K27-altered |
| MRS | magnetic resonance spectroscopy |
| MRI | magnetic resonance imaging |
| DRD2 | dopamine receptor D2 |
| ClpP | mitochondrial caseinolytic protease P |
| α-KG | α-ketoglutaric acid |
| TNT | tunneling nanotubes |
| ICD | immunogenic cell death |
| mEHT | modulated electrohyperthermia |
| TTF | Tumor Treating Fields |
| DCs | Dendritic cells |
| LB | Liquid biopsy |
| EVs | extracellular vesicles |
| CTCs | circulating tumor cells |
| CAR-T cells | chimeric antigen receptor T cells |
References
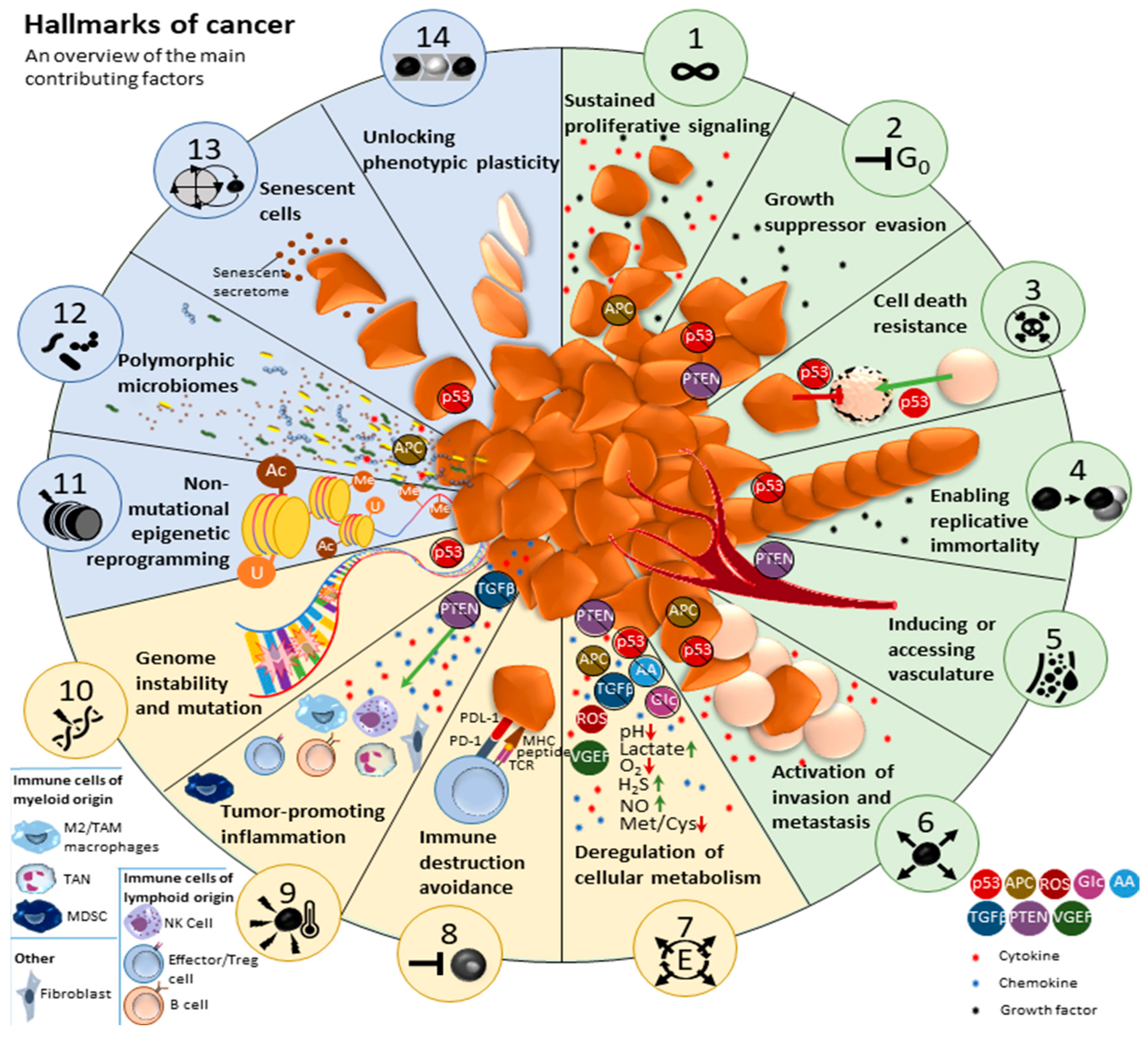
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Rouse, C.; Gittleman, H.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro-Oncology 2016, 18, 70–77. [Google Scholar] [CrossRef]
- Bilotta, M.T.; Antignani, A.; Fitzgerald, D.J. Managing the TME to improve the efficacy of cancer therapy. Front. Immunol. 2022, 13, 954992. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef]
- Suter, R.K.; Rodriguez-Blanco, J.; Ayad, N.G. Epigenetic pathways and plasticity in brain tumors. Neurobiol. Dis. 2020, 145, 105060. [Google Scholar] [CrossRef]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Schneider, M.; Giordano, F.A.; Kuner, T.; Wick, W.; Herrlinger, U.; Winkler, F. Disconnecting multicellular networks in brain tumours. Nat. Rev. Cancer 2022, 22, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Pericoli, G.; Galardi, A.; Paolini, A.; Petrilli, L.L.; Pepe, G.; Palma, A.; Colletti, M.; Ferretti, R.; Giorda, E.; Levi Mortera, S.; et al. Inhibition of exosome biogenesis affects cell motility in heterogeneous sub-populations of paediatric-type diffuse high-grade gliomas. Cell Biosci. 2023, 13, 207. [Google Scholar] [CrossRef]
- Taylor, K.R.; Barron, T.; Hui, A.; Spitzer, A.; Yalçin, B.; Ivec, A.E.; Geraghty, A.C.; Hartmann, G.G.; Arzt, M.; Gillespie, S.M.; et al. Glioma synapses recruit mechanisms of adaptive plasticity. Nature 2023, 623, 366–374. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Lerouge, L.; Ruch, A.; Pierson, J.; Thomas, N.; Barberi-Heyob, M. Non-targeted effects of radiation therapy for glioblastoma. Heliyon 2024, 10, e30813. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Fiore, S.; Sprenger, T.; Prix, L.; Schirrmacher, V.; Stuecker, W. Randomized Controlled Immunotherapy Clinical Trials for GBM Challenged. Cancers 2021, 13, 32. [Google Scholar] [CrossRef]
- Zhao, R.; Pan, Z.; Li, B.; Zhao, S.; Zhang, S.; Qi, Y.; Qiu, J.; Gao, Z.; Fan, Y.; Guo, Q.; et al. Comprehensive Analysis of the Tumor Immune Microenvironment Landscape in Glioblastoma Reveals Tumor Heterogeneity and Implications for Prognosis and Immunotherapy. Front. Immunol. 2022, 13, 820673. [Google Scholar] [CrossRef]
- Tanner, G.; Barrow, R.; Ajaib, S.; Al-Jabri, M.; Ahmed, N.; Pollock, S.; Finetti, M.; Rippaus, N.; Bruns, A.F.; Syed, K.; et al. IDHwt glioblastomas can be stratified by their transcriptional response to standard treatment, with implications for targeted therapy. Genome Biol. 2024, 25, 45. [Google Scholar] [CrossRef]
- De Simone, M.; Iaconetta, G.; Palermo, G.; Fiorindi, A.; Schaller, K.; De Maria, L. Clustering Functional Magnetic Resonance Imaging Time Series in Glioblastoma Characterization: A Review of the Evolution, Applications, and Potentials. Brain Sci. 2024, 14, 296. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Latzer, P.; Zelba, H.; Battke, F.; Reinhardt, A.; Shao, B.; Bartsch, O.; Rabsteyn, A.; Harter, J.; Schulze, M.; Okech, T.; et al. A real-world observation of patients with glioblastoma treated with a personalized peptide vaccine. Nat. Commun. 2024, 15, 6870. [Google Scholar] [CrossRef]
- Karimi-Sani, I.; Molavi, Z.; Naderi, S.; Mirmajidi, S.-H.; Zare, I.; Naeimzadeh, Y.; Mansouri, A.; Tajbakhsh, A.; Savardashtaki, A.; Sahebkar, A. Personalized mRNA vaccines in glioblastoma therapy: From rational design to clinical trials. J. Nanobiotechnol. 2024, 22, 601. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Van de Vliet, P.; Van Gool, S.; Sprenger, T.; Schirrmacher, V.; Stuecker, W. Individualized Multimodal Immunotherapy for Adults with IDH1 Wild-Type GBM: A Single Institute Experience. Cancers 2023, 15, 1194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shen, R.; Cheng, S.; Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 2019, 8, 2897–2907. [Google Scholar] [CrossRef]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.-C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Shi, T.; Zhu, J.; Zhang, X.; Mao, X. The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma. Cancers 2023, 15, 2613. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Ngo, A.T.L.; Le, H.M.; Trinh, N.T.H.; Jun, A.P.G.; Bach, T.Q.; Bui, H.T.H.; Hoang, V.T.; Bui, A.V.; Nguyen, L.T.; Hoang, D.M. Clinically relevant preservation conditions for mesenchymal stem/stromal cells derived from perinatal and adult tissue sources. J. Cell. Mol. Med. 2021, 25, 10747–10760. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef]
- Couturier, C.P.; Nadaf, J.; Li, Z.; Baig, S.; Riva, G.; Le, P.; Kloosterman, D.J.; Monlong, J.; Nkili Meyong, A.; Allache, R.; et al. Glioblastoma scRNA-seq shows treatment-induced, immune-dependent increase in mesenchymal cancer cells and structural variants in distal neural stem cells. Neuro-Oncology 2022, 24, 1494–1508. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liang, J.; Sun, H. The Network of Tumor Microtubes: An Improperly Reactivated Neural Cell Network With Stemness Feature for Resistance and Recurrence in Gliomas. Front. Oncol. 2022, 12, 921975. [Google Scholar] [CrossRef]
- Bielecka-Wajdman, A.M.; Lesiak, M.; Ludyga, T.; Sieroń, A.; Obuchowicz, E. Reversing glioma malignancy: A new look at the role of antidepressant drugs as adjuvant therapy for glioblastoma multiforme. Cancer Chemother. Pharmacol. 2017, 79, 1249–1256. [Google Scholar] [CrossRef]
- Hardee, M.E.; Dewhirst, M.W.; Agarwal, N.; Sorg, B.S. Novel Imaging Provides New Insights into Mechanisms of Oxygen Transport in Tumors. Curr. Mol. Med. 2009, 9, 435–441. [Google Scholar] [CrossRef]
- Lanzen, J.; Braun, R.D.; Klitzman, B.; Brizel, D.; Secomb, T.W.; Dewhirst, M.W. Direct Demonstration of Instabilities in Oxygen Concentrations within the Extravascular Compartment of an Experimental Tumor. Cancer Res. 2006, 66, 2219–2223. [Google Scholar] [CrossRef]
- Cárdenas-Navia, L.I.; Mace, D.; Richardson, R.A.; Wilson, D.F.; Shan, S.; Dewhirst, M.W. The Pervasive Presence of Fluctuating Oxygenation in Tumors. Cancer Res. 2008, 68, 5812–5819. [Google Scholar] [CrossRef]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta BBA—Rev. Cancer 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Lavogina, D.; Krõlov, M.K.; Vellama, H.; Modhukur, V.; Di Nisio, V.; Lust, H.; Eskla, K.-L.; Salumets, A.; Jaal, J. Inhibition of epigenetic and cell cycle-related targets in glioblastoma cell lines reveals that onametostat reduces proliferation and viability in both normoxic and hypoxic conditions. Sci. Rep. 2024, 14, 4303. [Google Scholar] [CrossRef]
- Lo Dico, A.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front. Oncol. 2018, 8, 249. [Google Scholar] [CrossRef]
- Aleyasin, H.; Karuppagounder, S.S.; Kumar, A.; Sleiman, S.; Basso, M.; Ma, T.; Siddiq, A.; Chinta, S.J.; Brochier, C.; Langley, B.; et al. Antihelminthic Benzimidazoles Are Novel HIF Activators That Prevent Oxidative Neuronal Death via Binding to Tubulin. Antioxid. Redox Signal. 2015, 22, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceño, E.; López-González, M.A. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Putta, C.L.; Eswar, K.; Rengan, A.K. Melatonin: Avenues in cancer therapy and its nanotechnological advancements. MedComm—Biomater. Appl. 2023, 2, e58. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Choi, W.S. Use of Melatonin in Cancer Treatment: Where Are We? Int. J. Mol. Sci. 2022, 23, 3779. [Google Scholar] [CrossRef]
- Doğanlar, O.; Doğanlar, Z.B.; Delen, E.; Doğan, A. The role of melatonin in angio-miR-associated inhibition of tumorigenesis and invasion in human glioblastoma tumour spheroids. Tissue Cell 2021, 73, 101617. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Kloosterman, D.J.; Erbani, J.; Boon, M.; Farber, M.; Handgraaf, S.M.; Ando-Kuri, M.; Sánchez-López, E.; Fontein, B.; Mertz, M.; Nieuwland, M.; et al. Macrophage-mediated myelin recycling fuels brain cancer malignancy. Cell 2024, 187, 5336–5356.e30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Jiang, Y.-G.; Peng, Y.; Koussougbo, K.S. Necroptosis: A novel therapeutic target for glioblastoma. Med. Hypotheses 2011, 76, 350–352. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.-Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Ahn, S.-H.; Park, H.; Ahn, Y.-H.; Kim, S.; Cho, M.-S.; Kang, J.L.; Choi, Y.-H. Necrotic cells influence migration and invasion of glioblastoma via NF-κB/AP-1-mediated IL-8 regulation. Sci. Rep. 2016, 6, 24552. [Google Scholar] [CrossRef] [PubMed]
- Di Ianni, N.; Musio, S.; Pellegatta, S. Altered Metabolism in Glioblastoma: Myeloid-Derived Suppressor Cell (MDSC) Fitness and Tumor-Infiltrating Lymphocyte (TIL) Dysfunction. Int. J. Mol. Sci. 2021, 22, 4460. [Google Scholar] [CrossRef]
- Mahalingam, D.; Goel, S.; Aparo, S.; Patel Arora, S.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160. [Google Scholar] [CrossRef]
- Scafidi, A.; Mogensen, F.L.-H.; Campus, E.; Pailas, A.; Neumann, K.; Legrave, N.; Bernardin, F.; Pereira, S.L.; Antony, P.M.A.; Nicot, N.; et al. Metformin impacts the differentiation of mouse bone marrow cells into macrophages affecting tumour immunity. Heliyon 2024, 10, e37792. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, L.; Zhang, H.; Zhang, Y.; Ju, H.; Wang, X.; Ren, H.; Zhu, X.; Dong, Y. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. Front. Immunol. 2022, 13, 1003651. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Ji, L.; Xu, D.; Wang, J.; Zou, J. TGF-β links glycolysis and immunosuppression in glioblastoma. Histol. Histopathol. 2021, 36, 1111–1124. [Google Scholar] [CrossRef]
- Poon, C.C.; Sarkar, S.; Yong, V.W.; Kelly, J.J.P. Glioblastoma-associated microglia and macrophages: Targets for therapies to improve prognosis. Brain 2017, 140, 1548–1560. [Google Scholar] [CrossRef]
- Geribaldi-Doldán, N.; Fernández-Ponce, C.; Quiroz, R.N.; Sánchez-Gomar, I.; Escorcia, L.G.; Velásquez, E.P.; Quiroz, E.N. The Role of Microglia in Glioblastoma. Front. Oncol. 2021, 10, 603495. [Google Scholar] [CrossRef] [PubMed]
- Pace, A.; Scirocchi, F.; Napoletano, C.; Zizzari, I.G.; D’Angelo, L.; Santoro, A.; Nuti, M.; Rahimi, H.; Rughetti, A. Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 6312. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef]
- Ohashi, T.; Aoki, M.; Tomita, H.; Akazawa, T.; Sato, K.; Kuze, B.; Mizuta, K.; Hara, A.; Nagaoka, H.; Inoue, N.; et al. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. 2017, 108, 1128–1134. [Google Scholar] [CrossRef]
- Leo, A.D.; Ugolini, A.; Yu, X.; Scirocchi, F.; Scocozza, D.; Peixoto, B.; Pace, A.; D’Angelo, L.; Liu, J.K.C.; Etame, A.B.; et al. Glucose-driven histone lactylation promotes the immunosuppressive activity of monocyte-derived macrophages in glioblastoma. Immunity 2024, 57, 1105–1123.e8. [Google Scholar] [CrossRef]
- Hao, Z.-N.; Tan, X.-P.; Zhang, Q.; Li, J.; Xia, R.; Ma, Z. Lactate and Lactylation: Dual Regulators of T-Cell-Mediated Tumor Immunity and Immunotherapy. Biomolecules 2024, 14, 1646. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Alban, T.J.; Grabowski, M.M.; Alvarado, A.G.; Otvos, B.; Bayik, D.; Roversi, G.; McGraw, M.; Huang, P.; Mohammadi, A.M.; et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight 2019, 4, e130748. [Google Scholar] [CrossRef] [PubMed]
- Yanchu, L.; Rong, P.; Rong, C.; Li, Z.; Xiaoyan, Y.; Feng, W. Ozone therapy for high-grade glioma: An overview. Front. Oncol. 2023, 13, 1161206. [Google Scholar] [CrossRef]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Investig. 2023, 133, e163446. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Riera-Domingo, C.; Audigé, A.; Granja, S.; Cheng, W.-C.; Ho, P.-C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, Hypoxia, and Metabolism–the Ménage à Trois of Cancer: Implications for Immunotherapy. Physiol. Rev. 2020, 100, 1–102. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Messiaen, J.; Jacobs, S.A.; De Smet, F. The tumor micro-environment in pediatric glioma: Friend or foe? Front. Immunol. 2023, 14, 1227126. [Google Scholar] [CrossRef]
- Paul, S.; Sa, G. Curcumin as an Adjuvant to Cancer Immunotherapy. Front. Oncol. 2021, 11, 675923. [Google Scholar] [CrossRef]
- Exley, M.A.; Garcia, S.; Zellander, A.; Zilberberg, J.; Andrews, D.W. Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma. J. Clin. Med. 2022, 11, 1069. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Y.; Li, Q.; Yao, J.; Yuan, X.; Zhang, Y.; Yin, X.; Saito, Y.; Fan, H.; Li, P.; et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients via activation of the macrophage histamine receptor H1. Cancer Cell 2022, 40, 36–52.e9. [Google Scholar] [CrossRef]
- du Chatinier, A.; Velilla, I.Q.; Meel, M.H.; Hoving, E.W.; Hulleman, E.; Metselaar, D.S. Microglia in pediatric brain tumors: The missing link to successful immunotherapy. Cell Rep. Med. 2023, 4, 101246. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.L.; Puigdelloses-Vallcorba, M.; Piñero, G.; Soni, N.; Thomason, W.; DeSisto, J.; Angione, A.; Tsankova, N.M.; Castro, M.G.; Schniederjan, M.; et al. Microglia and monocyte-derived macrophages drive progression of pediatric high-grade gliomas and are transcriptionally shaped by histone mutations. Immunity 2024, 57, 2669–2687.e6. [Google Scholar] [CrossRef]
- Sprooten, J.; Vanmeerbeek, I.; Datsi, A.; Govaerts, J.; Naulaerts, S.; Laureano, R.S.; Borràs, D.M.; Calvet, A.; Malviya, V.; Kuballa, M.; et al. Lymph node and tumor-associated PD-L1+ macrophages antagonize dendritic cell vaccines by suppressing CD8+ T cells. Cell Rep. Med. 2024, 5, 101377. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Jiang, P.; Zhang, P.; Mukthavaram, R.; Nomura, N.; Pingle, S.C.; Teng, D.; Chien, S.; Guo, F.; Kesari, S. Anti-cancer effects of nitrogen-containing bisphosphonates on human cancer cells. Oncotarget 2016, 7, 57932–57942. [Google Scholar] [CrossRef] [PubMed]
- Fournié, J.-J.; Sicard, H.; Poupot, M.; Bezombes, C.; Blanc, A.; Romagné, F.; Ysebaert, L.; Laurent, G. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell. Mol. Immunol. 2013, 10, 35–41. [Google Scholar] [CrossRef]
- da Fonseca, C.O.; Simão, M.; Lins, I.R.; Caetano, R.O.; Futuro, D.; Quirico-Santos, T. Efficacy of monoterpene perillyl alcohol upon survival rate of patients with recurrent glioblastoma. J. Cancer Res. Clin. Oncol. 2011, 137, 287–293. [Google Scholar] [CrossRef]
- Fonseca, C.O.D.; Teixeira, R.M.; Silva, J.C.T.; Fischer, J.D.S.D.G.; Meirelles, O.C.; Landeiro, J.A.; Quirico-Santos, T. Long-term Outcome in Patients with Recurrent Malignant Glioma Treated with Perillyl Alcohol Inhalation. Anticancer Res. 2013, 33, 5625–5631. [Google Scholar] [PubMed]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Zhong, H.; Lai, Y.; Zhang, R.; Daoud, A.; Feng, Q.; Zhou, J.; Shang, J. Low Dose Cyclophosphamide Modulates Tumor Microenvironment by TGF-β Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 957. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Font, L.; Arias-Ramos, N.; Lope-Piedrafita, S.; Julià-Sapé, M.; Pumarola, M.; Arús, C.; Candiota, A.P. Metronomic treatment in immunocompetent preclinical GL261 glioblastoma: Effects of cyclophosphamide and temozolomide. NMR Biomed. 2017, 30, e3748. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, N.; Palacín-Aliana, I.; Esteban-Rubio, S.; Madurga, R.; Rius-Rocabert, S.; Carrión-Navarro, J.; Presa, J.; Cuadrado-Castano, S.; Sánchez-Gómez, P.; García-Sastre, A.; et al. Newcastle Disease Virus (NDV) Oncolytic Activity in Human Glioma Tumors Is Dependent on CDKN2A-Type I IFN Gene Cluster Codeletion. Cells 2020, 9, 1405. [Google Scholar] [CrossRef]
- Chuntova, P.; Yamamichi, A.; Chen, T.; Narayanaswamy, R.; Ronseaux, S.; Hudson, C.; Tron, A.E.; Hyer, M.L.; Montoya, M.; Mende, A.L.; et al. Inhibition of D-2HG leads to upregulation of a proinflammatory gene signature in a novel HLA-A2/HLA-DR1 transgenic mouse model of IDH1R132H-expressing glioma. J. Immunother. Cancer 2022, 10, e004644. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Kaminska, B.; Czapski, B.; Guzik, R.; Król, S.K.; Gielniewski, B. Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules 2019, 24, 968. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Chen, S.; Zuo, M.; Xiang, Y.; Yuan, Y.; He, Y.; Zhang, S.; Liu, Y. Metabolic checkpoints in glioblastomas: Targets for new therapies and non-invasive detection. Front. Oncol. 2024, 14, 1462424. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Basto, D.; Trovisco, V.; Lopes, J.M.; Martins, A.; Pardal, F.; Soares, P.; Reis, R.M. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol. 2005, 109, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Keough, M.B.; Monje, M. Neural Signaling in Cancer. Annu. Rev. Neurosci. 2022, 45, 199–221. [Google Scholar] [CrossRef]
- Pan, Y.; Monje, M. Neuron–Glial Interactions in Health and Brain Cancer. Adv. Biol. 2022, 6, 2200122. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef]
- Barron, T.; Yalçın, B.; Mochizuki, A.; Cantor, E.; Shamardani, K.; Tlais, D.; Franson, A.; Lyons, S.; Mehta, V.; Jahan, S.M.; et al. GABAergic neuron-to-glioma synapses in diffuse midline gliomas. bioRxiv 2022, preprint. [Google Scholar] [CrossRef]
- Shanyue, S.; Xinyuan, C.; Nannan, D.; Miao, Z.; Xiaoru, L.; Lin, C.; Kai, S.; Yingchao, L. Gamma-aminobutyric acid-mediated neuro-immune interactions in glioblastoma: Implications for prognosis and immunotherapy response. Life Sci. 2024, 357, 123067. [Google Scholar] [CrossRef]
- Hanahan, D.; Monje, M. Cancer hallmarks intersect with neuroscience in the tumor microenvironment. Cancer Cell 2023, 41, 573–580. [Google Scholar] [CrossRef]
- Kumari, D.K.; Jha, D.A.M. Brain Tumors in Pediatric Patients. Int. J. Trends OncoScience 2024, 2, 14–22. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; Vitanza, N.A.; Crane, C.A. Immunotherapy for brain tumors: Understanding early successes and limitations. Expert Rev. Neurother. 2018, 18, 251–259. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, L.L.; Fuoco, C.; Palma, A.; Pasquini, L.; Pericoli, G.; Grabovska, Y.; Mackay, A.; Rossi, S.; Carcaboso, A.M.; Carai, A.; et al. Inter and intra-tumor heterogeneity of paediatric type diffuse high-grade gliomas revealed by single-cell mass cytometry. Front. Oncol. 2022, 12, 1016343. [Google Scholar] [CrossRef]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef]
- Czuba, É.; Deschuyter, M.; Entz-Werlé, N.; Noël, G.; Burckel, H. Overcoming the limits of pediatric brain tumor radiotherapy: The use of preclinical 3D models. Cancer/Radiothérapie 2024, 28, 424–434. [Google Scholar] [CrossRef]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suvà, M.L.; Vogel, H.; Monje, M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-Oncology 2019, 21, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.L.; Chen, Z.; Herting, C.J.; Grabovska, Y.; Szulzewsky, F.; Puigdelloses, M.; Monterroza, L.; Switchenko, J.; Wadhwani, N.R.; Cimino, P.J.; et al. Platelet-derived growth factor beta is a potent inflammatory driver in paediatric high-grade glioma. Brain 2021, 144, 53–69. [Google Scholar] [CrossRef]
- du Chatinier, A.; Meel, M.H.; Das, A.I.; Metselaar, D.S.; Waranecki, P.; Bugiani, M.; Breur, M.; Simonds, E.F.; Lu, E.D.; Weiss, W.A.; et al. Generation of immunocompetent syngeneic allograft mouse models for pediatric diffuse midline glioma. Neuro-Oncol. Adv. 2022, 4, vdac079. [Google Scholar] [CrossRef]
- Waibl Polania, J.; Hoyt-Miggelbrink, A.; Tomaszewski, W.H.; Wachsmuth, L.P.; Lorrey, S.J.; Wilkinson, D.S.; Lerner, E.; Woroniecka, K.; Finlay, J.B.; Ayasoufi, K.; et al. Antigen presentation by tumor-associated macrophages drives T cells from a progenitor exhaustion state to terminal exhaustion. Immunity 2025, 58, 232–246.e6. [Google Scholar] [CrossRef]
- Frederico, S.C.; Sharma, N.; Darling, C.; Taori, S.; Dubinsky, A.C.; Zhang, X.; Raphael, I.; Kohanbash, G. Myeloid cells as potential targets for immunotherapy in pediatric gliomas. Front. Pediatr. 2024, 12, 1346493. [Google Scholar] [CrossRef]
- Park, J.; Chung, C. Epigenetic and Metabolic Changes in Diffuse Intrinsic Pontine Glioma. Brain Tumor Res. Treat. 2023, 11, 86–93. [Google Scholar] [CrossRef]
- Yeom, K.W.; Lober, R.M.; Nelson, M.D.; Panigrahy, A.; Blüml, S. Citrate concentrations increase with hypoperfusion in pediatric diffuse intrinsic pontine glioma. J. Neurooncol. 2015, 122, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Bonner, E.R.; Feyen, O.; Domogalla, M.P.; Prix, L.; Schirrmacher, V.; Nazarian, J.; Stuecker, W. Addition of Multimodal Immunotherapy to Combination Treatment Strategies for Children with DIPG: A Single Institution Experience. Medicines 2020, 7, 29. [Google Scholar] [CrossRef]
- Venneti, S.; Kawakibi, A.R.; Ji, S.; Waszak, S.M.; Sweha, S.R.; Mota, M.; Pun, M.; Deogharkar, A.; Chung, C.; Tarapore, R.S.; et al. Clinical Efficacy of ONC201 in H3K27M-Mutant Diffuse Midline Gliomas Is Driven by Disruption of Integrated Metabolic and Epigenetic Pathways. Cancer Discov. 2023, 13, 2370–2393. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.R.; Persson, M.L.; Fish, C.J.; Findlay, I.J.; Mueller, S.; Nazarian, J.; Hulleman, E.; van der Lugt, J.; Duchatel, R.J.; Dun, M.D. A review of current therapeutics targeting the mitochondrial protease ClpP in diffuse midline glioma, H3 K27-altered. Neuro-Oncology 2024, 26, S136–S154. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, B.; Maswikiti, E.P.; Yu, Y.; Song, K.; Ma, C.; Han, X.; Ma, H.; Deng, X.; Yu, R.; et al. AMPK–a key factor in crosstalk between tumor cell energy metabolism and immune microenvironment? Cell Death Discov. 2024, 10, 237. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Kampers, L.F.C.; Van de Vliet, P.; Sprenger, T.; Schirrmacher, V.; Stücker, W. Dendritic cell vaccination for glioblastoma multiforme patients: Has a new milestone been reached? Transl. Cancer Res. 2023, 12, 2224–2228. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Van de Vliet, P.; Kampers, L.F.C.; Kosmal, J.; Sprenger, T.; Reich, E.; Schirrmacher, V.; Stuecker, W. Methods behind oncolytic virus-based DC vaccines in cancer: Toward a multiphase combined treatment strategy for Glioblastoma (GBM) patients. Methods Cell Biol. 2023, 183, 51–113. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Voloshin, T.; Kaynan, N.; Davidi, S.; Porat, Y.; Shteingauz, A.; Schneiderman, R.S.; Zeevi, E.; Munster, M.; Blat, R.; Tempel Brami, C.; et al. Tumor-treating fields (TTFields) induce immunogenic cell death resulting in enhanced antitumor efficacy when combined with anti-PD-1 therapy. Cancer Immunol. Immunother. 2020, 69, 1191–1204. [Google Scholar] [CrossRef]
- Meehan, B.; Rak, J.; Di Vizio, D. Oncosomes—Large and small: What are they, where they came from? J. Extracell. Vesicles 2016, 5, 33109. [Google Scholar] [CrossRef]
- Antonopoulos, M.; Gool, S.W.V.; Dionysiou, D.; Graf, N.; Stamatakos, G. Immune Phenotype Correlates With Survival in Patients With GBM Treated With Standard Temozolomide-based Therapy and Immunotherapy. Anticancer Res. 2019, 39, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Izumoto, S.; Tsuboi, A.; Oka, Y.; Suzuki, T.; Hashiba, T.; Kagawa, N.; Hashimoto, N.; Maruno, M.; Elisseeva, O.A.; Shirakata, T.; et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J. Neurosurg. JNS 2008, 108, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Galbo, P.M.; Ciesielski, M.J.; Figel, S.; Maguire, O.; Qiu, J.; Wiltsie, L.; Minderman, H.; Fenstermaker, R.A. Circulating CD9+/GFAP+/survivin+ exosomes in malignant glioma patients following survivin vaccination. Oncotarget 2017, 8, 114722–114735. [Google Scholar] [CrossRef]
- Grassl, N.; Poschke, I.; Lindner, K.; Bunse, L.; Mildenberger, I.; Boschert, T.; Jähne, K.; Green, E.W.; Hülsmeyer, I.; Jünger, S.; et al. A H3K27M-targeted vaccine in adults with diffuse midline glioma. Nat. Med. 2023, 29, 2586–2592. [Google Scholar] [CrossRef]
- Kleef, R.; Nagy, R.; Baierl, A.; Bacher, V.; Bojar, H.; McKee, D.L.; Moss, R.; Thoennissen, N.H.; Szász, M.; Bakacs, T. Low-dose ipilimumab plus nivolumab combined with IL-2 and hyperthermia in cancer patients with advanced disease: Exploratory findings of a case series of 131 stage IV cancers—A retrospective study of a single institution. Cancer Immunol. Immunother. 2021, 70, 1393–1403. [Google Scholar] [CrossRef]
- Mitchell, A.P.; Goldstein, D.A. Cost Savings and Increased Access With Ultra-Low-Dose Immunotherapy. J. Clin. Oncol. 2023, 41, 170–172. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Van Gool, S.; Stuecker, W. Individualized Multimodal Immunotherapy (IMI): Scientific Rationale and Clinical Experience from a Single Institution. Biomedicines 2024, 12, 754. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- DePeaux, K.; Delgoffe, G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021, 21, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Yang, X.; Ye, S.; Huang, L.; Mu, W. Antigen escape in CAR-T cell therapy: Mechanisms and overcoming strategies. Biomed. Pharmacother. 2024, 178, 117252. [Google Scholar] [CrossRef]
- Dariš, B.; Verboten, M.T.; Knez, Ž.; Ferk, P. Cannabinoids in cancer treatment: Therapeutic potential and legislation. Bosn. J. Basic Med. Sci. 2019, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.; Fischer, O.M.; Ullrich, A. Cannabinoids Induce Cancer Cell Proliferation via Tumor Necrosis Factor α-Converting Enzyme (TACE/ADAM17)-Mediated Transactivation of the Epidermal Growth Factor Receptor. Cancer Res. 2004, 64, 1943–1950. [Google Scholar] [CrossRef]
- Xiong, X.; Chen, S.; Shen, J.; You, H.; Yang, H.; Yan, C.; Fang, Z.; Zhang, J.; Cai, X.; Dong, X.; et al. Cannabis suppresses antitumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct. Target. Ther. 2022, 7, 99. [Google Scholar] [CrossRef]
- Taha, T.; Meiri, D.; Talhamy, S.; Wollner, M.; Peer, A.; Bar-Sela, G. Cannabis Impacts Tumor Response Rate to Nivolumab in Patients with Advanced Malignancies. Oncologist 2019, 24, 549–554. [Google Scholar] [CrossRef]
- Bar-Sela, G.; Cohen, I.; Campisi-Pinto, S.; Lewitus, G.M.; Oz-Ari, L.; Jehassi, A.; Peer, A.; Turgeman, I.; Vernicova, O.; Berman, P.; et al. Cannabis Consumption Used by Cancer Patients during Immunotherapy Correlates with Poor Clinical Outcome. Cancers 2020, 12, 2447. [Google Scholar] [CrossRef]
- Dain, L.; Zhu, G. Nucleic acid immunotherapeutics and vaccines: A promising approach to glioblastoma multiforme treatment. Int. J. Pharm. 2023, 638, 122924. [Google Scholar] [CrossRef]
- Lan, Y.; Li, X.; Liu, B.; Lu, J.; Zuo, B.; Wang, Y.; Cao, S.; Fu, X.; Yue, Q.; Luo, X.; et al. Framework nucleic acid-based nanoparticles enhance temozolomide sensitivity in glioblastoma. Drug Resist. Updates 2024, 76, 101122. [Google Scholar] [CrossRef]
- Phillips, R.E.; Soshnev, A.A.; Allis, C.D. Epigenomic Reprogramming as a Driver of Malignant Glioma. Cancer Cell 2020, 38, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Vorasidenib: First Approval. Drugs 2024, 84, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; French, P.J.; Brat, D.; Tonn, J.C.; Touat, M.; Ellingson, B.M.; Young, R.J.; Pallud, J.; von Deimling, A.; Sahm, F.; et al. The biological significance of tumor grade, age, enhancement, and extent of resection in IDH-mutant gliomas: How should they inform treatment decisions in the era of IDH inhibitors? Neuro-Oncology 2024, 26, 1805–1822. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.M.; Daniels, D.J. Epigenetic-Targeted Treatments for H3K27M-Mutant Midline Gliomas. Adv. Exp. Med. Biol. 2021, 1283, 73–84. [Google Scholar] [CrossRef]
- Keane, L.; Cheray, M.; Saidi, D.; Kirby, C.; Friess, L.; Gonzalez-Rodriguez, P.; Gerdes, M.E.; Grabert, K.; McColl, B.W.; Joseph, B. Inhibition of microglial EZH2 leads to anti-tumoral effects in pediatric diffuse midline gliomas. Neuro-Oncol. Adv. 2021, 3, vdab096. [Google Scholar] [CrossRef]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the gut-brain-axis: Implications for new therapeutic design in the CNS. EBioMedicine 2022, 77, 103908. [Google Scholar] [CrossRef]
- Zhang, H.; Hong, Y.; Wu, T.; Ben, E.; Li, S.; Hu, L.; Xie, T. Role of gut microbiota in regulating immune checkpoint inhibitor therapy for glioblastoma. Front. Immunol. 2024, 15, 1401967. [Google Scholar] [CrossRef]
- Shireman, J.M.; Ammanuel, S.; Eickhoff, J.C.; Dey, M. Sexual dimorphism of the immune system predicts clinical outcomes in glioblastoma immunotherapy: A systematic review and meta-analysis. Neuro-Oncol. Adv. 2022, 4, vdac082. [Google Scholar] [CrossRef]
- Lee, J.; Nicosia, M.; Hong, E.S.; Silver, D.J.; Li, C.; Bayik, D.; Watson, D.C.; Lauko, A.; Kay, K.E.; Wang, S.Z.; et al. Sex-Biased T-cell Exhaustion Drives Differential Immune Responses in Glioblastoma. Cancer Discov. 2023, 13, 2090–2105. [Google Scholar] [CrossRef]
- Bonner, E.R.; Bornhorst, M.; Packer, R.J.; Nazarian, J. Liquid biopsy for pediatric central nervous system tumors. Npj Precis. Oncol. 2018, 2, 29. [Google Scholar] [CrossRef]
- Aibaidula, A.; Fain, C.E.; Garcia, L.C.; Wier, A.; Bouchal, S.M.; Bauman, M.M.; Jung, M.-Y.; Sarkaria, J.N.; Johnson, A.J.; Parney, I.F. Spectral flow cytometry identifies distinct nonneoplastic plasma extracellular vesicle phenotype in glioblastoma patients. Neuro-Oncol. Adv. 2023, 5, vdad082. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Okuma, Y. Perspective on immune oncology with liquid biopsy, peripheral blood mononuclear cells, and microbiome with non-invasive biomarkers in cancer patients. Clin. Transl. Oncol. 2018, 20, 966–974. [Google Scholar] [CrossRef]
- Pan, C.; Diplas, B.H.; Chen, X.; Wu, Y.; Xiao, X.; Jiang, L.; Geng, Y.; Xu, C.; Sun, Y.; Zhang, P.; et al. Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol. 2019, 137, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Bounajem, M.T.; Karsy, M.; Jensen, R.L. Liquid biopsies for the diagnosis and surveillance of primary pediatric central nervous system tumors: A review for practicing neurosurgeons. Neurosurg. Focus FOC 2020, 48, E8. [Google Scholar] [CrossRef] [PubMed]
- Müller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef]
- Hutóczki, G.; Virga, J.; Birkó, Z.; Klekner, A. Novel Concepts of Glioblastoma Therapy Concerning Its Heterogeneity. Int. J. Mol. Sci. 2021, 22, 10005. [Google Scholar] [CrossRef]
- Del Bene, M.; Osti, D.; Faletti, S.; Beznoussenko, G.V.; DiMeco, F.; Pelicci, G. Extracellular vesicles: The key for precision medicine in glioblastoma. Neuro-Oncology 2022, 24, 184–196. [Google Scholar] [CrossRef]
- Panditharatna, E.; Kilburn, L.B.; Aboian, M.S.; Kambhampati, M.; Gordish-Dressman, H.; Magge, S.N.; Gupta, N.; Myseros, J.S.; Hwang, E.I.; Kline, C.; et al. Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy. Clin. Cancer Res. 2018, 24, 5850–5859. [Google Scholar] [CrossRef]
- Verduin, M.; Compter, I.; Steijvers, D.; Postma, A.A.; Eekers, D.B.P.; Anten, M.M.; Ackermans, L.; ter Laan, M.; Leijenaar, R.T.H.; van de Weijer, T.; et al. Noninvasive Glioblastoma Testing: Multimodal Approach to Monitoring and Predicting Treatment Response. Dis. Markers 2018, 2018, 2908609. [Google Scholar] [CrossRef]
- Eibl, R.H.; Schneemann, M. Liquid biopsy and glioblastoma. Explor. Target. Anti-Tumor Ther. 2023, 4, 28–41. [Google Scholar] [CrossRef]
- Malara, N.; Donato, G. Blood-Brain Barrier Breakdown by Combined Detection of Circulating Tumor and Endothelial Cells in Liquid Biopsy. In Liquid Biopsy; IntechOpen: London, UK, 2019; pp. 127–148. [Google Scholar]
- Zaky, W.; Ragoonanan, D.; Batth, I.; Dao, L.; Wang, J.; Xia, X.; Daw, N.C.; Gill, J.B.; Khatua, S.; Li, S. Automated Capture and Analysis of Circulating Tumor Cells in Pediatric, Adolescent and Young Adult Patients with Central Nervous System Tumors. Cancers 2023, 15, 3853. [Google Scholar] [CrossRef]
- Patel, J.; Aittaleb, R.; Doherty, R.; Gera, A.; Lau, B.; Messinger, D.; Wadden, J.; Franson, A.; Saratsis, A.; Koschmann, C. Liquid biopsy in H3K27M diffuse midline glioma. Neuro-Oncology 2024, 26, S101–S109. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Sun, Q.; Deng, G.; Zhang, H.; Xu, Y.; Li, Y.; Huang, S.; Li, Y.; Ye, Z.; Wang, Y.; et al. Identifying circulating glioma cells and their clusters as diagnostic markers by a novel detection platform. Clin. Transl. Med. 2021, 11, e318. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Wollmann, K.; Salviano-Silva, A.; Drexler, R.; Maire, C.L.; Kaul, M.G.; Reimer, R.; Schüller, U.; Heinemann, S.; Kolbe, K.; et al. Circulating extracellular vesicles as biomarker for diagnosis, prognosis, and monitoring in glioblastoma patients. Neuro-Oncology 2024, 26, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Ling, A.L.; Reardon, D.A.; Chiocca, E.A. Lessons learned from phase 3 trials of immunotherapy for glioblastoma: Time for longitudinal sampling? Neuro-Oncology 2024, 26, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Skretteberg, M.A.; Johannesen, T.B.; Brandal, P.; Vik-Mo, E.O.; Helseth, E.; Langmoen, I.A. Real-world validity of randomized controlled phase III trials in newly diagnosed glioblastoma: To whom do the results of the trials apply? Neuro-Oncol. Adv. 2021, 3, vdab008. [Google Scholar] [CrossRef]
- Alomari, S.; Zhang, I.; Hernandez, A.; Kraft, C.Y.; Raj, D.; Kedda, J.; Tyler, B. Drug Repurposing for Glioblastoma and Current Advances in Drug Delivery—A Comprehensive Review of the Literature. Biomolecules 2021, 11, 1870. [Google Scholar] [CrossRef]
- Ntafoulis, I.; Koolen, S.L.W.; Leenstra, S.; Lamfers, M.L.M. Drug Repurposing, a Fast-Track Approach to Develop Effective Treatments for Glioblastoma. Cancers 2022, 14, 3705. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kampers, L.F.C.; Metselaar, D.S.; Vinci, M.; Scirocchi, F.; Veldhuijzen van Zanten, S.; Eyrich, M.; Biassoni, V.; Hulleman, E.; Karremann, M.; Stücker, W.; et al. The Complexity of Malignant Glioma Treatment. Cancers 2025, 17, 879. https://doi.org/10.3390/cancers17050879
Kampers LFC, Metselaar DS, Vinci M, Scirocchi F, Veldhuijzen van Zanten S, Eyrich M, Biassoni V, Hulleman E, Karremann M, Stücker W, et al. The Complexity of Malignant Glioma Treatment. Cancers. 2025; 17(5):879. https://doi.org/10.3390/cancers17050879
Chicago/Turabian StyleKampers, Linde F. C., Dennis S. Metselaar, Maria Vinci, Fabio Scirocchi, Sophie Veldhuijzen van Zanten, Matthias Eyrich, Veronica Biassoni, Esther Hulleman, Michael Karremann, Wilfried Stücker, and et al. 2025. "The Complexity of Malignant Glioma Treatment" Cancers 17, no. 5: 879. https://doi.org/10.3390/cancers17050879
APA StyleKampers, L. F. C., Metselaar, D. S., Vinci, M., Scirocchi, F., Veldhuijzen van Zanten, S., Eyrich, M., Biassoni, V., Hulleman, E., Karremann, M., Stücker, W., & Van Gool, S. W. (2025). The Complexity of Malignant Glioma Treatment. Cancers, 17(5), 879. https://doi.org/10.3390/cancers17050879