
Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy
Simple Summary
Abstract
1. Introduction
2. Immune System and Immunotherapy
2.1. Immune Checkpoints
2.1.1. Immune Checkpoint Inhibitors (ICIs) and Mechanism of Action
2.1.2. Preclinical and Clinical Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | ICI Target | FDA-Approved Indications | Ref. |
|---|---|---|---|
| Ipilimumab | CTLA-4 | Metastatic melanoma | [125] |
| Nivolumab | PD-1 | Metastatic melanoma | [91] |
| Metastatic RCC | [91] | ||
| Hodgkins’s lymphoma | [92] | ||
| HNSCC | [93] | ||
| HCC | [94] | ||
| ESCC | [95] | ||
| NSCLC | [126] | ||
| Pembrolizumab | PD-1 | NSCLC | [98,99] |
| Melanoma | [98,99] | ||
| HNSCC | [100] | ||
| GC | [102] | ||
| CC | [103,104] | ||
| UC | [105] | ||
| TNBC | [107] | ||
| Cemiplimab | PD-1 | Metastatic CSCC | [109] |
| Atezolizumab | PD-L1 | UC | [113] |
| Metastatic non-squamous and squamous NSCLC | [114,115] | ||
| SCLC | [116] | ||
| Melanoma | [117] | ||
| HCC | [118] | ||
| Durvalumab | PD-L1 | Locally advanced or metastatic urothelial carcinoma | [119] |
| NSCLC | [127] | ||
| Avelumab | PD-L1 | Merkel cell carcinoma MCC | [121] |
| Urothelial carcinoma | [122] | ||
| RCC | [123,124] |
2.2. Cytokines and Cytokine-Based Therapies Against Cancers
2.3. Oncolytic Virus-Based Therapies and Vaccines Against Cancers
2.4. Adoptive Cell Therapy (ACT)
2.5. Bispecific Immune-Checkpoint Blockade Antibodies
3. Combination of Immunotherapies
4. Immune-Related Adverse Events (IrAEs)
4.1. Autoreactive T Cells
4.2. Autoreactive B Cells
4.3. Cytokine Storm
5. Resistance to Immunotherapy
5.1. Primary Resistance
5.1.1. Tumor Mutational Burden and Neoantigen Expression in Immunotherapy Response and Resistance Mechanisms
5.1.2. Antigen Processing and Presentation Dysfunction and ICI Therapy Resistance
5.1.3. Immunosuppressive Dynamics in the Tumor Microenvironment and ICI Therapy Resistance
5.1.4. T-Cell Exhaustion and Resistance to Immune-Checkpoint Blockade
5.1.5. Deregulated Signaling Pathways in Immune Evasion and ICI Therapy Resistance
5.2. Acquired Resistance
5.2.1. Interferon-Driven Immune Response and Resistance Mechanisms
5.2.2. Immunoediting and Selection Pressure as Mechanisms of Acquired Resistance
5.2.3. Alternative Immune Checkpoint Activation and Acquired Resistance Mechanisms
5.2.4. Impaired CD8+ T-Cell Memory and Acquired Resistance
6. Therapeutic Strategies for Overcoming Drug Resistance with ICIs
6.1. Enhancing Tumor Infiltration by T Cells
6.2. Bispecific T-Cell Engagers as Immunotherapeutics
6.3. Epigenetic Modulation to Reshape the Tumor Microenvironment
6.4. Improving the Tumor Immune Microenvironment
6.5. Increasing Tumor Immunogenicity and T-Cell Priming
6.5.1. Oncolytic Virus Therapies and Vaccines Against Cancers
6.5.2. Chemotherapy
6.5.3. Radiotherapy
6.6. Combination with Other Targeted Therapies
7. Comprehensive Biomarkers in Immune-Checkpoint Inhibitor Therapies
7.1. PD-L1 Expression
7.2. Tumor Mutational Burden
7.3. Microsatellite Instability and Mismatch Repair Deficiency
7.4. Cytokines and Transcription Factors
7.5. Tumor-Infiltrating Lymphocytes
7.6. Microbiome and Immunotherapy
8. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Adoptive cell therapy |
| AI | Artificial intelligence |
| APCs | Antigen-presenting cells |
| ATR | Ataxia Telangiectasia inhibitors and Rad3-related |
| APP | Antigen processing and presentation |
| β2M | Beta-2 microglobulin |
| B-ALL | B-cell progenitor acute lymphoblastic leukemia |
| BCG | Bacille Calmette-Guérin |
| BdC | Bladder cancer |
| BLA | Biologics license application |
| BTC | Biliary tract cancer |
| CC | Cervical cancer |
| ccRCC | Clear cell renal cell carcinoma |
| CD-28 | Cluster of Differentiation 28 |
| CDK4/6i | Cyclin-Dependent Kinase 4/6 inhibitors |
| CML | Chronic myelogenous leukemia |
| CHK1 | Checkpoint Kinase 1 |
| CRC | Colorectal cancer |
| CSCC | Cutaneous squamous cell carcinoma |
| CSF1R | Colony-stimulating factor 1 receptor |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| CXCL | Chemokine (C-X-C motif) ligand |
| DCs | Dendritic cells |
| dMMR | Deficient mismatch repair |
| EndC | Endometrial carcinoma |
| ESCC | Esophageal squamous cell carcinoma |
| FDA | Food and Drug Administration |
| FGF-2 | Fibroblast growth factor -2 |
| GC | Gastric cancer |
| G-CSF | Granulocyte colony-stimulating factor |
| GMCSF | Granulocyte-macrophage colony-stimulating factor |
| HCC | Hepatocellular carcinoma |
| HLA | Human leukocyte antigen |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| ICD | Immunogenic cell death |
| ICIs | Immune-checkpoint inhibitors |
| IDO | Indoleamine 2,3-dioxygenase |
| IFNs | Interferons |
| IFN-α | Interferon alpha |
| IHC | Immunohistochemistry |
| IL | Interleukin |
| ILCs | Innate Lymphoid Cells |
| ITIM | Immunoreceptor tyrosine-based inhibition motif |
| ITSM | Immunoreceptor tyrosine-based switch motif |
| irAEs | Immune-related adverse events |
| JAK | Janus kinase |
| LAG3 | Lymphocyte Activating Gene- 3 |
| LCK | Lymphocyte-specific protein tyrosine kinase |
| LCMV | Chronic lymphocytic choriomeningitis virus |
| LOH | Loss of heterozygosity |
| Mab | Monoclonal antibodies |
| MAPK | Mitogen-activated protein kinase pathway |
| MCC | Merkel cell carcinoma |
| MDSCs | Myeloid-derived suppressive cells |
| MHCs | Major histocompatibility complexes |
| MPM | Malignant pleural mesothelioma |
| MSI-H | High microsatellite instability |
| mut/Mb | Mutations per megabase |
| NK | natural killer cells |
| NSCLC | Non-small-cell lung carcinoma |
| OS | Overall survival |
| OVC | Ovarian cancer |
| PCa | Prostate cancer |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PFS | Progression-free survival |
| PI3K | Phosphatidylinositol 3-kinase pathway |
| PM | Pleural mesothelioma |
| PP2A | Protein Phosphatase 2A |
| PARPi | Polymerase inhibitors |
| PTEN | Phosphatase and tensin homolog |
| RCC | Renal cell carcinoma |
| STAT | Signal transducers and activators of transcription |
| SHP | Src homology region containing protein tyrosine phosphatase |
| TAAs | Tumor-associated antigens |
| TAP | Transporters associated with neoantigen presentation |
| TAMs | Tumor-associated macrophages |
| TCR | T-cell receptor |
| TPOAb | Thyroid peroxidase antibodies |
| TGAb | Anti-thyroglobulin antibodies |
| TGFβ | Transforming growth factor-beta |
| TIGIT | T-cell immunoglobulin and ITIM domain |
| TILs | Tumor-Infiltrating Lymphocytes |
| TIM3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TLR | Toll-like receptor |
| TMB | Tumors mutational burden |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| TOX | Thymocyte selection-associated high-mobility group box gene |
| Tregs | Regulatory T cells |
| T-VEC | Talimogene laherparepvec |
| UBC | Urothelial bladder cancer |
| UC | Urothelial carcinoma |
| VEGF | Vascular endothelial growth factor |
| Wnt | Wingless-related integration site |
| ZAP-70 | Zeta-chain-associated protein kinase-70 |
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R.R. Immunotherapies and Combination Strategies for Immuno-Oncology. Int. J. Mol. Sci. 2020, 21, 5009. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ribas, A. Releasing the Brakes on Cancer Immunotherapy. N. Engl. J. Med. 2015, 373, 1490–1492. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Toor, S.M.; Sasidharan Nair, V.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2020, 65, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paluch, C.; Santos, A.M.; Anzilotti, C.; Cornall, R.J.; Davis, S.J. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front. Immunol. 2018, 9, 2306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Wang, Y.; Zhao, W.; Li, H.; Zhang, L.; Li, X.; Zhang, T.; Zhang, H.; Huang, H.; et al. Application of immune checkpoint targets in the anti-tumor novel drugs and traditional Chinese medicine development. Acta Pharm. Sin. B 2021, 11, 2957–2972. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Guilarte, L.; Ramírez, M.A.; Andrade, C.A.; Kalergis, A.M. LAG-3 Contribution to T Cell Downmodulation during Acute Respiratory Viral Infections. Viruses 2023, 15, 147. [Google Scholar] [CrossRef]
- Hasan, M.F.; Croom-Perez, T.J.; Oyer, J.L.; Dieffenthaller, T.A.; Robles-Carrillo, L.D.; Eloriaga, J.E.; Kumar, S.; Andersen, B.W.; Copik, A.J. TIGIT Expression on Activated NK Cells Correlates with Greater Anti-Tumor Activity but Promotes Functional Decline upon Lung Cancer Exposure: Implications for Adoptive Cell Therapy and TIGIT-Targeted Therapies. Cancers 2023, 15, 2712. [Google Scholar] [CrossRef]
- Yang, S.; Cao, B.; Zhou, G.; Zhu, L.; Wang, L.; Zhang, L.; Kwok, H.F.; Zhang, Z.; Zhao, Q. Targeting B7-H3 Immune Checkpoint With Chimeric Antigen Receptor-Engineered Natural Killer Cells Exhibits Potent Cytotoxicity Against Non-Small Cell Lung Cancer. Front. Pharmacol. 2020, 11, 1089. [Google Scholar] [CrossRef]
- Shekari, N.; Shanehbandi, D.; Kazemi, T.; Zarredar, H.; Baradaran, B.; Jalali, S.A. VISTA and its ligands: The next generation of promising therapeutic targets in immunotherapy. Cancer Cell Int. 2023, 23, 265. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, Y.; Mao, R.; Su, Z.; Zhang, J. BTLA/HVEM Signaling: Milestones in Research and Role in Chronic Hepatitis B Virus Infection. Front. Immunol. 2019, 10, 617. [Google Scholar] [CrossRef]
- Lazaridou, M.F.; Massa, C.; Handke, D.; Mueller, A.; Friedrich, M.; Subbarayan, K.; Tretbar, S.; Dummer, R.; Koelblinger, P.; Seliger, B. Identification of microRNAs Targeting the Transporter Associated with Antigen Processing TAP1 in Melanoma. J. Clin. Med. 2020, 9, 2690. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. In Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy; Xu, J., Ed.; Springer: Singapore, 2020; pp. 33–59. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Feng, Y.; Roy, A.; Masson, E.; Chen, T.T.; Humphrey, R.; Weber, J.S. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin. Cancer Res. 2013, 19, 3977–3986. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 034002. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.D.; Le Gros, G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol. Cell Biol. 1999, 77, 1–10. [Google Scholar] [CrossRef]
- Bretscher, P. The two-signal model of lymphocyte activation twenty-one years later. Immunol. Today 1992, 13, 74–76. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Walker, L.S. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Brunner, M.C.; Chambers, C.A.; Chan, F.K.; Hanke, J.; Winoto, A.; Allison, J.P. CTLA-4-Mediated inhibition of early events of T cell proliferation. J. Immunol. 1999, 162, 5813–5820. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef]
- van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.T.; Bradshaw, J.; Cleaveland, J.S.; Linsley, P.S. Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J. Biol. Chem. 1995, 270, 25107–25114. [Google Scholar] [CrossRef]
- Alegre, M.L.; Noel, P.J.; Eisfelder, B.J.; Chuang, E.; Clark, M.R.; Reiner, S.L.; Thompson, C.B. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 1996, 157, 4762–4770. [Google Scholar] [CrossRef]
- Egen, J.G.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Pentcheva-Hoang, T.; Egen, J.G.; Wojnoonski, K.; Allison, J.P. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 2004, 21, 401–413. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Mellor, A.L. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J. Immunol. 2004, 172, 4100–4110. [Google Scholar] [CrossRef]
- Park, J.; Kwon, M.; Shin, E.C. Immune checkpoint inhibitors for cancer treatment. Arch. Pharm. Res. 2016, 39, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Sage, P.T.; Paterson, A.M.; Lovitch, S.B.; Sharpe, A.H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014, 41, 1026–1039. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Lewis, A.L.; Chaft, J.; Girotra, M.; Fischer, G.W. Immune checkpoint inhibitors: A narrative review of considerations for the anaesthesiologist. Br. J. Anaesth. 2020, 124, 251–260. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Blair, P.J.; Riley, J.L.; Levine, B.L.; Lee, K.P.; Craighead, N.; Francomano, T.; Perfetto, S.J.; Gray, G.S.; Carreno, B.M.; June, C.H. CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-X(L) induction. J. Immunol. 1998, 160, 12–15. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef]
- Ribatti, D. From the discovery of monoclonal antibodies to their therapeutic application: An historical reappraisal. Immunol. Lett. 2014, 161, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, D.; Oszmiana, A.; Finch, D.K.; Strickland, I.; Schofield, D.J.; Lowe, D.C.; Sleeman, M.A.; Davis, D.M. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell-mediated antibody-dependent cellular cytotoxicity. Blood 2013, 121, 4694–4702. [Google Scholar] [CrossRef]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 14. [Google Scholar]
- Liebl, M.C.; Hofmann, T.G. Identification of responders to immune checkpoint therapy: Which biomarkers have the highest value? J. Eur. Acad. Dermatol. Venereol. 2019, 33 (Suppl. 8), 52–56. [Google Scholar] [CrossRef]
- Borgeaud, M.; Sandoval, J.; Obeid, M.; Banna, G.; Michielin, O.; Addeo, A.; Friedlaender, A. Novel targets for immune-checkpoint inhibition in cancer. Cancer Treat. Rev. 2023, 120, 102614. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhan, H.; Ye, Y.; Yang, J.; Zhang, M.; Li, J.; Zhuang, Y. Current Progress and Future Perspectives of Immune Checkpoint in Cancer and Infectious Diseases. Front. Genet. 2021, 12, 785153. [Google Scholar] [CrossRef]
- Lee, J.B.; Kim, H.R.; Ha, S.J. Immune Checkpoint Inhibitors in 10 Years: Contribution of Basic Research and Clinical Application in Cancer Immunotherapy. Immune Netw. 2022, 22, e2. [Google Scholar] [CrossRef]
- Dhasmana, A.; Dhasmana, S.; Haque, S.; Cobos, E.; Yallapu, M.M.; Chauhan, S.C. Next-generation immune checkpoint inhibitors as promising functional molecules in cancer therapeutics. Cancer Metastasis Rev. 2023, 42, 597–600. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Górska, M.; Polityńska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef]
- Malik, P.; Rani, R.; Solanki, R.; Patel, V.H.; Mukherjee, T.K. Understanding the feasibility of chemotherapeutic and immunotherapeutic targets against non-small cell lung cancers: An update of resistant responses and recent combinatorial therapies. Explor. Target. Anti-Tumor Ther. 2023, 4, 850–895. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Li, Z.Z.; Zhong, N.N.; Cao, L.M.; Liu, B.; Bu, L.L. Charting new frontiers: Co-inhibitory immune checkpoint proteins in therapeutics, biomarkers, and drug delivery systems in cancer care. Transl. Oncol. 2023, 38, 101794. [Google Scholar] [CrossRef] [PubMed]
- Gopal Krishnan, P.D.; Golden, E.; Woodward, E.A.; Pavlos, N.J.; Blancafort, P. Rab GTPases: Emerging Oncogenes and Tumor Suppressive Regulators for the Editing of Survival Pathways in Cancer. Cancers 2020, 12, 259. [Google Scholar] [CrossRef]
- Radhakrishna, H.; Donaldson, J.G. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 1997, 139, 49–61. [Google Scholar] [CrossRef]
- Yang, Y.; Kong, B.; Jung, Y.; Park, J.B.; Oh, J.M.; Hwang, J.; Cho, J.Y.; Kweon, D.H. Soluble N-Ethylmaleimide-Sensitive Factor Attachment Protein Receptor-Derived Peptides for Regulation of Mast Cell Degranulation. Front. Immunol. 2018, 9, 725. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, M.; Zhang, Y.Y.; Zhao, S.Z.; Gu, S. The endosomal sorting complex required for transport repairs the membrane to delay cell death. Front. Oncol. 2022, 12, 1007446. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Nyåkern, M.; Tabellini, G.; Bortul, R.; Tazzari, P.L.; Evangelisti, C.; Cocco, L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006, 20, 911–928. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Xiao, H.; Wang, H.; Xu, X. Recent Advances in PROTACs for Drug Targeted Protein Research. Int. J. Mol. Sci. 2022, 23, 10328. [Google Scholar] [CrossRef] [PubMed]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. USA 2017, 114, E4223–E4232. [Google Scholar] [CrossRef]
- Callahan, M.K.; Wolchok, J.D. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 41–53. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Camacho, L.H.; Antonia, S.; Sosman, J.; Kirkwood, J.M.; Gajewski, T.F.; Redman, B.; Pavlov, D.; Bulanhagui, C.; Bozon, V.A.; Gomez-Navarro, J.; et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J. Clin. Oncol. 2009, 27, 1075–1081. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Kazandjian, D.; Suzman, D.L.; Blumenthal, G.; Mushti, S.; He, K.; Libeg, M.; Keegan, P.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non-Small Cell Lung Cancer with Progression on or After Platinum-Based Chemotherapy. Oncologist 2016, 21, 634–642. [Google Scholar] [CrossRef]
- Ramchandren, R.; Domingo-Domènech, E.; Rueda, A.; Trněný, M.; Feldman, T.A.; Lee, H.J.; Provencio, M.; Sillaber, C.; Cohen, J.B.; Savage, K.J.; et al. Nivolumab for Newly Diagnosed Advanced-Stage Classic Hodgkin Lymphoma: Safety and Efficacy in the Phase II CheckMate 205 Study. J. Clin. Oncol. 2019, 37, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Borel, C.; Jung, A.C.; Burgy, M. Immunotherapy Breakthroughs in the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2691. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.Y.; Chin, K.; Kadowaki, S.; Ahn, M.J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Wright, K. FDA Approves Nivolumab Plus Ipilimumab for Previously Untreated Unresectable Malignant Pleural Mesothelioma. Oncology 2020, 34, 502–503. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Kang, S.P.; Gergich, K.; Lubiniecki, G.M.; de Alwis, D.P.; Chen, C.; Tice, M.A.B.; Rubin, E.H. Pembrolizumab KEYNOTE-001: An adaptive study leading to accelerated approval for two indications and a companion diagnostic. Ann. Oncol. 2017, 28, 1388–1398. [Google Scholar] [CrossRef]
- Raedler, L.A. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96–100. [Google Scholar]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Borcoman, E.; Le Tourneau, C. Keynote-158 study, FDA granted accelerated approval of pembrolizumab for the treatment of patients with advanced PD-L1-positive cervical cancer. Ann. Transl. Med. 2020, 8, 1611. [Google Scholar] [CrossRef] [PubMed]
- Ghatalia, P.; Zibelman, M.; Geynisman, D.M.; Plimack, E. Approved checkpoint inhibitors in bladder cancer: Which drug should be used when? Ther. Adv. Med. Oncol. 2018, 10, 1758835918788310. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Heeke, A.L.; Tan, A.R. Checkpoint inhibitor therapy for metastatic triple-negative breast cancer. Cancer Metastasis Rev. 2021, 40, 537–547. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H., Jr.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients With Microsatellite Instability-High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Keeping, S.; Xu, Y.; Chen, C.I.; Cope, S.; Mojebi, A.; Kuznik, A.; Konidaris, G.; Ayers, D.; Sasane, M.; Allen, R.; et al. Comparative efficacy of cemiplimab versus other systemic treatments for advanced cutaneous squamous cell carcinoma. Future Oncol. 2021, 17, 611–627. [Google Scholar] [CrossRef]
- Huang, Q.; Zheng, Y.; Gao, Z.; Yuan, L.; Sun, Y.; Chen, H. Comparative Efficacy and Safety of PD-1/PD-L1 Inhibitors for Patients with Solid Tumors: A Systematic Review and Bayesian Network Meta-analysis. J. Cancer 2021, 12, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Mathieu, L.; Shah, S.; Pai-Scherf, L.; Larkins, E.; Vallejo, J.; Li, X.; Rodriguez, L.; Mishra-Kalyani, P.; Goldberg, K.B.; Kluetz, P.G.; et al. FDA Approval Summary: Atezolizumab and Durvalumab in Combination with Platinum-Based Chemotherapy in Extensive Stage Small Cell Lung Cancer. Oncologist 2021, 26, 433–438. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Syed, Y.Y. Durvalumab: First Global Approval. Drugs 2017, 77, 1369–1376. [Google Scholar] [CrossRef]
- Rimini, M.; Fornaro, L.; Lonardi, S.; Niger, M.; Lavacchi, D.; Pressiani, T.; Lucchetti, J.; Giordano, G.; Pretta, A.; Tamburini, E.; et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer: An early exploratory analysis of real-world data. Liver Int. 2023, 43, 1803–1812. [Google Scholar] [CrossRef]
- Walker, J.W.; Lebbé, C.; Grignani, G.; Nathan, P.; Dirix, L.; Fenig, E.; Ascierto, P.A.; Sandhu, S.; Munhoz, R.; Benincasa, E.; et al. Efficacy and safety of avelumab treatment in patients with metastatic Merkel cell carcinoma: Experience from a global expanded access program. J. Immunother. Cancer 2020, 8, e000313. [Google Scholar] [CrossRef]
- Apolo, A.B.; Ellerton, J.A.; Infante, J.R.; Agrawal, M.; Gordon, M.S.; Aljumaily, R.; Gourdin, T.; Dirix, L.; Lee, K.-W.; Taylor, M.H.; et al. Avelumab as second-line therapy for metastatic, platinum-treated urothelial carcinoma in the phase Ib JAVELIN Solid Tumor study: 2-year updated efficacy and safety analysis. J. Immunother. Cancer 2020, 8, e001246. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D.; et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018, 19, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Basudan, A.M. The Role of Immune Checkpoint Inhibitors in Cancer Therapy. Clin. Pract. 2022, 13, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Mansh, M. Ipilimumab and cancer immunotherapy: A new hope for advanced stage melanoma. Yale J. Biol. Med. 2011, 84, 381–389. [Google Scholar]
- Rolfo, C.; Caglevic, C.; Santarpia, M.; Araujo, A.; Giovannetti, E.; Gallardo, C.D.; Pauwels, P.; Mahave, M. Immunotherapy in NSCLC: A Promising and Revolutionary Weapon. Adv. Exp. Med. Biol. 2017, 995, 97–125. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Spira, A.; Raben, D.; Planchard, D.; Cho, B.C.; Özgüroğlu, M.; Daniel, D.; Villegas, A.; Vicente, D.; Hui, R.; et al. Outcomes with durvalumab by tumour PD-L1 expression in unresectable, stage III non-small-cell lung cancer in the PACIFIC trial. Ann. Oncol. 2020, 31, 798–806. [Google Scholar] [CrossRef]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Tura, S.; Baccarani, M.; Zuffa, E.; Russo, D.; Fanin, R.; Zaccaria, A.; Fiacchini, M. Interferon alfa-2a as compared with conventional chemotherapy for the treatment of chronic myeloid leukemia. N. Engl. J. Med. 1994, 330, 820–825. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Liang, Y.; Tang, H.; Guo, J.; Qiu, X.; Yang, Z.; Ren, Z.; Sun, Z.; Bian, Y.; Xu, L.; Xu, H.; et al. Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat. Commun. 2018, 9, 4586. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T.; et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Tang, A.; Harding, F. The challenges and molecular approaches surrounding interleukin-2-based therapeutics in cancer. Cytokine X 2018, 1, 100001. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Zídek, Z.; Anzenbacher, P.; Kmonícková, E. Current status and challenges of cytokine pharmacology. Br. J. Pharmacol. 2009, 157, 342–361. [Google Scholar] [CrossRef]
- Pires, I.S.; Hammond, P.T.; Irvine, D.J. Engineering Strategies for Immunomodulatory Cytokine Therapies—Challenges and Clinical Progress. Adv. Ther. 2021, 4, 2100035. [Google Scholar] [CrossRef] [PubMed]
- Chamie, K.; Chang, S.S.; Gonzalgo, M.; Kramolowsky, E.V.; Sexton, W.J.; Bhar, P.; Reddy, S.K.; Soon-Shiong, P. Final Clinical Results of Pivotal Trial of IL-15RαFc Superagonist N-803 with BCG in BCG-Unresponsive CIS and Papillary Nonmuscle-Invasive Bladder Cancer (NMIBC); American Society of Clinical Oncology: Alexandria, VA, USA, 2022. [Google Scholar]
- Chang, S.S.; Chamie, K.; Gonzalgo, M.L.; Kramolowsky, E.V.; Sexton, W.J.; Reddy, S.K.; Bhar, P.; Huang, M.; Soon-Shiong, P. Positive Efficacy and Safety Phase 3 Results in Both CIS and Papillary Cohorts BCG-Unresponsive Nonmuscle Invasive Bladder Cancer (NMIBC) After IL-15RαFc Superagonist N-803 (Anktiva) and BCG Infusion; American Society of Clinical Oncology: Alexandria, VA, USA, 2022. [Google Scholar]
- Zhang, P.; Ding, Y. Novel Therapeutic Strategies for BCG-unresponsive Non-muscle Invasive Bladder Cancer. Ann. Urol. Oncol. 2022, 5, 59–67. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Cerullo, V.; Koski, A.; Vähä-Koskela, M.; Hemminki, A. Chapter eight--Oncolytic adenoviruses for cancer immunotherapy: Data from mice, hamsters, and humans. Adv. Cancer Res. 2012, 115, 265–318. [Google Scholar] [CrossRef]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Ribas, A.; Puzanov, I.; Vanderwalde, A.M.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.S.; et al. Efficacy analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016, 34, 9568. [Google Scholar] [CrossRef]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wu, K.; Visconte, V.; Anwer, F.; Calton, C.M.; Carew, J.S.; Nawrocki, S.T. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia 2018, 32, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.; Surendran, A.; Wedge, M.E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef]
- Olszanski, A.J. Principles of immunotherapy. J. Natl. Compr. Canc Netw. 2015, 13, 670–672. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Lee, J.B.; Kang, H.; Fang, L.; D’Souza, C.; Adeyi, O.; Zhang, L. Developing Allogeneic Double-Negative T Cells as a Novel Off-the-Shelf Adoptive Cellular Therapy for Cancer. Clin. Cancer Res. 2019, 25, 2241–2253. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, W.D. Graft-versus-host disease. Nat. Rev. Immunol. 2007, 7, 340–352. [Google Scholar] [CrossRef]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy—More Than a Pipe Dream. Front. Immunol. 2021, 11, 618427. [Google Scholar] [CrossRef]
- Ehrmann, D.A. Polycystic ovary syndrome. N. Engl. J. Med. 2005, 352, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Eshhar, Z. The T-body approach: Redirecting T cells with antibody specificity. In Therapeutic Antibodies; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 329–342. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Chen, Y.J.; Abila, B.; Mostafa Kamel, Y. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int. 2022, 22, 2. [Google Scholar] [CrossRef]
- Kodumudi, K.N.; Siegel, J.; Weber, A.M.; Scott, E.; Sarnaik, A.A.; Pilon-Thomas, S. Immune Checkpoint Blockade to Improve Tumor Infiltrating Lymphocytes for Adoptive Cell Therapy. PLoS ONE 2016, 11, e0153053. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.; Zhu, A.; Ngai, D.; McGee, E.; Chintala, N.; Messinger, J.; Cheema, W.; et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. J. Clin. Oncol. 2019, 37, 2511. [Google Scholar] [CrossRef]
- Zhang, T.; Lin, Y.; Gao, Q. Bispecific antibodies targeting immunomodulatory checkpoints for cancer therapy. Cancer Biol. Med. 2023, 20, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.A.; Najmi, A.; Ahsan, W.; Zoghebi, K. Targeting PD-1/PD-L-1 immune checkpoint inhibition for cancer immunotherapy: Success and challenges. Front. Immunol. 2024, 15, 1383456. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Sumrow, B.J.; Stahl, K.; Shah, K.; Liu, D.; Li, J.; Hao, S.S.; De Costa, A.; Kaul, S.; Bendell, J.; et al. Development and Preliminary Clinical Activity of PD-1-Guided CTLA-4 Blocking Bispecific DART Molecule. Cell Rep. Med. 2020, 1, 100163. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1+ Activated T Cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef]
- Pang, X.; Huang, Z.; Zhong, T.; Zhang, P.; Wang, Z.M.; Xia, M.; Li, B. Cadonilimab, a tetravalent PD-1/CTLA-4 bispecific antibody with trans-binding and enhanced target binding avidity. MAbs 2023, 15, 2180794. [Google Scholar] [CrossRef]
- Luke, J.J.; Patel, M.R.; Blumenschein, G.R.; Hamilton, E.; Chmielowski, B.; Ulahannan, S.V.; Connolly, R.M.; Santa-Maria, C.A.; Wang, J.; Bahadur, S.W.; et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: A phase 1 trial. Nat. Med. 2023, 29, 2814–2824. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Limagne, E.; Richard, C.; Thibaudin, M.; Fumet, J.D.; Truntzer, C.; Lagrange, A.; Favier, L.; Coudert, B.; Ghiringhelli, F. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology 2019, 8, e1564505. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncology. Annu. Meet. 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Lacal, P.M.; Martire, M.; Navarra, P.; Graziani, G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol. Res. 2022, 175, 105997. [Google Scholar] [CrossRef]
- Xia, B.; Herbst, R.S. Immune checkpoint therapy for non-small-cell lung cancer: An update. Immunotherapy 2016, 8, 279–298. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Q.; Wang, R.; Li, Y.; Sun, Y.; Yang, L. Targeting DNA Damage Repair for Immune Checkpoint Inhibition: Mechanisms and Potential Clinical Applications. Front. Oncol. 2021, 11, 648687. [Google Scholar] [CrossRef]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Gajewski, T.F. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin. Cancer Res. 2015, 21, 4774–4779. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Chennamadhavuni, A.; Abushahin, L.; Jin, N.; Presley, C.J.; Manne, A. Risk Factors and Biomarkers for Immune-Related Adverse Events: A Practical Guide to Identifying High-Risk Patients and Rechallenging Immune Checkpoint Inhibitors. Front. Immunol. 2022, 13, 779691. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Hammer, C.; Guardino, E.; Chandler, G.S.; Albert, M.L. Mechanisms of immune-related adverse events associated with immune checkpoint blockade: Using germline genetics to develop a personalized approach. Genome Med. 2019, 11, 39. [Google Scholar] [CrossRef]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, R.; Cheema, A.; Khan, T.; Amirpour, A.; Paul, A.; Chaughtai, S.; Patel, S.; Patel, T.; Bramson, J.; Gupta, V.; et al. Adverse Effects of Immune Checkpoint Inhibitors (Programmed Death-1 Inhibitors and Cytotoxic T-Lymphocyte-Associated Protein-4 Inhibitors): Results of a Retrospective Study. J. Clin. Med. Res. 2019, 11, 225–236. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- González-Rodríguez, E.; Rodríguez-Abreu, D. Immune Checkpoint Inhibitors: Review and Management of Endocrine Adverse Events. Oncologist 2016, 21, 804–816. [Google Scholar] [CrossRef]
- Lee, H.; Hodi, F.S.; Giobbie-Hurder, A.; Ott, P.A.; Buchbinder, E.I.; Haq, R.; Tolaney, S.; Barroso-Sousa, R.; Zhang, K.; Donahue, H.; et al. Characterization of Thyroid Disorders in Patients Receiving Immune Checkpoint Inhibition Therapy. Cancer Immunol. Res. 2017, 5, 1133–1140. [Google Scholar] [CrossRef]
- Faje, A. Immunotherapy and hypophysitis: Clinical presentation, treatment, and biologic insights. Pituitary 2016, 19, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Cukier, P.; Santini, F.C.; Scaranti, M.; Hoff, A.O. Endocrine side effects of cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, T331–T347. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Chiec, L.; Mohindra, N.; Gentzler, R.; Patel, J.; Giles, F. A case of pembrolizumab-induced type-1 diabetes mellitus and discussion of immune checkpoint inhibitor-induced type 1 diabetes. Cancer Immunol. Immunother. 2017, 66, 25–32. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef]
- Darnell, E.P.; Mooradian, M.J.; Baruch, E.N.; Yilmaz, M.; Reynolds, K.L. Immune-Related Adverse Events (irAEs): Diagnosis, Management, and Clinical Pearls. Curr. Oncol. Rep. 2020, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Carriero, F.; Rubino, V.; Leone, S.; Montanaro, R.; Brancaleone, V.; Ruggiero, G.; Terrazzano, G. Regulatory TR3-56 Cells in the Complex Panorama of Immune Activation and Regulation. Cells 2023, 12, 2841. [Google Scholar] [CrossRef]
- Nandi, D.; Pathak, S.; Verma, T.; Singh, M.; Chattopadhyay, A.; Thakur, S.; Raghavan, A.; Gokhroo, A.; Vijayamahantesh. T cell costimulation, checkpoint inhibitors and anti-tumor therapy. J. Biosci. 2020, 45, 50. [Google Scholar] [CrossRef]
- Johnson, P.C.; Abramson, J.S. Engineered T Cells: CAR T Cell Therapy and Beyond. Curr. Oncol. Rep. 2022, 24, 23–31. [Google Scholar] [CrossRef]
- Lee, D.J.; Lee, H.J., Jr.; Farmer, J.R.; Reynolds, K.L. Mechanisms Driving Immune-Related Adverse Events in Cancer Patients Treated with Immune Checkpoint Inhibitors. Curr. Cardiol. Rep. 2021, 23, 98. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Vera Aguilera, J.; Chintakuntlawar, A.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef]
- Yin, Q.; Wu, L.; Han, L.; Zheng, X.; Tong, R.; Li, L.; Bai, L.; Bian, Y. Immune-related adverse events of immune checkpoint inhibitors: A review. Front. Immunol. 2023, 14, 1167975. [Google Scholar] [CrossRef]
- Lozano, A.X.; Chaudhuri, A.A.; Nene, A.; Bacchiocchi, A.; Earland, N.; Vesely, M.D.; Usmani, A.; Turner, B.E.; Steen, C.B.; Luca, B.A.; et al. T cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat. Med. 2022, 28, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; Wistuba-Hamprecht, K.; Yuan, J.; Postow, M.A.; Wong, P.; Capone, M.; Madonna, G.; Khammari, A.; Schilling, B.; Sucker, A.; et al. Increases in Absolute Lymphocytes and Circulating CD4+ and CD8+ T Cells Are Associated with Positive Clinical Outcome of Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 4848–4858. [Google Scholar] [CrossRef]
- Tietze, J.K.; Angelova, D.; Heppt, M.V.; Reinholz, M.; Murphy, W.J.; Spannagl, M.; Ruzicka, T.; Berking, C. The proportion of circulating CD45RO+CD8+ memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur. J. Cancer 2017, 75, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; Wistuba-Hamprecht, K.; Geukes Foppen, M.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef]
- MacLennan, I.C. Autoimmunity. Deletion of autoreactive B cells. Curr. Biol. 1995, 5, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Chen, C.; Zhou, H.; Liu, B.; Ma, Q. GTB-PPI: Predict Protein-protein Interactions Based on L1-regularized Logistic Regression and Gradient Tree Boosting. Genom. Proteom. Bioinform. 2020, 18, 582–592. [Google Scholar] [CrossRef]
- de Moel, E.C.; Rozeman, E.A.; Kapiteijn, E.H.; Verdegaal, E.M.E.; Grummels, A.; Bakker, J.A.; Huizinga, T.W.J.; Haanen, J.B.; Toes, R.E.M.; van der Woude, D. Autoantibody Development under Treatment with Immune-Checkpoint Inhibitors. Cancer Immunol. Res. 2019, 7, 6–11. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; Yang, J.Y.H.; Ghazanfar, S.; Kefford, R.F.; et al. Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1-Based Immunotherapy. Clin. Cancer Res. 2019, 25, 1557–1563. [Google Scholar] [CrossRef]
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J. Transl. Med. 2018, 16, 94. [Google Scholar] [CrossRef]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the era of immune checkpoint inhibitors: Friend or foe? Nat. Rev. Rheumatol. 2021, 17, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Link, V.M.; Lima-Junior, D.S.; Delaleu, J.; Bouladoux, N.; Han, S.J.; Collins, N.; Belkaid, Y. Immune checkpoint inhibitors unleash pathogenic immune responses against the microbiota. Proc. Natl. Acad. Sci. USA 2022, 119, e2200348119. [Google Scholar] [CrossRef]
- Nayak, L.; Iwamoto, F.M.; LaCasce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular pathways: Next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, S.; Tempora, P.; Melaiu, O.; Lucarini, V.; Cifaldi, L.; Locatelli, F.; Fruci, D. Targeting the antigen processing and presentation pathway to overcome resistance to immune checkpoint therapy. Front. Immunol. 2022, 13, 948297. [Google Scholar] [CrossRef]
- Žilionytė, K.; Bagdzevičiūtė, U.; Mlynska, A.; Urbštaitė, E.; Paberalė, E.; Dobrovolskienė, N.; Krasko, J.A.; Pašukonienė, V. Functional antigen processing and presentation mechanism as a prerequisite factor of response to treatment with dendritic cell vaccines and anti-PD-1 in preclinical murine LLC1 and GL261 tumor models. Cancer Immunol. Immunother. 2022, 71, 2691–2700. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Ferrone, S. HLA Class I Antigen Processing Machinery Defects in Cancer Cells-Frequency, Functional Significance, and Clinical Relevance with Special Emphasis on Their Role in T Cell-Based Immunotherapy of Malignant Disease. Methods Mol. Biol. 2020, 2055, 325–350. [Google Scholar] [CrossRef]
- Hulpke, S.; Tampé, R. The MHC I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem. Sci. 2013, 38, 412–420. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Saveanu, L.; Carroll, O.; Hassainya, Y.; van Endert, P. Complexity, contradictions, and conundrums: Studying post-proteasomal proteolysis in HLA class I antigen presentation. Immunol. Rev. 2005, 207, 42–59. [Google Scholar] [CrossRef]
- Seliger, B.; Höhne, A.; Knuth, A.; Bernhard, H.; Ehring, B.; Tampé, R.; Huber, C. Reduced membrane major histocompatibility complex class I density and stability in a subset of human renal cell carcinomas with low TAP and LMP expression. Clin. Cancer Res. 1996, 2, 1427–1433. [Google Scholar]
- Ling, A.; Löfgren-Burström, A.; Larsson, P.; Li, X.; Wikberg, M.L.; Öberg, Å.; Stenling, R.; Edin, S.; Palmqvist, R. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology 2017, 6, e1356143. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Reichert, T.E.; Kunkel, M.; Gooding, W.; Whiteside, T.L.; Ferrone, S.; Seliger, B. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: Association with clinical outcome. Clin. Cancer Res. 2005, 11, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Dissemond, J.; Kothen, T.; Mörs, J.; Weimann, T.K.; Lindeke, A.; Goos, M.; Wagner, S.N. Downregulation of tapasin expression in progressive human malignant melanoma. Arch. Dermatol. Res. 2003, 295, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Shionoya, Y.; Kanaseki, T.; Miyamoto, S.; Tokita, S.; Hongo, A.; Kikuchi, Y.; Kochin, V.; Watanabe, K.; Horibe, R.; Saijo, H.; et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology 2017, 6, e1274476. [Google Scholar] [CrossRef]
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8+ T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 279. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Cathro, H.P.; Smolkin, M.E.; Theodorescu, D.; Jo, V.Y.; Ferrone, S.; Frierson, H.F., Jr. Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol. Immunother. 2010, 59, 465–472. [Google Scholar] [CrossRef]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef]
- Peters, S.; Paz-Ares, L.; Herbst, R.S.; Reck, M. Addressing CPI resistance in NSCLC: Targeting TAM receptors to modulate the tumor microenvironment and future prospects. J. Immunother. Cancer 2022, 10, e004863. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Nishihara, R.; Yang, J.; Mima, K.; da Silva, A.; Shi, Y.; Inamura, K.; Cao, Y.; Song, M.; Nowak, J.A.; et al. Tumour CD274 (PD-L1) expression and T cells in colorectal cancer. Gut 2017, 66, 1463–1473. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Noyes, D.; Bag, A.; Oseni, S.; Semidey-Hurtado, J.; Cen, L.; Sarnaik, A.A.; Sondak, V.K.; Adeegbe, D. Tumor-associated Tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2022, 10, e004605. [Google Scholar] [CrossRef]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Zhou, K.; Li, S.; Zhao, Y.; Cheng, K. Mechanisms of drug resistance to immune checkpoint inhibitors in non-small cell lung cancer. Front. Immunol. 2023, 14, 1127071. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, Y.; Wang, J.; Gu, J. Defects in Macrophage Reprogramming in Cancer Therapy: The Negative Impact of PD-L1/PD-1. Front. Immunol. 2021, 12, 690869. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef]
- Ozbay Kurt, F.G.; Lasser, S.; Arkhypov, I.; Utikal, J.; Umansky, V. Enhancing immunotherapy response in melanoma: Myeloid-derived suppressor cells as a therapeutic target. J. Clin. Investig. 2023, 133, e170762. [Google Scholar] [CrossRef]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef]
- Youn, J.I.; Park, S.M.; Park, S.; Kim, G.; Lee, H.J.; Son, J.; Hong, M.H.; Ghaderpour, A.; Baik, B.; Islam, J.; et al. Peripheral natural killer cells and myeloid-derived suppressor cells correlate with anti-PD-1 responses in non-small cell lung cancer. Sci. Rep. 2020, 10, 9050. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Ghonim, M.A.; Ibba, S.V.; Tarhuni, A.F.; Errami, Y.; Luu, H.H.; Dean, M.J.; El-Bahrawy, A.H.; Wyczechowska, D.; Benslimane, I.A.; Del Valle, L.; et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J. Immunother. Cancer 2021, 9, e001643. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Gil, M.; Komorowski, M.P.; Seshadri, M.; Rokita, H.; McGray, A.J.; Opyrchal, M.; Odunsi, K.O.; Kozbor, D. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J. Immunol. 2014, 193, 5327–5337. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Sui, J.; Geng, S.; Muvaffak, A.; Bai, M.; Fuhlbrigge, R.C.; Lo, A.; Yammanuru, A.; Hubbard, L.; Sheehan, J.; et al. Humanization of an anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol. Cancer Ther. 2012, 11, 2451–2461. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef]
- Moser, M. Regulation of Th1/Th2 development by antigen-presenting cells in vivo. Immunobiology 2001, 204, 551–557. [Google Scholar] [CrossRef]
- Rabin, R.L.; Park, M.K.; Liao, F.; Swofford, R.; Stephany, D.; Farber, J.M. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J. Immunol. 1999, 162, 3840–3850. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, Y.; Fang, J.; Liu, W.; Chen, J.; Liu, Z.; Xu, Q. Targeting STAT3 Abrogates Tim-3 Upregulation of Adaptive Resistance to PD-1 Blockade on Regulatory T Cells of Melanoma. Front. Immunol. 2021, 12, 654749. [Google Scholar] [CrossRef]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef]
- Schöffski, P.; Tan, D.S.W.; Martín, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer 2022, 10, e003776. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Ikubo, J.; Yoshikawa, H.; Maenaka, K.; Ishimaru, N.; Kosako, H.; et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 2022, 55, 912–924.e8. [Google Scholar] [CrossRef] [PubMed]
- Datar, I.; Sanmamed, M.F.; Wang, J.; Henick, B.S.; Choi, J.; Badri, T.; Dong, W.; Mani, N.; Toki, M.; Mejías, L.D.; et al. Expression Analysis and Significance of PD-1, LAG-3, and TIM-3 in Human Non-Small Cell Lung Cancer Using Spatially Resolved and Multiparametric Single-Cell Analysis. Clin. Cancer Res. 2019, 25, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [Google Scholar] [CrossRef]
- Hosseinkhani, N.; Shadbad, M.A.; Asghari Jafarabadi, M.; Karim Ahangar, N.; Asadzadeh, Z.; Mohammadi, S.M.; Lotfinejad, P.; Alizadeh, N.; Brunetti, O.; Fasano, R.; et al. A Systematic Review and Meta-Analysis on the Significance of TIGIT in Solid Cancers: Dual TIGIT/PD-1 Blockade to Overcome Immune-Resistance in Solid Cancers. Int. J. Mol. Sci. 2021, 22, 10389. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Kim, K.; Park, S.; Park, S.Y.; Kim, G.; Park, S.M.; Cho, J.W.; Kim, D.H.; Park, Y.M.; Koh, Y.W.; Kim, H.R.; et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020, 12, 22. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Wang, X.; He, Q.; Shen, H.; Xia, A.; Tian, W.; Yu, W.; Sun, B. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 2019, 71, 731–741. [Google Scholar] [CrossRef]
- Keniry, M.; Parsons, R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008, 27, 5477–5485. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- von Locquenghien, M.; Rozalén, C.; Celià-Terrassa, T. Interferons in cancer immunoediting: Sculpting metastasis and immunotherapy response. J. Clin. Investig. 2021, 131, e143296. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Takeda, K.; Nakayama, M.; Hayakawa, Y.; Kojima, Y.; Ikeda, H.; Imai, N.; Ogasawara, K.; Okumura, K.; Thomas, D.M.; Smyth, M.J. IFN-γ is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat. Commun. 2017, 8, 14607. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Matijevich, K.; Gajewski, T.F. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J. Immunother. Cancer 2019, 7, 295. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, W.; Yang, W.; Zhou, M.; Liu, F. Acquired resistance for immune checkpoint inhibitors in cancer immunotherapy: Challenges and prospects. Aging 2022, 14, 1048–1064. [Google Scholar] [CrossRef]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 2018, 3, e120360. [Google Scholar] [CrossRef]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [PubMed]
- Karasarides, M.; Cogdill, A.P.; Robbins, P.B.; Bowden, M.; Burton, E.M.; Butterfield, L.H.; Cesano, A.; Hammer, C.; Haymaker, C.L.; Horak, C.E.; et al. Hallmarks of Resistance to Immune-Checkpoint Inhibitors. Cancer Immunol. Res. 2022, 10, 372–383. [Google Scholar] [CrossRef]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef]
- Song, M.; Chen, X.; Wang, L.; Zhang, Y. Future of anti-PD-1/PD-L1 applications: Combinations with other therapeutic regimens. Chin. J. Cancer Res. 2018, 30, 157–172. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Y.; Chlewicki, L.K.; Zhang, Y.; Guo, J.; Liang, W.; Wang, J.; Wang, X.; Fu, Y.X. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 2016, 29, 285–296. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Young, A.; Blake, S.J.; Hill, G.R.; Yagita, H.; Teng, M.W.; Korman, A.J.; Smyth, M.J. Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res. 2016, 76, 6266–6277. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T., Jr.; et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12, eaay8456. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Kudchadkar, R.R.; DeConti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Kapp, K.; Volz, B.; Oswald, D.; Wittig, B.; Baumann, M.; Schmidt, M. Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology 2019, 8, e1659096. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Blake, S.J.; Ching, A.L.; Kenna, T.J.; Galea, R.; Large, J.; Yagita, H.; Steptoe, R.J. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS ONE 2015, 10, e0119483. [Google Scholar] [CrossRef] [PubMed]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Bartlett, N.L.; Herrera, A.F.; Domingo-Domenech, E.; Mehta, A.; Forero-Torres, A.; Garcia-Sanz, R.; Armand, P.; Devata, S.; Izquierdo, A.R.; Lossos, I.S.; et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2020, 136, 2401–2409. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Tien, F.M.; Lu, H.H.; Lin, S.Y.; Tsai, H.C. Epigenetic remodeling of the immune landscape in cancer: Therapeutic hurdles and opportunities. J. Biomed. Sci. 2023, 30, 3. [Google Scholar] [CrossRef]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun. 2018, 9, 248. [Google Scholar] [CrossRef]
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 2000, 165, 7017–7024. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Ebine, K.; Kumar, K.; Pham, T.N.; Shields, M.A.; Collier, K.A.; Shang, M.; DeCant, B.T.; Urrutia, R.; Hwang, R.F.; Grimaldo, S.; et al. Interplay between interferon regulatory factor 1 and BRD4 in the regulation of PD-L1 in pancreatic stellate cells. Sci. Rep. 2018, 8, 13225. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.E.; Avila, D.E.; Spallanzani, R.G.; Ziblat, A.; Fuertes, M.B.; Lapyckyj, L.; Croci, D.O.; Rabinovich, G.A.; Domaica, C.I.; Zwirner, N.W. Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J. Leukoc. Biol. 2012, 91, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, M.; Cui, M.; Niu, C.; Xu, J.; Zhou, L.; Li, W.; Gao, Y.; Kong, W.; Cui, J.; et al. Epigenetic suppression of the antitumor cytotoxicity of NK cells by histone deacetylase inhibitor valproic acid. Am. J. Cancer Res. 2016, 6, 600–614. [Google Scholar]
- Zhu, S.; Denman, C.J.; Cobanoglu, Z.S.; Kiany, S.; Lau, C.C.; Gottschalk, S.M.; Hughes, D.P.; Kleinerman, E.S.; Lee, D.A. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm. Res. 2015, 32, 779–792. [Google Scholar] [CrossRef]
- Lao, Y.; Shen, D.; Zhang, W.; He, R.; Jiang, M. Immune Checkpoint Inhibitors in Cancer Therapy-How to Overcome Drug Resistance? Cancers 2022, 14, 3575. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef]
- Youngblood, B.; Noto, A.; Porichis, F.; Akondy, R.S.; Ndhlovu, Z.M.; Austin, J.W.; Bordi, R.; Procopio, F.A.; Miura, T.; Allen, T.M.; et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J. Immunol. 2013, 191, 540–544. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Y.; Weng, S.; Xu, H.; Li, L.; Han, X. A New Trend in Cancer Treatment: The Combination of Epigenetics and Immunotherapy. Front. Immunol. 2022, 13, 809761. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenetics 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A. The Role of PI3K Inhibition in the Treatment of Breast Cancer, Alone or Combined with Immune Checkpoint Inhibitors. Front. Mol. Biosci. 2021, 8, 648663. [Google Scholar] [CrossRef]
- Heuser, C.; Renner, K.; Kreutz, M.; Gattinoni, L. Targeting lactate metabolism for cancer immunotherapy—A matter of precision. Semin. Cancer Biol. 2023, 88, 32–45. [Google Scholar] [CrossRef]
- Navarro, C.; Ortega, Á.; Santeliz, R.; Garrido, B.; Chacín, M.; Galban, N.; Vera, I.; De Sanctis, J.B.; Bermúdez, V. Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches. Pharmaceutics 2022, 14, 1303. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, Y.; Wu, X.; Liu, Z.; Liu, X. A novel strategy to fuel cancer immunotherapy: Targeting glucose metabolism to remodel the tumor microenvironment. Front. Oncol. 2022, 12, 931104. [Google Scholar] [CrossRef]
- Li, H.; Zhao, A.; Li, M.; Shi, L.; Han, Q.; Hou, Z. Targeting T-cell metabolism to boost immune checkpoint inhibitor therapy. Front. Immunol. 2022, 13, 1046755. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.; Cammarata, I.; Martire, C.; Focaccetti, C.; Piconese, S.; Buccilli, M.; Mancone, C.; Buzzacchino, F.; Berrios, J.R.G.; D’Alessandris, N.; et al. Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun. Biol. 2020, 3, 85. [Google Scholar] [CrossRef]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed]
- van der Most, R.G.; Currie, A.J.; Mahendran, S.; Prosser, A.; Darabi, A.; Robinson, B.W.; Nowak, A.K.; Lake, R.A. Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: A role for cycling TNFR2-expressing effector-suppressor T cells in limiting effective chemotherapy. Cancer Immunol. Immunother. 2009, 58, 1219–1228. [Google Scholar] [CrossRef]
- Procureur, A.; Simonaggio, A.; Bibault, J.E.; Oudard, S.; Vano, Y.A. Enhance the Immune Checkpoint Inhibitors Efficacy with Radiotherapy Induced Immunogenic Cell Death: A Comprehensive Review and Latest Developments. Cancers 2021, 13, 678. [Google Scholar] [CrossRef]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef]
- Takeda, Y.; Kataoka, K.; Yamagishi, J.; Ogawa, S.; Seya, T.; Matsumoto, M. A TLR3-Specific Adjuvant Relieves Innate Resistance to PD-L1 Blockade without Cytokine Toxicity in Tumor Vaccine Immunotherapy. Cell Rep. 2017, 19, 1874–1887. [Google Scholar] [CrossRef]
- Peña-Romero, A.C.; Orenes-Piñero, E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers 2022, 14, 1681. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Day, D.; Nicholls, S.J.; Segelov, E. Immune Checkpoint Inhibitor Therapy in Oncology: Current Uses and Future Directions: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022, 4, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Cardona, Z.; Sosman, J.A.; Chandra, S.; Huang, W. Endocrine side effects of immune checkpoint inhibitors. Front. Endocrinol. 2023, 14, 1157805. [Google Scholar] [CrossRef]
- Lin, X.; Zong, C.; Zhang, Z.; Fang, W.; Xu, P. Progresses in biomarkers for cancer immunotherapy. MedComm 2023, 4, e387. [Google Scholar] [CrossRef]
- Böger, C.; Behrens, H.M.; Krüger, S.; Röcken, C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology 2017, 6, e1293215. [Google Scholar] [CrossRef]
- Lei, Y.; Li, X.; Huang, Q.; Zheng, X.; Liu, M. Progress and Challenges of Predictive Biomarkers for Immune Checkpoint Blockade. Front. Oncol. 2021, 11, 617335. [Google Scholar] [CrossRef]
- Gutic, B.; Bozanovic, T.; Mandic, A.; Dugalic, S.; Todorovic, J.; Stanisavljevic, D.; Dugalic, M.G.; Sengul, D.; Detanac, D.A.; Sengul, I.; et al. Programmed cell death-1 and its ligands: Current knowledge and possibilities in immunotherapy. Clinics 2023, 78, 100177. [Google Scholar] [CrossRef]
- Ritprajak, P.; Azuma, M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral. Oncol. 2015, 51, 221–228. [Google Scholar] [CrossRef]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef]
- Suzuki, A.; Itami, S.; Ohishi, M.; Hamada, K.; Inoue, T.; Komazawa, N.; Senoo, H.; Sasaki, T.; Takeda, J.; Manabe, M. Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation. Cancer Res. 2003, 63, 674–681. [Google Scholar]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Maleki Vareki, S.; Garrigós, C.; Duran, I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef]
- Cho, J.; Kim, S.J.; Park, W.-Y.; Kim, J.; Woo, J.; Kim, G.; Yoon, S.E.; Ko, Y.H.; Kim, W.S. Immune subtyping of extranodal NK/T-cell lymphoma: A new biomarker and an immune shift during disease progression. Mod. Pathol. 2020, 33, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Walker, J.; Andrew Williams, J.; Bellmunt, J. The evolving role of PD-L1 testing in patients with metastatic urothelial carcinoma. Cancer Treat. Rev. 2020, 82, 101925. [Google Scholar] [CrossRef] [PubMed]
- Morihiro, T.; Kuroda, S.; Kanaya, N.; Kakiuchi, Y.; Kubota, T.; Aoyama, K.; Tanaka, T.; Kikuchi, S.; Nagasaka, T.; Nishizaki, M.; et al. PD-L1 expression combined with microsatellite instability/CD8+ tumor infiltrating lymphocytes as a useful prognostic biomarker in gastric cancer. Sci. Rep. 2019, 9, 4633. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L. Nivolumab versus docetaxel in previously treated patients with advanced non–small-cell lung cancer: Two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Fumet, J.D.; Truntzer, C.; Yarchoan, M.; Ghiringhelli, F. Tumour mutational burden as a biomarker for immunotherapy: Current data and emerging concepts. Eur. J. Cancer 2020, 131, 40–50. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e4. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Wang, F.; Wei, X.L.; Wang, F.H.; Xu, N.; Shen, L.; Dai, G.H.; Yuan, X.L.; Chen, Y.; Yang, S.J.; Shi, J.H.; et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann. Oncol. 2019, 30, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D.; et al. Prevalence of High Tumor Mutational Burden and Association With Survival in Patients With Less Common Solid Tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Berland, L.; Heeke, S.; Humbert, O.; Macocco, A.; Long-Mira, E.; Lassalle, S.; Lespinet-Fabre, V.; Lalvée, S.; Bordone, O.; Cohen, C.; et al. Current views on tumor mutational burden in patients with non-small cell lung cancer treated by immune checkpoint inhibitors. J. Thorac. Dis. 2019, 11, S71–S80. [Google Scholar] [CrossRef]
- Panda, A.; Mehnert, J.M.; Hirshfield, K.M.; Riedlinger, G.; Damare, S.; Saunders, T.; Kane, M.; Sokol, L.; Stein, M.N.; Poplin, E.; et al. Immune Activation and Benefit From Avelumab in EBV-Positive Gastric Cancer. J. Natl. Cancer Inst. 2018, 110, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Randrian, V.; Evrard, C.; Tougeron, D. Microsatellite Instability in Colorectal Cancers: Carcinogenesis, Neo-Antigens, Immuno-Resistance and Emerging Therapies. Cancers 2021, 13, 3063. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef]
- Ganesan, S.; Mehnert, J. Biomarkers for Response to Immune Checkpoint Blockade. Annu. Rev. Cancer Biol. 2020, 4, 331–351. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Muyas, F.; Hauck, H.; Smith, O.C.; Sacks, N.J.; Ely, Z.A.; Jaeger, A.M.; Rideout, W.M., 3rd; Zhang, D.; Bhutkar, A.; et al. Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity. Nat. Genet. 2023, 55, 1686–1695. [Google Scholar] [CrossRef]
- Manca, P.; Corti, F.; Intini, R.; Mazzoli, G.; Miceli, R.; Germani, M.M.; Bergamo, F.; Ambrosini, M.; Cristarella, E.; Cerantola, R.; et al. Tumour mutational burden as a biomarker in patients with mismatch repair deficient/microsatellite instability-high metastatic colorectal cancer treated with immune checkpoint inhibitors. Eur. J. Cancer 2023, 187, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Pilard, C.; Ancion, M.; Delvenne, P.; Jerusalem, G.; Hubert, P.; Herfs, M. Cancer immunotherapy: It’s time to better predict patients’ response. Br. J. Cancer 2021, 125, 927–938. [Google Scholar] [CrossRef]
- Thompson, N.A.; Stewart, G.D.; Welsh, S.J.; Doherty, G.J.; Robinson, M.J.; Neville, B.A.; Vervier, K.; Harris, S.R.; Adams, D.J.; Dalchau, K.; et al. The MITRE trial protocol: A study to evaluate the microbiome as a biomarker of efficacy and toxicity in cancer patients receiving immune checkpoint inhibitor therapy. BMC Cancer 2022, 22, 99. [Google Scholar] [CrossRef]
- Hommes, J.W.; Verheijden, R.J.; Suijkerbuijk, K.P.M.; Hamann, D. Biomarkers of Checkpoint Inhibitor Induced Immune-Related Adverse Events-A Comprehensive Review. Front. Oncol. 2020, 10, 585311. [Google Scholar] [CrossRef]
- Xue, T.; Yan, K.; Cai, Y.; Sun, J.; Chen, Z.; Chen, X.; Wu, W. Prognostic significance of CD163+ tumor-associated macrophages in colorectal cancer. World J. Surg. Oncol. 2021, 19, 186. [Google Scholar] [CrossRef]
- Fujimura, T.; Sato, Y.; Tanita, K.; Kambayashi, Y.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; Funakoshi, T.; Hata, H.; et al. Serum levels of soluble CD163 and CXCL5 may be predictive markers for immune-related adverse events in patients with advanced melanoma treated with nivolumab: A pilot study. Oncotarget 2018, 9, 15542–15551. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Shao, Y.; He, W.; Hu, W.; Xu, Y.; Chen, J.; Wu, C.; Jiang, J. Prognostic Role of Tumor-Infiltrating Lymphocytes in Lung Cancer: A Meta-Analysis. Cell Physiol. Biochem. 2015, 37, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hou, X.; Huang, S.; Tai, R.; Lei, L.; Li, S.; Abuliz, A.; Wang, G.; Yang, S. Biomarkers related to immune checkpoint inhibitors therapy. Biomed. Pharmacother. 2022, 147, 112470. [Google Scholar] [CrossRef]
- Yi, M.; Qin, S.; Chu, Q.; Wu, K. The role of gut microbiota in immune checkpoint inhibitor therapy. Hepatobiliary Surg. Nutr. 2018, 7, 481–483. [Google Scholar] [CrossRef]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol. 2020, 11, 814. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Zhang, S.; Dong, L. Gut microbiota-mediated immunomodulation in tumor. J. Exp. Clin. Cancer Res. 2021, 40, 221. [Google Scholar] [CrossRef]
- Shim, J.A.; Ryu, J.H.; Jo, Y.; Hong, C. The role of gut microbiota in T cell immunity and immune mediated disorders. Int. J. Biol. Sci. 2023, 19, 1178–1191. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Däster, S.R.; et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, P.; Tsantsaridou, A.; Papasotiriou, I.; Toloudi, M.; Chatziioannou, M.; Giamouzis, G. Bacterial and fungal microflora in surgically removed lung cancer samples. J. Cardiothorac. Surg. 2011, 6, 137. [Google Scholar] [CrossRef]
- Ramírez-Labrada, A.G.; Isla, D.; Artal, A.; Arias, M.; Rezusta, A.; Pardo, J.; Gálvez, E.M. The Influence of Lung Microbiota on Lung Carcinogenesis, Immunity, and Immunotherapy. Trends Cancer 2020, 6, 86–97. [Google Scholar] [CrossRef]
- Picardo, S.L.; Coburn, B.; Hansen, A.R. The microbiome and cancer for clinicians. Crit. Rev. Oncol./Hematol. 2019, 141, 1–12. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef]
- Sommariva, M.; Le Noci, V.; Bianchi, F.; Camelliti, S.; Balsari, A.; Tagliabue, E.; Sfondrini, L. The lung microbiota: Role in maintaining pulmonary immune homeostasis and its implications in cancer development and therapy. Cell. Mol. Life Sci. 2020, 77, 2739–2749. [Google Scholar] [CrossRef]
- McLean, A.E.B.; Kao, S.C.; Barnes, D.J.; Wong, K.K.H.; Scolyer, R.A.; Cooper, W.A.; Kohonen-Corish, M.R.J. The emerging role of the lung microbiome and its importance in non-small cell lung cancer diagnosis and treatment. Lung Cancer 2022, 165, 124–132. [Google Scholar] [CrossRef]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef]
- Bingula, R.; Filaire, M.; Radosevic-Robin, N.; Bey, M.; Berthon, J.Y.; Bernalier-Donadille, A.; Vasson, M.P.; Filaire, E. Desired Turbulence? Gut-Lung Axis, Immunity, and Lung Cancer. J. Oncol. 2017, 2017, 5035371. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [PubMed]


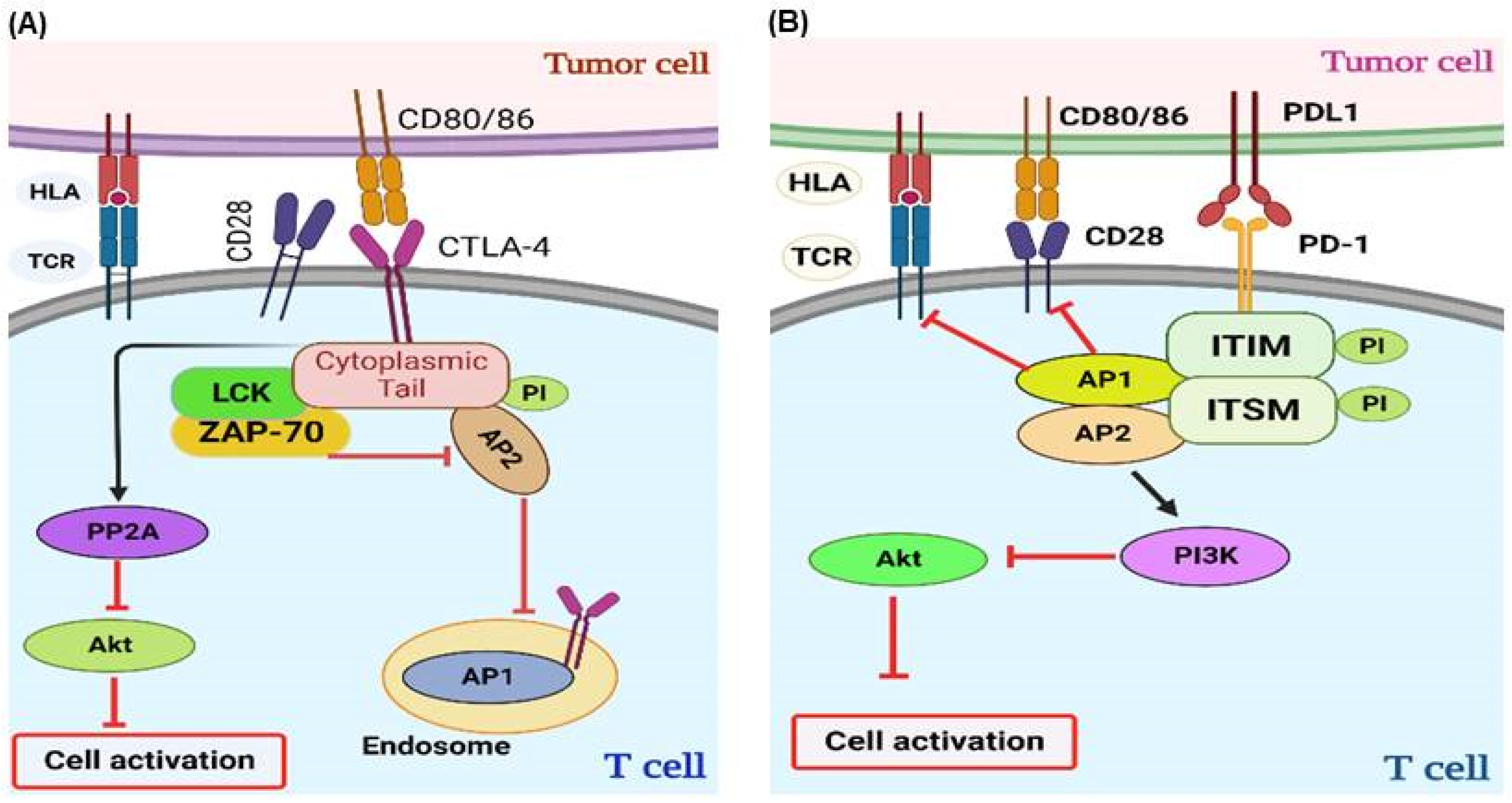

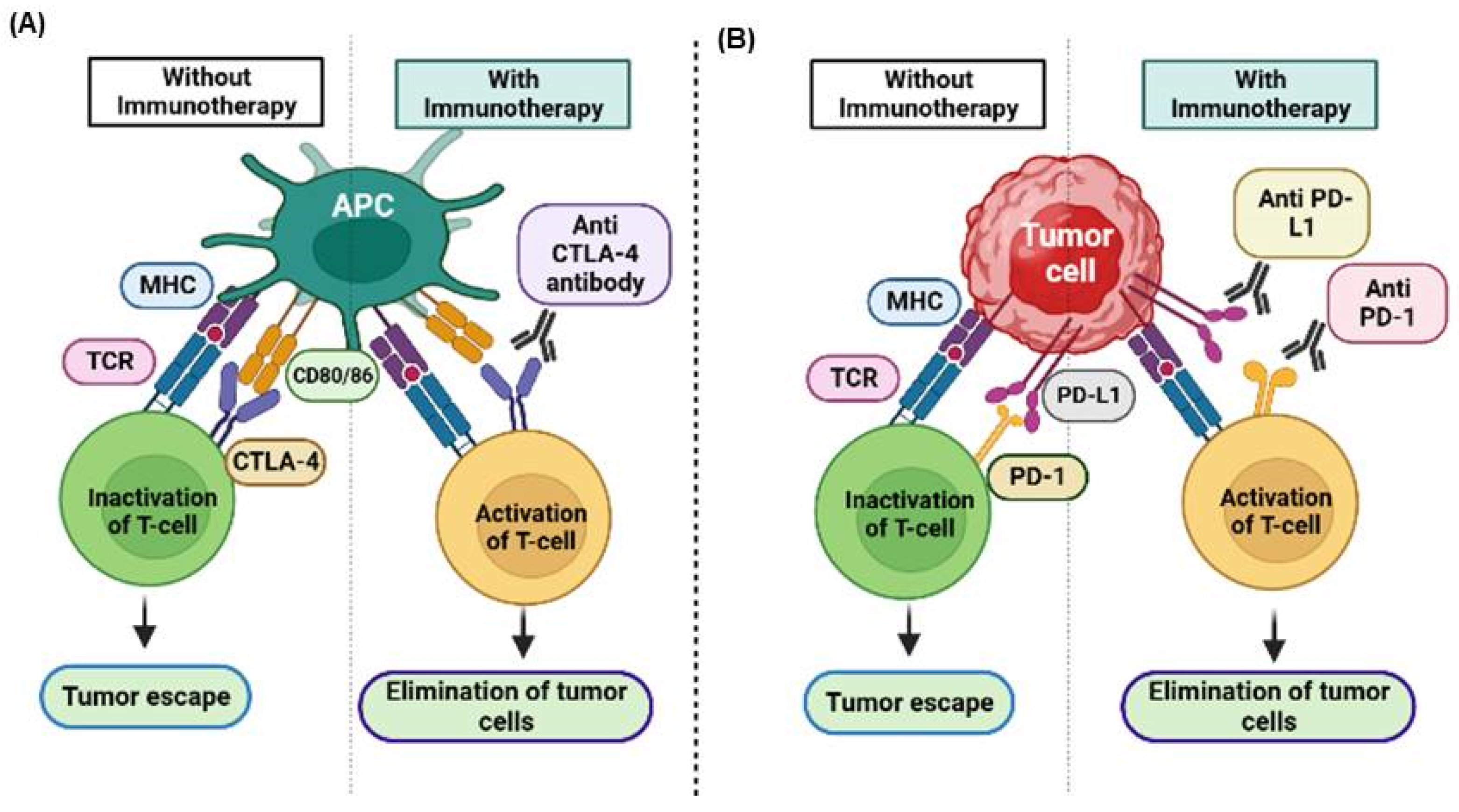
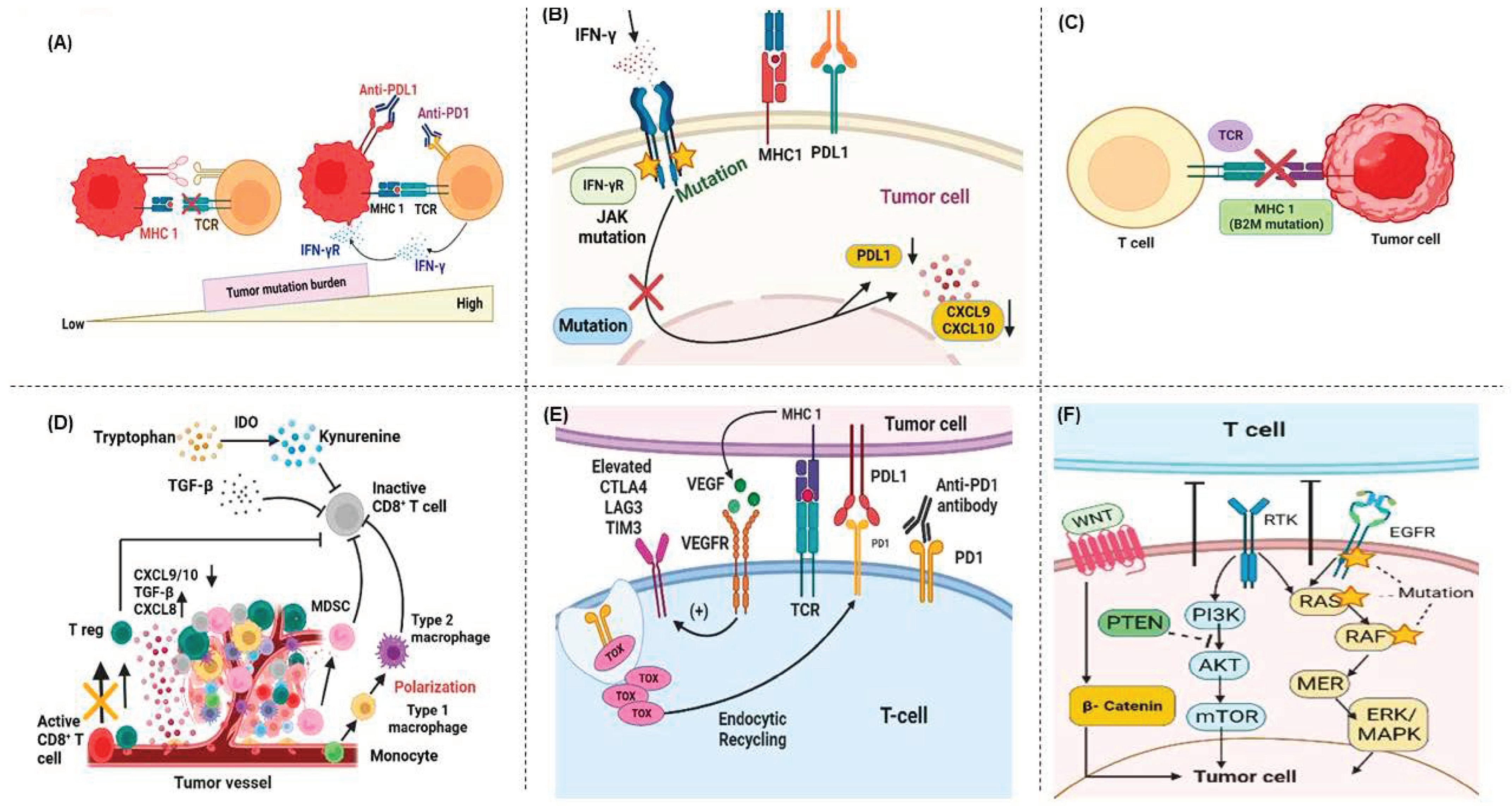
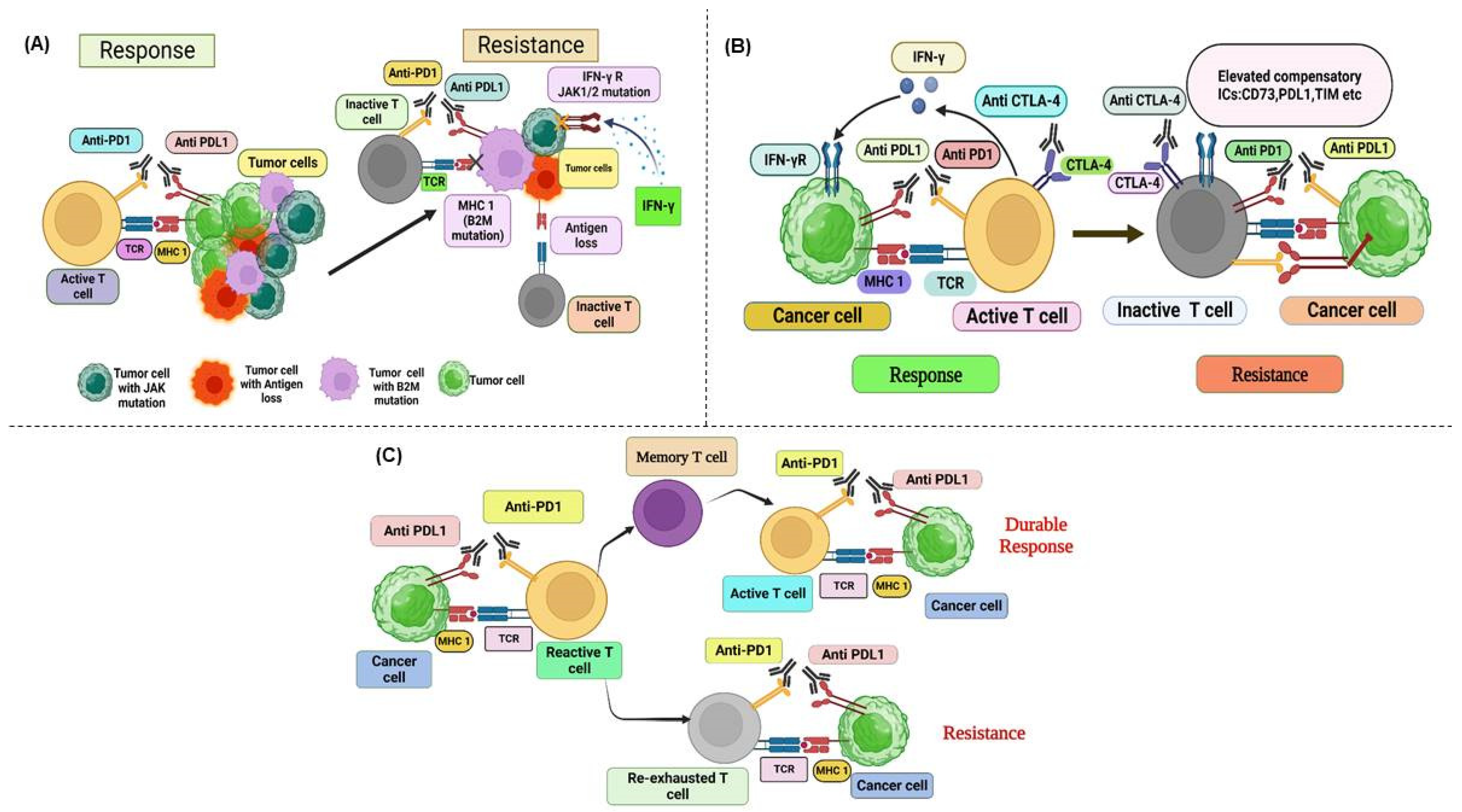
| Mechanism | Type of Strategy | Example | Ref. |
|---|---|---|---|
| Enhancing T-Cell Infiltration | Co-stimulatory agonist | Ab-guided LIGHT fusion protein + anti-PD-L1 Ab | [352] |
| Overcoming T-cell exhaustion | Blockade of alternate co-inhibitory ICIs | Nivolumab + Anti-TIM3 blocking Ab | [183,184] |
| Anti-TIGIT + Anti-PD-1 Ab | [184] | ||
| Co-stimulatory agonists including 4-1BB, OX40, CD40, GITR, and ICOS | Anti-PD-1 Ab + Agonistic anti-CD40 Ab | [353] | |
| Inhibition of epigenetic modifiers | Epigenetic regulation | DNA methyltransferase inhibitor and histone deacetylase inhibitors + anti-PD-1 and anti-CTLA-4 Ab | [354] |
| Improving the Tumor Immune Microenvironment | Cytokine or chemokine receptor blockade | Anti-PD-1 Ab + CSF1R blockade | [273] |
| Anti-PD-1 Ab + CXCR2 mAb blockade | [282] | ||
| CXCR4 mAb blockade | [292] | ||
| IDO inhibitor | Anti-PD-L1 & anti- CTLA-4 Ab + IDO inhibitor | [355] | |
| Inhibition of adenosinergic pathway | Anti-PD-1 Ab + CD73 inhibitor | ||
| Anti-PD-1 Ab + A2AR antagonist | [356] | ||
| Inhibition of TGF-β | Anti-PD-1 Ab + TGF-β blockade | [357] | |
| VEGF inhibitors | Bevacizumab + atezolizumab + chemotherapy | [115] | |
| PI3K inhibitor | Anti-CTLA-4 Ab + Anti-PD-1 Ab + Selective PI3K blockade | [358] | |
| T-Cell Priming Enhancement | Cancer vaccines | Nivolumab + Multipeptide vaccine | [359] |
| Ipilimumab + T-VEC | [146] | ||
| Pembrolizumab + T-VEC | [147] | ||
| Chemotherapy | Carboplatin & paclitaxel + Pembrolizumab | [360] | |
| Radiotherapy | Pembrolizumab + radiotherapy | [361] | |
| TLR agonist | Anti-PD-L1 Ab + TLR3-RNA agonist | ||
| Anti-PD-L1/PD-1 Ab + TLR9 agonist | [362] | ||
| IFN-α | anti-PD-L1-IFN-α fusion protein | [132] | |
| Combination with other therapies | Oncogenic pathway inhibitor | Targeting MAPK pathway with BRAF inhibitor (vemurafenib) + Anti-PD-1/PD-L1 Ab | [363] |
| BRAF, ERK inhibitors + Anti-PD-1 Ab | [364] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almawash, S. Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy. Cancers 2025, 17, 880. https://doi.org/10.3390/cancers17050880
Almawash S. Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy. Cancers. 2025; 17(5):880. https://doi.org/10.3390/cancers17050880
Chicago/Turabian StyleAlmawash, Saud. 2025. "Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy" Cancers 17, no. 5: 880. https://doi.org/10.3390/cancers17050880
APA StyleAlmawash, S. (2025). Revolutionary Cancer Therapy for Personalization and Improved Efficacy: Strategies to Overcome Resistance to Immune Checkpoint Inhibitor Therapy. Cancers, 17(5), 880. https://doi.org/10.3390/cancers17050880