
LiverSCA 2.0: An Enhanced Comprehensive Cell Atlas for Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma

 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample and Data Processing
2.2. Quality Control of Data
2.3. Batch Effect Correction
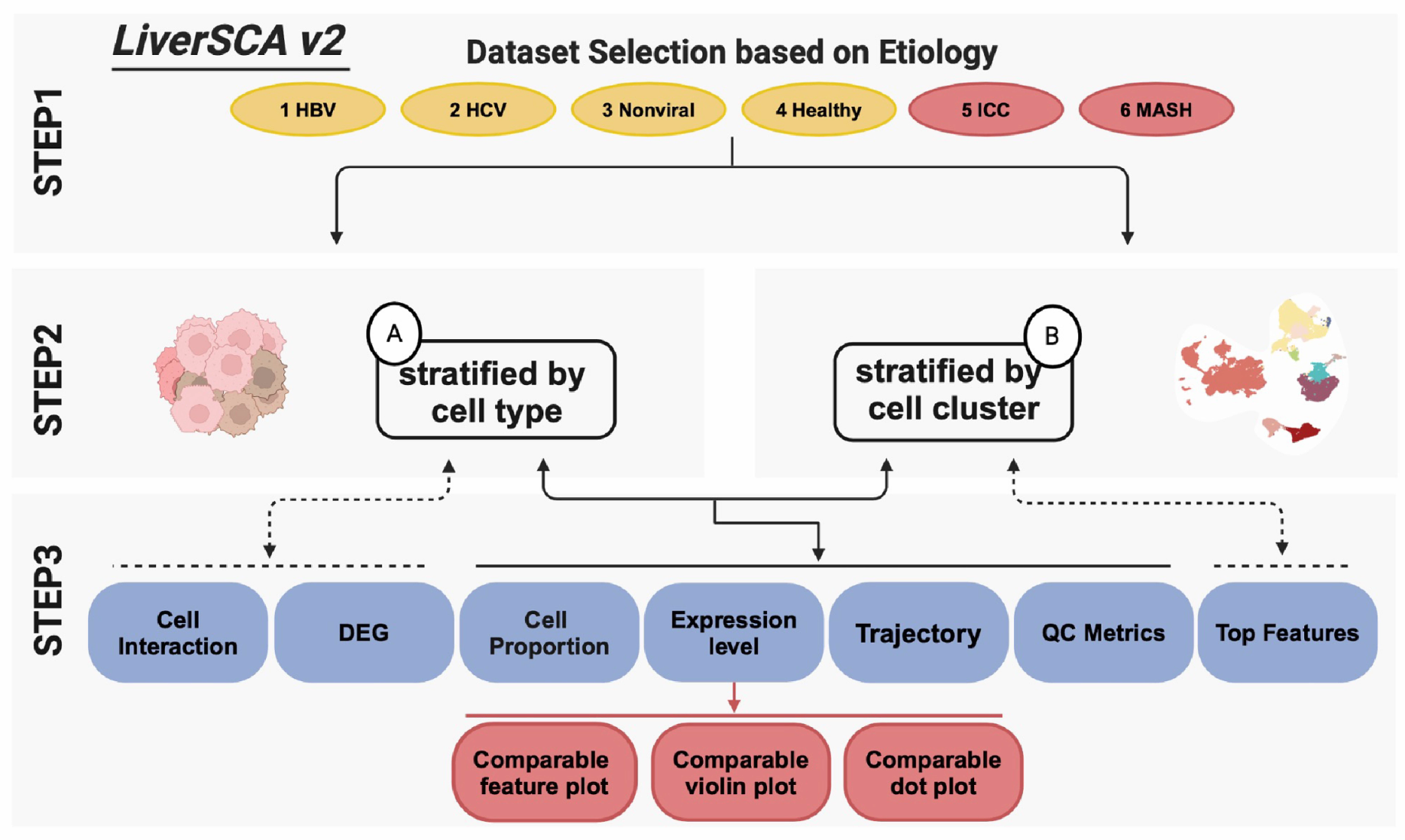
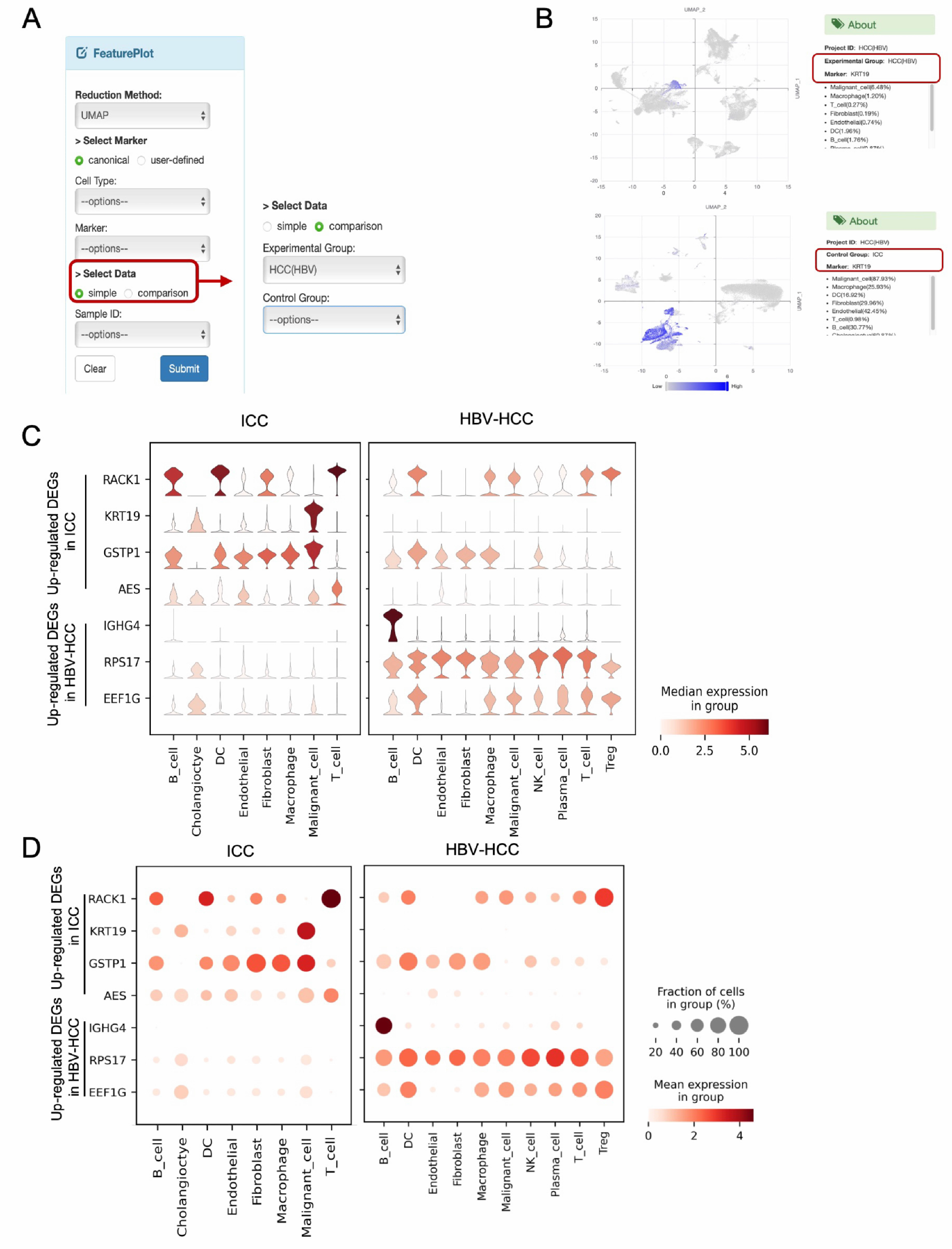
2.4. Website Building
3. Results
3.1. Overview of New Data
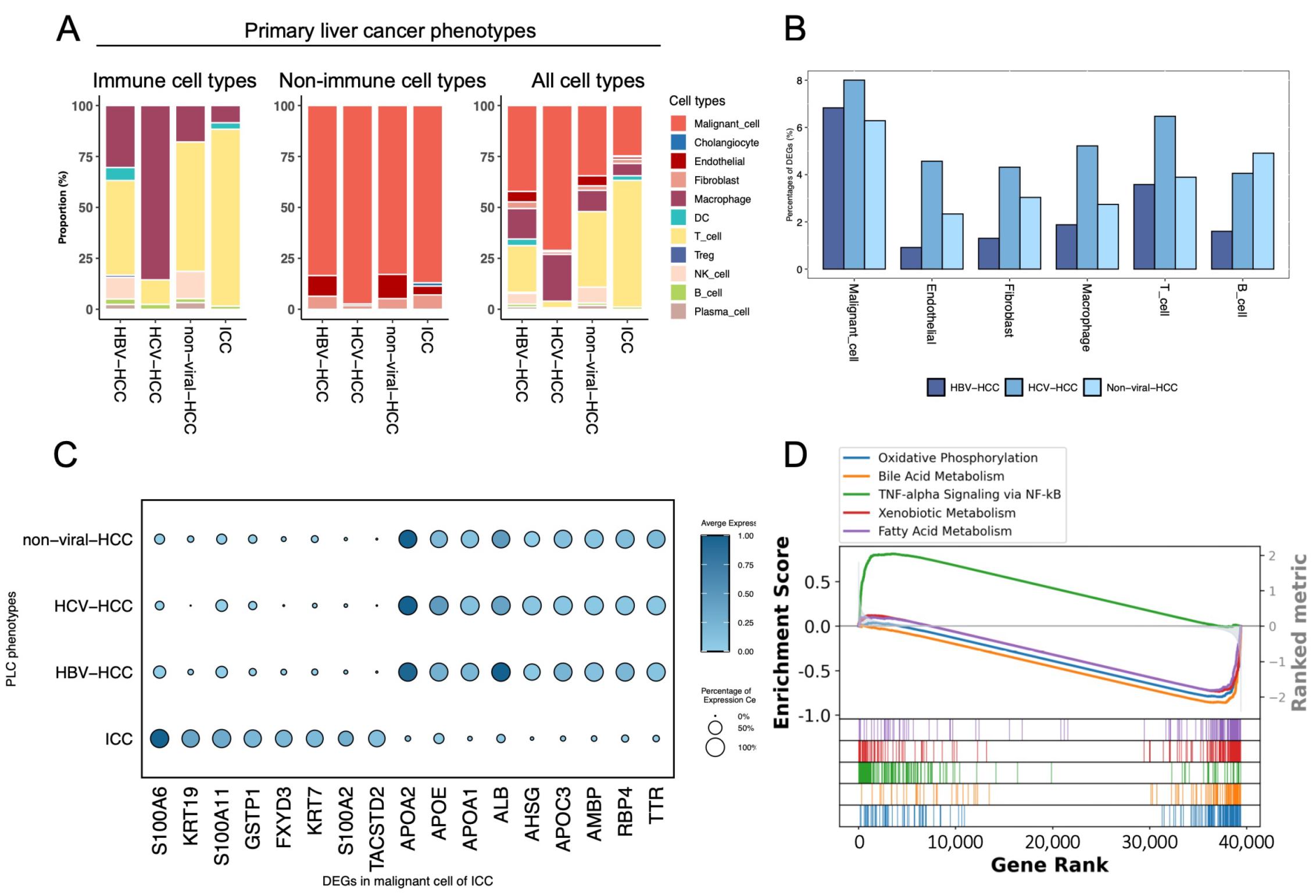
3.2. Comparison of Four Different PLC Phenotypes
3.3. Comparative Visualization Approaches
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PLC | Primary liver cancer |
| HCC | Hepatocellular carcinoma |
| ICC | Intrahepatic cholangiocarcinoma |
| MASLD | Metabolic dysfunction fatty liver disease |
| HBV | Hepatitis B viruses |
| HCV | Hepatitis C viruses |
| ICIs | Immune-checkpoint inhibitors |
| TKIs | Tyrosine kinase inhibitors |
| ScRNA-seq | Single-cell RNA sequencing |
| LSECs | Liver sinusoid endothelial cells |
| TME | Tumor microenvironment |
| TAMs | Tumor-associated macrophages |
| GSEA | Gene set enrichment analysis |
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma (primer). Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Willoughby, C.E.; Singal, A.G.; Greten, T.F.; Heikenwälder, M.; El-Serag, H.B.; Finn, R.S.; Friedman, S.L. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: Pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 487–503. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Pinyol, R.; Kelley, R.K.; El-Khoueiry, A.; Reeves, H.L.; Wang, X.W.; Gores, G.J.; Villanueva, A. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat. Cancer 2022, 3, 386–401. [Google Scholar] [CrossRef]
- Moris, D.; Palta, M.; Kim, C.; Allen, P.J.; Morse, M.A.; Lidsky, M.E. Advances in the treatment of intrahepatic cholangiocarcinoma: An overview of the current and future therapeutic landscape for clinicians. CA Cancer J. Clin. 2023, 73, 198–222. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Komuta, M.; Roskams, T.; Gores, G.J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 512–522. [Google Scholar] [CrossRef]
- Brindley, P.J.; Bachini, M.; Ilyas, S.I.; Khan, S.A.; Loukas, A.; Sirica, A.E.; Teh, B.T.; Wongkham, S.; Gores, G.J. Cholangiocarcinoma. Nat. Rev. Dis. Prim. 2021, 7, 65. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef]
- Lok, A.S.; Seeff, L.B.; Morgan, T.R.; Di Bisceglie, A.M.; Sterling, R.K.; Curto, T.M.; Everson, G.T.; Lindsay, K.L.; Lee, W.M.; Bonkovsky, H.L.; et al. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology 2009, 136, 138–148. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, J.; Li, H.Y.; Wang, Z.H.; Wu, J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim. Biophys. Acta (Bba)—Rev. Cancer 2020, 1874, 188441. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bhalla, S.; Kaur, D.; Raghava, G.P. CancerLivER: A database of liver cancer gene expression resources and biomarkers. Database 2020, 2020, baaa012. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.; Li, Q.; Feng, W.; Jia, X.; Zhou, R.; Huang, Y.; Li, Y.; Hu, Z.; Hu, X.; et al. GepLiver: An integrative liver expression atlas spanning developmental stages and liver disease phases. Sci. Data 2023, 10, 376. [Google Scholar] [CrossRef]
- Long, R.; Suoangbaji; Ng, I.O.L.; Ho, D.W.H. LiverSCA: A comprehensive and user-friendly cell atlas in human hepatocellular carcinoma. Comput. Struct. Biotechnol. J. 2024, 23, 2740–2745. [Google Scholar] [CrossRef] [PubMed]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; Von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yang, A.; Quan, C.; Pan, Y.; Zhang, H.; Li, Y.; Gao, C.; Lu, H.; Wang, X.; Cao, P.; et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat. Commun. 2022, 13, 4594. [Google Scholar] [CrossRef]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 2020, 183, 377–394. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Zhang, Q.; Tsui, Y.M.; Zhang, V.X.; Lu, A.J.; Lee, J.M.F.; Lee, E.; Cheung, G.C.H.; Li, P.M.; Cheung, E.T.Y.; Chia, N.H.; et al. Reciprocal interactions between malignant cells and macrophages enhance cancer stemness and M2 polarization in HBV-associated hepatocellular carcinoma. Theranostics 2024, 14, 892. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef]
- Ma, L.; Heinrich, S.; Wang, L.; Keggenhoff, F.L.; Khatib, S.; Forgues, M.; Kelly, M.; Hewitt, S.M.; Saif, A.; Hernandez, J.M.; et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat. Commun. 2022, 13, 7533. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.; Dora, E.; Henderson, B.; Luu, N.; Portman, J.; Matchett, K.; Brice, M.; Marwick, J.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.r.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Song, G.; Shi, Y.; Zhang, M.; Goswami, S.; Afridi, S.; Meng, L.; Ma, J.; Chen, Y.; Lin, Y.; Zhang, J.; et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020, 6, 90. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef]
- Hernandez–Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the Microenvironment in the Pathogenesis and Treatment of Hepatocellular Carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.H. Epigenetics of hepatocellular carcinoma. Clin. Transl. Med. 2019, 8, 13. [Google Scholar] [CrossRef]
- Cheng, K.; Cai, N.; Zhu, J.; Yang, X.; Liang, H.; Zhang, W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun. 2022, 42, 1112–1140. [Google Scholar] [CrossRef]
- Zhang, M.x.; Gan, W.; Jing, C.y.; Zheng, S.s.; Yi, Y.; Zhang, J.; Xu, X.; Lin, J.j.; Zhang, B.h.; Qiu, S.j. S100A11 promotes cell proliferation via P38/MAPK signaling pathway in intrahepatic cholangiocarcinoma. Mol. Carcinog. 2019, 58, 19–30. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Etiology | Sample ID | No. Patients | Tumor Cell | Hepato-Cyte | Cholan-Giocyte | LSEC | Fibro-Blast | Macro-Phage | Dendritic Cell | T Cell | NK Cell | B Cell | Plasma Cell | Total Cell Count |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ICC | ICC_N1 | 5 | 9464 | 0 | 184 | 354 | 393 | 1476 | 260 | 651 | 0 | 172 | 0 | 12,954 |
| ICC_N2 | 3 | 934 | 0 | 0 | 169 | 448 | 1085 | 674 | 25,505 | 0 | 283 | 0 | 29,098 | |
| MASH | MASH_N1 | 10 | 0 | 13 | 0 | 35 | 0 | 3799 | 84 | 27,935 | 17,771 | 2526 | 192 | 52,355 |
| MASH_N2 | 5 | 0 | 138 | 0 | 257 | 35 | 1336 | 28 | 3503 | 961 | 505 | 130 | 6893 | |
| Healthy | healthy_N1 | 5 | 0 | 3732 | 0 | 817 | 40 | 1191 | 0 | 1185 | 843 | 127 | 503 | 8438 |
| HBV | HBV_N1 | 1 | 2598 | 0 | 0 | 853 | 437 | 845 | 58 | 97 | 3 | 16 | 2 | 4909 |
| HBV_N2 | 5 | 4818 | 0 | 0 | 1149 | 883 | 3300 | 466 | 3851 | 1174 | 381 | 364 | 16,386 | |
| HBV_N3 | 9 | 1135 | 0 | 0 | 2119 | 1257 | 431 | 196 | 2144 | 908 | 65 | 191 | 8446 | |
| HBV_N4 | 9 | 25,578 | 0 | 0 | 42 | 10 | 7607 | 1826 | 12,952 | 2180 | 618 | 361 | 51,174 | |
| HCV | HCV_N1 | 2 | 5249 | 0 | 0 | 44 | 95 | 1703 | 0 | 240 | 0 | 47 | 0 | 7378 |
| Non_viral | non_viral_N1 | 1 | 11,165 | 0 | 0 | 743 | 475 | 510 | 0 | 71 | 0 | 1 | 8 | 12,973 |
| non_viral_N2 | 3 | 2518 | 0 | 0 | 584 | 493 | 2315 | 0 | 1441 | 199 | 51 | 119 | 7720 | |
| non_viral_N3 | 5 | 3758 | 0 | 0 | 1173 | 119 | 2259 | 0 | 17,315 | 3744 | 502 | 838 | 29,718 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suoangbaji, T.; Long, R.; Ng, I.O.-L.; Mak, L.L.-Y.; Ho, D.W.-H. LiverSCA 2.0: An Enhanced Comprehensive Cell Atlas for Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancers 2025, 17, 890. https://doi.org/10.3390/cancers17050890
Suoangbaji T, Long R, Ng IO-L, Mak LL-Y, Ho DW-H. LiverSCA 2.0: An Enhanced Comprehensive Cell Atlas for Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancers. 2025; 17(5):890. https://doi.org/10.3390/cancers17050890
Chicago/Turabian StyleSuoangbaji, Tina, Renwen Long, Irene Oi-Lin Ng, Loey Lung-Yi Mak, and Daniel Wai-Hung Ho. 2025. "LiverSCA 2.0: An Enhanced Comprehensive Cell Atlas for Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma" Cancers 17, no. 5: 890. https://doi.org/10.3390/cancers17050890
APA StyleSuoangbaji, T., Long, R., Ng, I. O.-L., Mak, L. L.-Y., & Ho, D. W.-H. (2025). LiverSCA 2.0: An Enhanced Comprehensive Cell Atlas for Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancers, 17(5), 890. https://doi.org/10.3390/cancers17050890