
QSAR-Based Drug Repurposing and RNA-Seq Metabolic Networks Highlight Treatment Opportunities for Hepatocellular Carcinoma Through Pyrimidine Starvation

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Transcriptomic Dataset Retrieval
2.2. Gene Essentiality Analysis
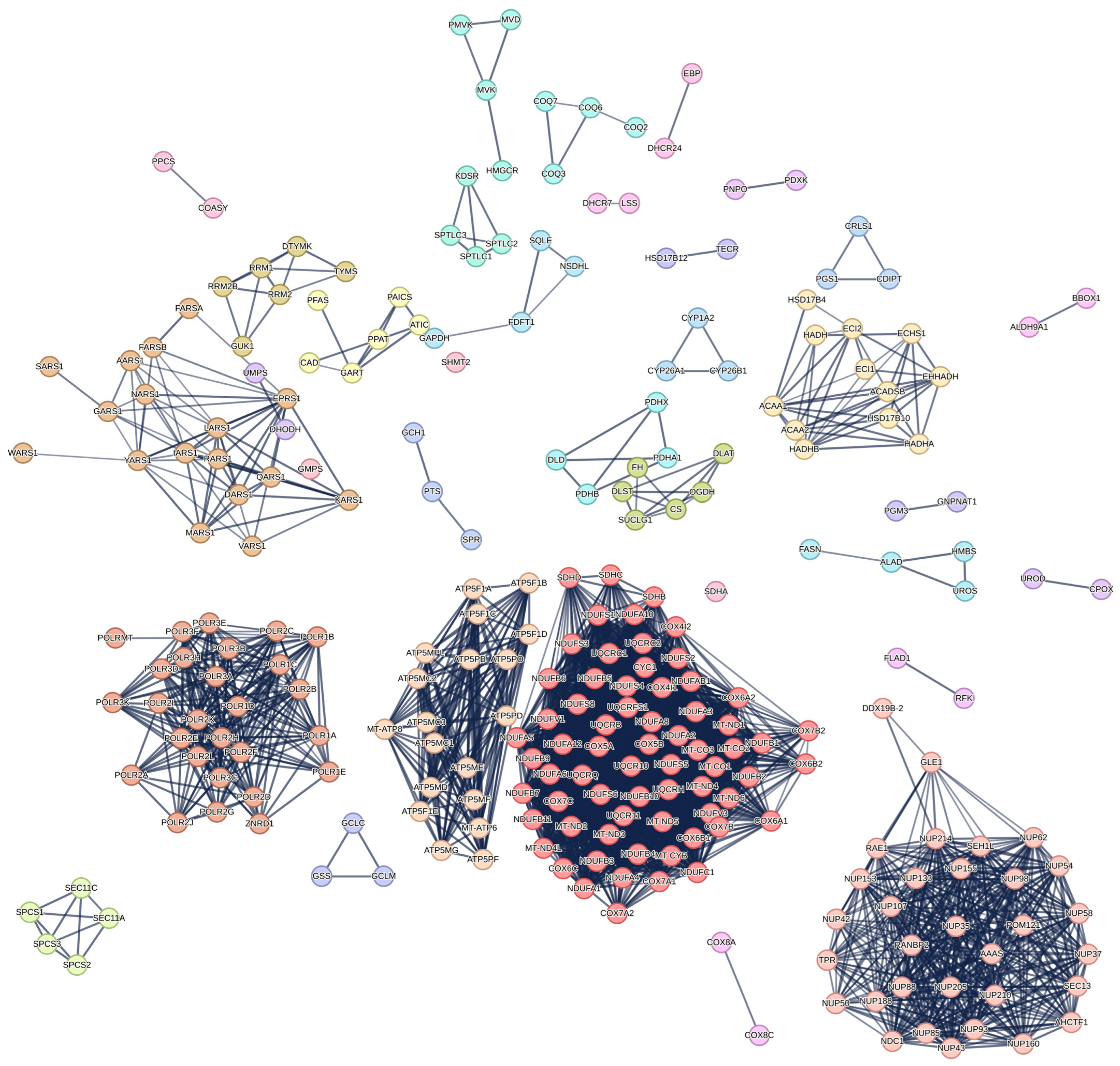
2.3. Functional Enrichment of Lethal Genes
2.4. Identification and Comparison of Differentially Expressed Genes
2.5. External Validation
2.6. ChEMBL Data Collection
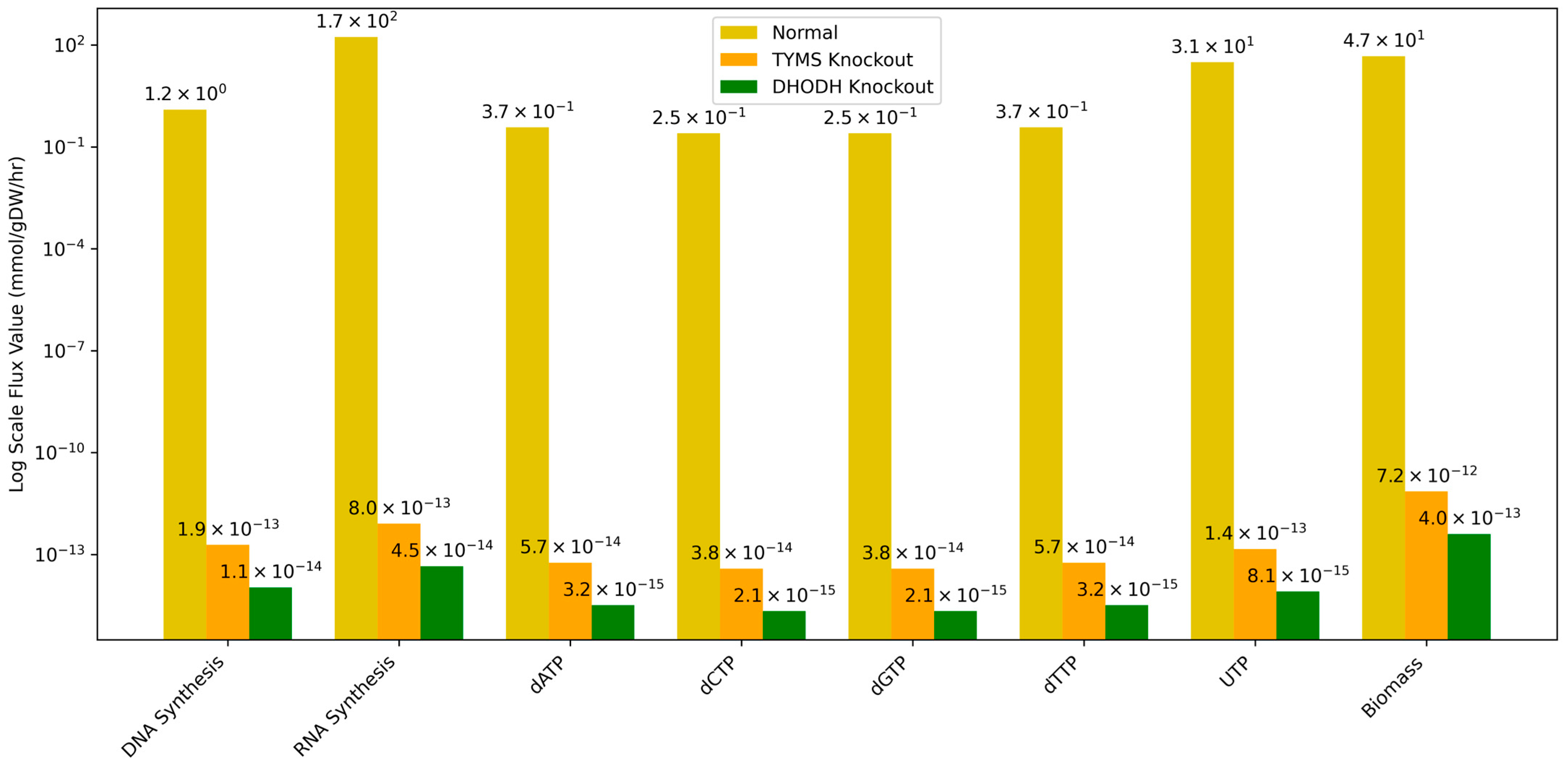
2.7. Flux Balance Analysis and Single Gene Knockout
2.8. D Structure Generation and Molecular Descriptors Calculation
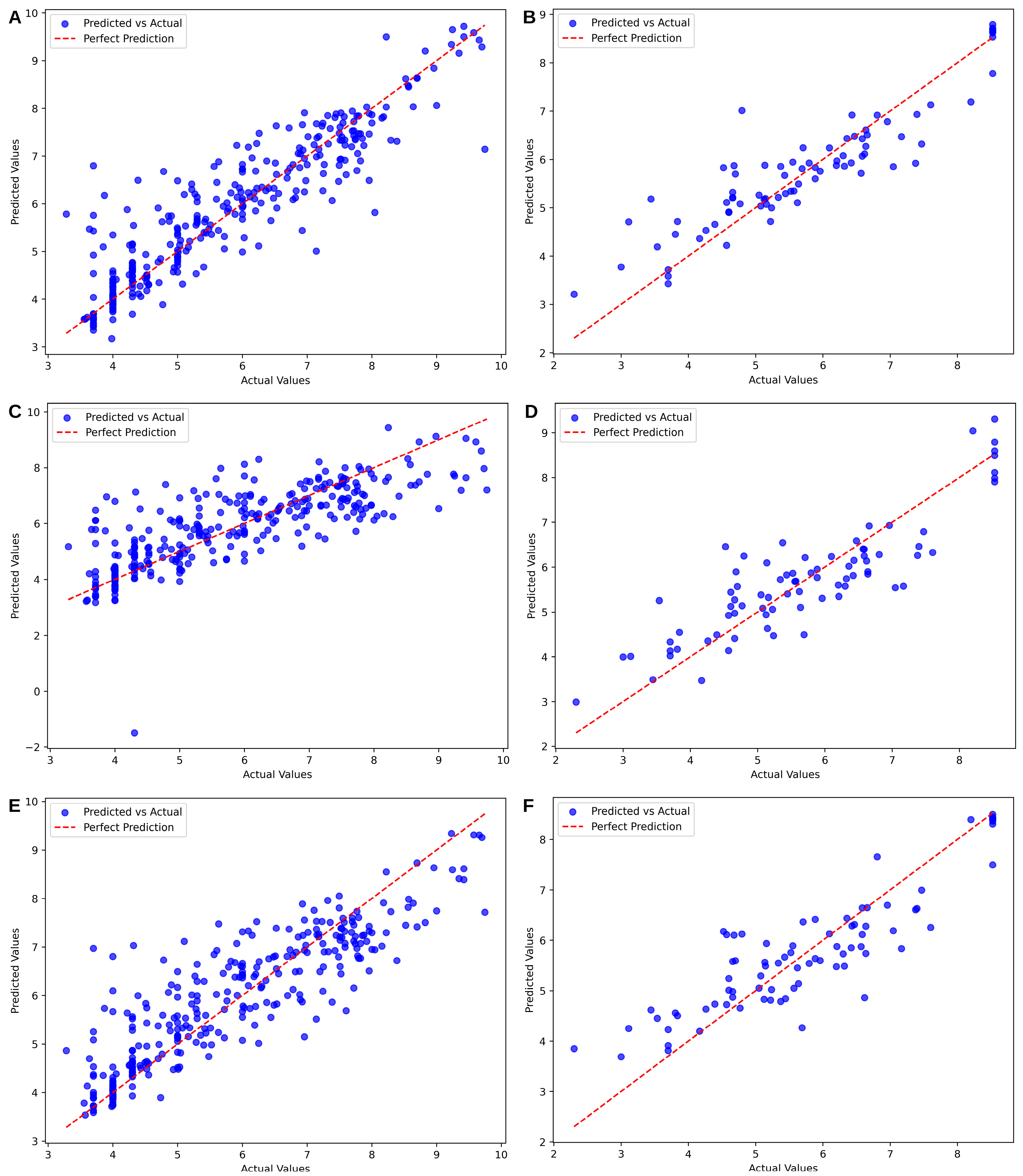
2.9. Feature Selection and QSAR Model Training
2.10. DrugBank Database Collection
2.11. Molecular Docking
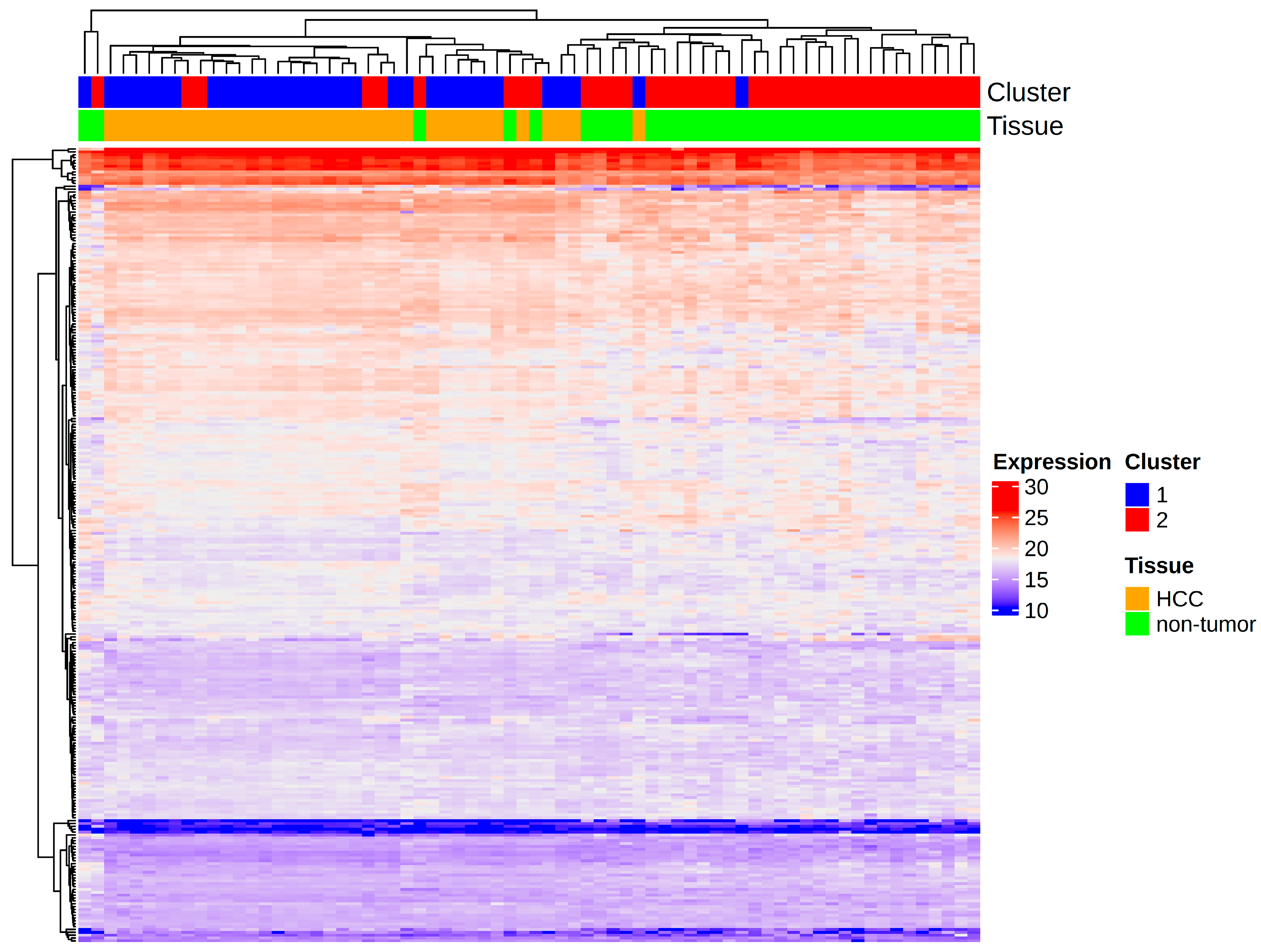
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular Carcinoma |
| QSAR | Quantitative Structure-Activity Relationship |
| BCLC | Barcelona Clinic Liver Cancer |
| AFP | Alpha-Fetoprotein |
| ALBI | Albumin-Bilirubin Index |
| MELD | Model for End-Stage Liver Disease |
| TACE | Transarterial Chemoembolization |
| TKI | Tyrosine Kinase Inhibitor |
| TMB | Tumor Mutational Burden |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| gMCS | Genetic Minimal Cut Set |
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Qazi, A.S. Introduction and Overview of Cancer Therapeutics. Cancer Treat. Res. 2023, 185, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC Strategy for Prognosis Prediction and Treatment Recommendation: The 2022 Update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Ntellas, P.; Chau, I. Updates on Systemic Therapy for Hepatocellular Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e430028. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chen, K.; Zhang, X.; Li, G.; Yuan, K.; Lin, L.; Wu, D.; Wang, J.; Yu, Z.; Chen, Z. Potentiating Sorafenib Efficacy against Hepatocellular Carcinoma via a Carrier-Free Nanomedicine of Artesunate Prodrug. Smart Mater. Med. 2024, 5, 114–126. [Google Scholar] [CrossRef]
- Zheng, S.; Chan, S.W.; Liu, F.; Liu, J.; Chow, P.K.H.; Toh, H.C.; Zheng, S.; Chan, S.W.; Liu, F.; Liu, J.; et al. Hepatocellular Carcinoma: Current Drug Therapeutic Status, Advances and Challenges. Cancers 2024, 16, 1582. [Google Scholar] [CrossRef]
- Orda, M.A.; Fowler, P.M.P.T.; Tayo, L.L. Modular Hub Genes in DNA Microarray Suggest Potential Signaling Pathway Interconnectivity in Various Glioma Grades. Biology 2024, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Mailem, R.C.; Tayo, L.L. Drug Repurposing Using Gene Co-Expression and Module Preservation Analysis in Acute Respiratory Distress Syndrome (ARDS), Systemic Inflammatory Response Syndrome (SIRS), Sepsis, and COVID-19. Biology 2022, 11, 1827. [Google Scholar] [CrossRef] [PubMed]
- Talubo, N.D.D.; Tsai, P.W.; Tayo, L.L. Comprehensive RNA-Seq Gene Co-Expression Analysis Reveals Consistent Molecular Pathways in Hepatocellular Carcinoma across Diverse Risk Factors. Biology 2024, 13, 765. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New Functionalities in the TCGAbiolinks Package for the Study and Integration of Cancer Data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor Package for Integrative Analysis of TCGA Data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327. [Google Scholar] [CrossRef] [PubMed]
- Valcárcel, L.V.; San José-Enériz, E.; Ordoñez, R.; Apaolaza, I.; Olaverri-Mendizabal, D.; Barrena, N.; Valcárcel, A.; Garate, L.; San Miguel, J.; Pineda-Lucena, A.; et al. An Automated Network-Based Tool to Search for Metabolic Vulnerabilities in Cancer. Nat. Commun. 2024, 15, 8685. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Hutchison, G.R. Fast, Efficient Fragment-Based Coordinate Generation for Open Babel. J. Cheminform. 2019, 11, 49. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A Molecular Descriptor Calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, M.; Kang, Y.; Leung, E.L.H.; Lei, T.; Shen, C.; Jiang, D.; Wang, Z.; Cao, D.; Hou, T. Do We Need Different Machine Learning Algorithms for QSAR Modeling? A Comprehensive Assessment of 16 Machine Learning Algorithms on 14 QSAR Data Sets. Brief. Bioinform. 2021, 22, bbaa321. [Google Scholar] [CrossRef]
- Kamboj, S.; Rajput, A.; Rastogi, A.; Thakur, A.; Kumar, M. Targeting Non-Structural Proteins of Hepatitis C Virus for Predicting Repurposed Drugs Using QSAR and Machine Learning Approaches. Comput. Struct. Biotechnol. J. 2022, 20, 3422–3438. [Google Scholar] [CrossRef]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res 2024, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Stork, C.; Mathai, N.; Kirchmair, J. Computational Prediction of Frequent Hitters in Target-Based and Cell-Based Assays. Artif. Intell. Life Sci. 2021, 1, 100007. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Zeng, T.; Zuo, Z.; Luo, Y.; Zhao, Y.; Yu, Y.; Chen, Q. A Novel Series of Human Dihydroorotate Dehydrogenase Inhibitors Discovered by in Vitro Screening: Inhibition Activity and Crystallographic Binding Mode. FEBS Open Bio 2019, 9, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Jansson, A.; Sim, D.; Larsson, A.; Nordlund, P. Structural Analyses of Human Thymidylate Synthase Reveal a Site That May Control Conformational Switching between Active and Inactive States. J. Biol. Chem. 2017, 292, 13449–13458. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and Surveillance for Hepatocellular Carcinoma: New Trends. J. Hepatol. 2020, 72, 250. [Google Scholar] [CrossRef]
- Ladd, A.D.; Duarte, S.; Sahin, I.; Zarrinpar, A. Mechanisms of Drug Resistance in HCC. Hepatology 2024, 79, 926–940. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer Metabolism: Looking Forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, W.; Bian, Y.; Li, Y.; Cong, L. Multifaceted Roles of Aerobic Glycolysis and Oxidative Phosphorylation in Hepatocellular Carcinoma. PeerJ 2023, 11, e14797. [Google Scholar] [CrossRef]
- Wang, W.; Cui, J.; Ma, H.; Lu, W.; Huang, J. Targeting Pyrimidine Metabolism in the Era of Precision Cancer Medicine. Front. Oncol. 2021, 11, 684961. [Google Scholar] [CrossRef]
- Siddiqui, A.; Ceppi, P. A Non-Proliferative Role of Pyrimidine Metabolism in Cancer. Mol. Metab. 2020, 35, 100962. [Google Scholar] [CrossRef]
- Zhu, W.P.; Liu, Z.Y.; Zhao, Y.M.; He, X.G.; Pan, Q.; Zhang, N.; Zhou, J.M.; Wang, L.R.; Wang, M.; Zhan, D.H.; et al. Dihydropyrimidine Dehydrogenase Predicts Survival and Response to Interferon-α in Hepatocellular Carcinoma. Cell Death Dis. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and Cancer: Promising Prospects to Be Explored. Cancer Metab. 2021, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Miao, M.; Wu, Y.; Pan, D.; Wu, Q.; Kang, Z.; Zeng, J.; Zhong, G.; Liu, C.; Wang, J. DHODH Inhibition Represents a Therapeutic Strategy and Improves Abiraterone Treatment in Castration-Resistant Prostate Cancer. Oncogene 2024, 43, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Fan, R.; Zhao, T.; Lin, R.; Zhuang, J.; Deng, A.; Meng, S.; Hou, Z.; Wei, G. A Novel Mitochondria-Targeting DHODH Inhibitor Induces Robust Ferroptosis and Alleviates Immune Suppression. Pharmacol. Res. 2024, 202, 107115. [Google Scholar] [CrossRef]
- Sykes, D.B. The Emergence of Dihydroorotate Dehydrogenase (DHODH) as a Therapeutic Target in Acute Myeloid Leukemia. Expert. Opin. Ther. Targets 2018, 22, 893. [Google Scholar] [CrossRef]
- Li, X.; Yu, Q.; Zhao, R.; Guo, X.; Liu, C.; Zhang, K.; Zhang, W.; Liu, J.; Yu, J.; Wang, S.; et al. Designer Exosomes for Targeted Delivery of a Novel Therapeutic Cargo to Enhance Sorafenib-Mediated Ferroptosis in Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 898156. [Google Scholar] [CrossRef]
- Bhaskar, L.V.K.S.; Saikrishna, L. Molecular Markers for Treatment Response and Toxicity of Gemcitabine. In Breaking Tolerance to Pancreatic Cancer Unresponsiveness to Chemotherapy; Academic Press: Cambridge, MA, USA, 2019; pp. 175–195. [Google Scholar] [CrossRef]
- Geng, Y.; Xie, L.; Wang, Y.; Wang, Y. Unveiling the Oncogenic Significance of Thymidylate Synthase in Human Cancers. medRxiv 2024. [Google Scholar] [CrossRef]
- Guijarro, M.V.; Nawab, A.; Dib, P.; Burkett, S.; Luo, X.; Feely, M.; Nasri, E.; Seifert, R.P.; Kaye, F.J.; Zajac-Kaye, M. TYMS Promotes Genomic Instability and Tumor Progression in Ink4a/Arf Null Background. Oncogene 2023, 42, 1926–1939. [Google Scholar] [CrossRef]
- Wang, L.; Shi, C.; Yu, J.; Xu, Y. FOXM1-Induced TYMS Upregulation Promotes the Progression of Hepatocellular Carcinoma. Cancer Cell Int. 2022, 22, 47. [Google Scholar] [CrossRef]
- Siddiqui, A.; Gollavilli, P.N.; Schwab, A.; Vazakidou, M.E.; Ersan, P.G.; Ramakrishnan, M.; Pluim, D.; Coggins, S.A.; Saatci, O.; Annaratone, L.; et al. Thymidylate Synthase Maintains the De-Differentiated State of Triple Negative Breast Cancers. Cell Death Differ. 2019, 26, 2223–2236. [Google Scholar] [CrossRef] [PubMed]
- Mteremko, D.; Chilongola, J.; Paluch, A.S.; Chacha, M. Targeting Human Thymidylate Synthase: Ensemble-Based Virtual Screening for Drug Repositioning and the Role of Water. J. Mol. Graph. Model. 2023, 118, 108348. [Google Scholar] [CrossRef]
- Ajmal, A.; Mahmood, A.; Hayat, C.; Hakami, M.A.; Alotaibi, B.S.; Umair, M.; Abdalla, A.N.; Li, P.; He, P.; Wadood, A.; et al. Computer-Assisted Drug Repurposing for Thymidylate Kinase Drug Target in Monkeypox Virus. Front. Cell Infect. Microbiol. 2023, 13, 1159389. [Google Scholar] [CrossRef] [PubMed]
- Berber, B.; Doluca, O. A Comprehensive Drug Repurposing Study for COVID19 Treatment: Novel Putative Dihydroorotate Dehydrogenase Inhibitors Show Association to Serotonin–Dopamine Receptors. Brief. Bioinform. 2021, 22, 1023–1037. [Google Scholar] [CrossRef]
- Abdelhameid, M.K.; Zaki, I.; Mohammed, M.R.; Mohamed, K.O. Design, Synthesis, and Cytotoxic Screening of Novel Azole Derivatives on Hepatocellular Carcinoma (HepG2 Cells). Bioorg. Chem. 2020, 101, 103995. [Google Scholar] [CrossRef]
- Sainas, S.; Pippione, A.C.; Giorgis, M.; Lupino, E.; Goyal, P.; Ramondetti, C.; Buccinnà, B.; Piccinini, M.; Braga, R.C.; Andrade, C.H.; et al. Design, Synthesis, Biological Evaluation and X-Ray Structural Studies of Potent Human Dihydroorotate Dehydrogenase Inhibitors Based on Hydroxylated Azole Scaffolds. Eur. J. Med. Chem. 2017, 129, 287–302. [Google Scholar] [CrossRef]
- Xiong, J.-X.; Li, Y.-T.; Tan, X.-Y.; Chen, T.; Liu, B.-H.; Fu, L. Targeting PRSS23 with Tipranavir Induces Gastric Cancer Stem Cell Apoptosis and Inhibits Growth of Gastric Cancer via the MKK3/P38 MAPK-IL24 Pathway. Acta Pharmacol. Sin. 2023, 45, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, Q.; Zhang, L.; Wang, J.; Shu, M.; Huang, K.; Hu, Y.; Lin, Z. Virtual Screening Identifies Tipranavir As a SIRT1 Inhibitor With Anti-Hepatocarcinoma Effect. Future Med. Chem. 2023, 15, 437–451. [Google Scholar] [CrossRef]
- Alkhouri, N.; Imawari, M.; Izumi, N.; Osaki, Y.; Ochiai, T.; Kano, T.; Bentley, R.; Trevisani, F. Lusutrombopag Is Safe and Efficacious for Treatment of Thrombocytopenia in Patients With and Without Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2020, 18, 2600–2608.e1. [Google Scholar] [CrossRef]
- Kong, D.; Jiang, Y.; Miao, X.; Wu, Z.; Liu, H.; Gong, W. Tadalafil Enhances the Therapeutic Efficacy of BET Inhibitors in Hepatocellular Carcinoma through Activating Hippo Pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166267. [Google Scholar] [CrossRef]
- Chhonker, S.K.; Rawat, D.; Koiri, R.K. Repurposing PDE5 Inhibitor Tadalafil and Sildenafil as Anticancer Agent against Hepatocellular Carcinoma via Targeting Key Events of Glucose Metabolism and Multidrug Resistance. J. Biochem. Mol. Toxicol. 2022, 36, e23100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, X.; Han, C.; Wu, L. Structure-Based Virtual Screening towards the Discovery of Novel Thrombin Inhibitors with Anti-HCC Activities. Front. Chem. 2024, 12, 1451574. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.-S.; Han, X.-Y.; Zhou, Z.-Y.; Li, S.-Y.; Zhu, J.-Y.; Zhang, L.; Xue, S.-T. Design and Synthesis of Dabigatran Etexilate Derivatives with Inhibiting Thrombin Activity for Hepatocellular Carcinoma Treatment. Biomed. Pharmacother. 2024, 170, 116018. [Google Scholar] [CrossRef] [PubMed]
- Tabatabai, E.; Khazaei, M.; Asgharzadeh, F.; Nazari, S.E.; Shakour, N.; Fiuji, H.; Ziaeemehr, A.; Mostafapour, A.; Parizadeh, M.R.; Nouri, M.; et al. Inhibition of Angiotensin II Type 1 Receptor by Candesartan Reduces Tumor Growth and Ameliorates Fibrosis in Colorectal Cancer. EXCLI J. 2021, 20, 863. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Normal (mmol/gDW/h) | TYMS Knockout (mmol/gDW/h) | DHODH Knockout (mmol/gDW/h) |
|---|---|---|---|
| DNA Synthesis | 1.25 × 100 | 1.91 × 10−13 | 1.07 × 10−14 |
| RNA Synthesis | 1.72 × 102 | 8.05 × 10−13 | 4.51 × 10−14 |
| dATP | 3.74 × 10−1 | 5.74 × 10−14 | 3.22 × 10−15 |
| dCTP | 2.49 × 10−1 | 3.82 × 10−14 | 2.14 × 10−15 |
| dGTP | 2.49 × 10−1 | 3.82 × 10−14 | 2.14 × 10−15 |
| dTTP | 3.74 × 10−1 | 5.74 × 10−14 | 3.22 × 10−15 |
| UTP | 3.10 × 101 | 1.45 × 10−13 | 8.12 × 10−15 |
| Biomass | 4.67 × 101 | 7.16 × 10−12 | 4.02 × 10−13 |
| Algorithm | Gene | Model Parameters | MAE | RMSE | R2 | PCC | p-Value |
|---|---|---|---|---|---|---|---|
| SVM | DHODH | Kernel: rbf, C:10, gamma: 0.01 | 0.4501 | 0.6709 | 0.8210 | 0.9080 | 3.705 × 10−119 |
| TYMS | Kernel: rbf, C: 10, gamma: 0.01 | 0.3976 | 0.6306 | 0.8101 | 0.9044 | 1.3818 × 10−30 | |
| Ridge Regression | DHODH | Alpha: 1.0, Intercept: True | 0.4885 | 0.6989 | 0.7667 | 0.8757 | 2.2515 × 10−26 |
| TYMS | Alpha: 0.1, Intercept: True | 1.0000 | 1.0000 | 0.6022 | 0.7836 | 4.6468 × 10−19 | |
| XGBoost | DHODH | Estimators: 200, max depth: 5, learning rate: 0.1 | 0.5835 | 0.7639 | 0.7679 | 0.8781 | 2.809 × 10−101 |
| TYMS | Estimators: 200, max depth: 5, learning rate: 0.1 | 0.4946 | 0.7033 | 0.7638 | 0.8771 | 1.4818 × 10−26 |
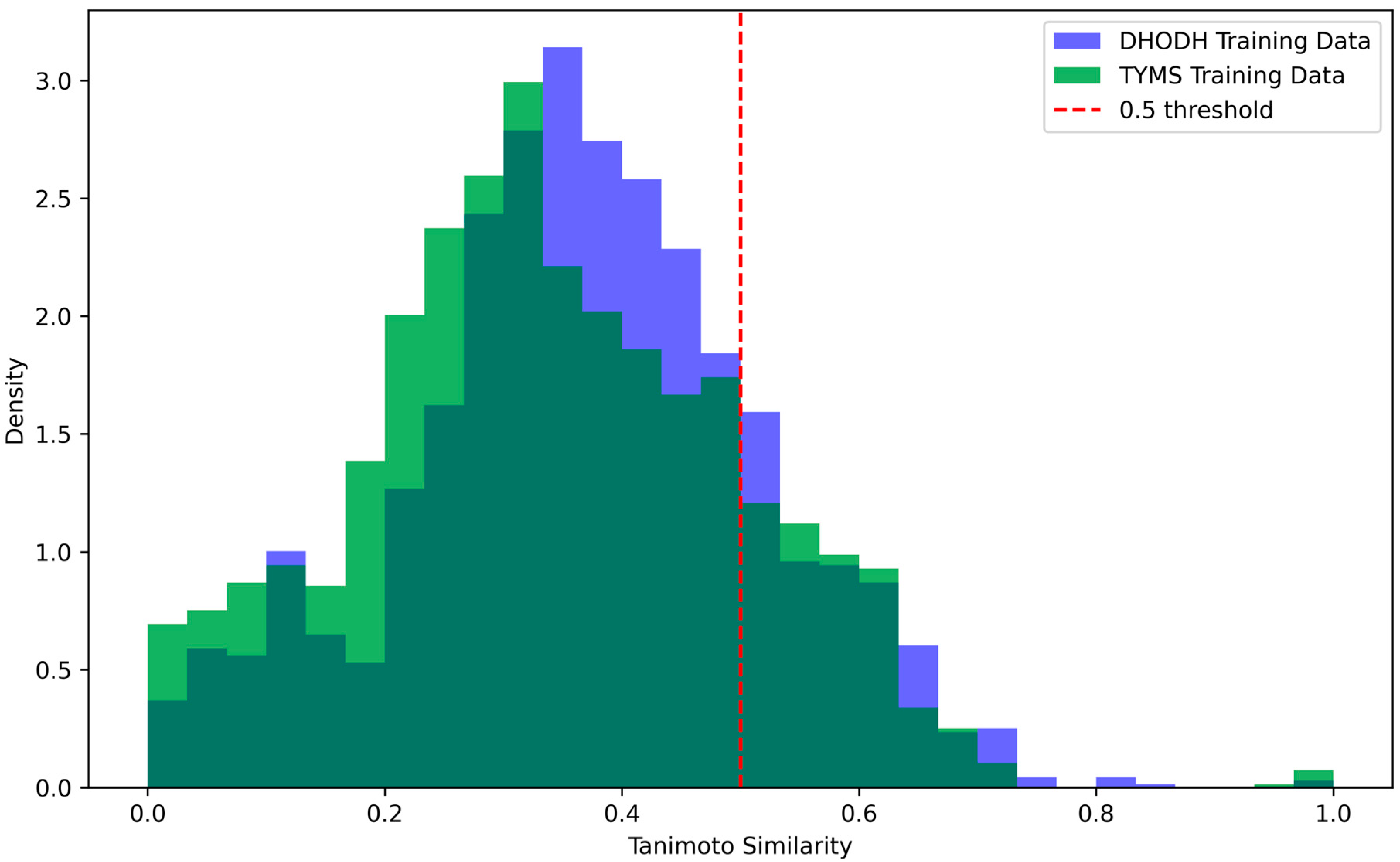
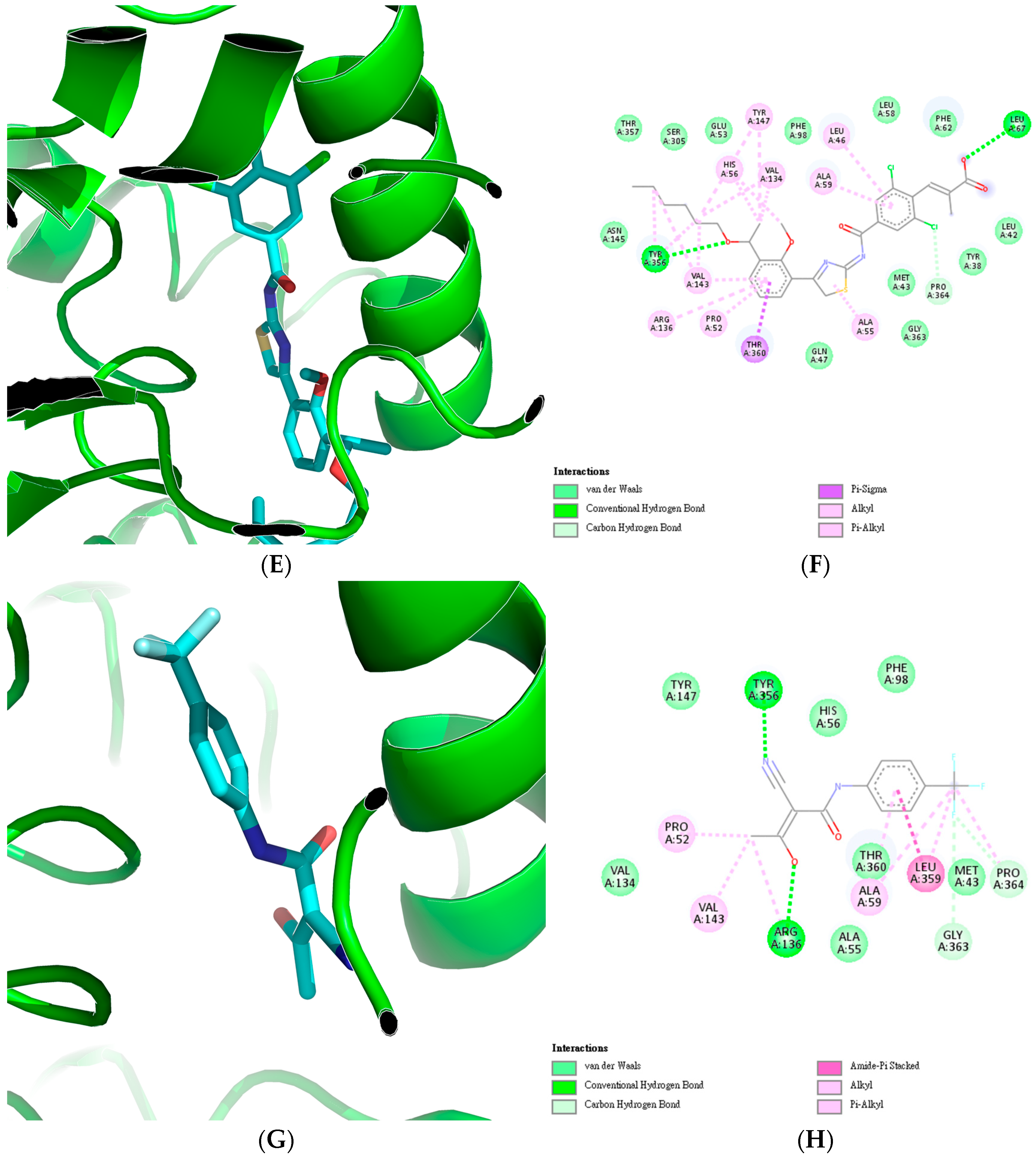
| DrugBank ID | Generic Name | Predicted pIC50 | Binding Energy (kcal/mol) | Tanimoto Similarity | Approved for Cancer | Approved for HCC |
|---|---|---|---|---|---|---|
| DB04865 | Omacetaxine mepesuccinate | 8.2252 | −7.6 | 0.67 | Yes | No |
| DB01076 | Atorvastatin | 7.9997 | −6 | 0.64 | No | No |
| DB00275 | Olmesartan | 7.9988 | −10.3 | 0.53 | No | No |
| DB17472 | Pirtobrutinib | 7.9837 | −13.1 | 0.55 | Yes | No |
| DB09063 | Ceritinib | 7.9404 | −9.3 | 0.54 | Yes | No |
| DB00762 | Irinotecan | 7.8653 | −10.7 | 0.72 | Yes | No |
| DB15569 | Sotorasib | 7.8416 | −6.8 | 0.69 | Yes | No |
| DB00278 | Argatroban | 7.6664 | −9.2 | 0.54 | No | No |
| DB01089 | Deserpidine | 7.6316 | −9.5 | 0.63 | No | No |
| DB13055 | Oteseconazole | 7.5823 | −12 | 0.59 | No | No |
| DB01603 | Meticillin | 7.5724 | −7.4 | 0.55 | No | No |
| DB15444 | Elexacaftor | 7.4930 | −8.2 | 0.67 | No | No |
| DB08911 | Trametinib | 7.4893 | −8.3 | 0.70 | Yes | No |
| DB11431 | Moxidectin | 7.4381 | 9.5 | 0.54 | No | No |
| DB12548 | Sparsentan | 7.3431 | −7.8 | 0.62 | No | No |
| DB11691 | Naldemedine | 7.3320 | −8.2 | 0.76 | No | No |
| DB00932 | Tipranavir | 7.3318 | −11.4 | 0.56 | No | No |
| DB13125 | Lusutrombopag | 7.3279 | −11 | 0.56 | No | No |
| DB15031 | Daridorexant | 7.3097 | −8.6 | 0.58 | No | No |
| DB01112 | Cefuroxime | 7.2844 | −9.9 | 0.61 | No | No |
| DB00255 | Teriflunomide 1 | 6.3896 | −9.8 | 1 | Yes | No |
| DrugBank ID | Generic Name | Predicted pIC50 | Binding Energy (kcal/mol) | Tanimoto Similarity | Approved for Cancer | Approved for HCC |
|---|---|---|---|---|---|---|
| DB09053 | Ibrutinib | 8.0347 | −9.9 | 0.55 | Yes | No |
| DB13125 | Lusutrombopag | 7.8837 | −7.7 | 0.54 | No | No |
| DB13783 | Acemetacin | 7.7353 | −8 | 0.52 | No | No |
| DB15031 | Daridorexant | 7.6796 | −8.3 | 0.58 | No | No |
| DB09330 | Osimertinib | 7.6676 | −7.6 | 0.52 | Yes | No |
| DB15568 | Adagrasib | 7.5618 | −9.2 | 0.64 | Yes | No |
| DB00762 | Irinotecan | 7.5322 | −10.2 | 0.70 | Yes | No |
| DB09063 | Ceritinib | 7.5320 | −8 | 0.51 | Yes | No |
| DB00820 | Tadalafil | 7.5070 | −9.9 | 0.61 | No | No |
| DB00845 | Clofazimine | 7.5037 | −8.5 | 0.53 | No | No |
| DB08881 | Vemurafenib | 7.4767 | −8.8 | 0.51 | Yes | No |
| DB16390 | Mobocertinib | 7.4689 | −7.8 | 0.61 | Yes | No |
| DB00328 | Indomethacin | 7.4268 | −8.5 | 0.50 | No | No |
| DB14840 | Ripretinib | 7.4179 | −8.9 | 0.54 | Yes | No |
| DB08903 | Bedaquiline | 7.4054 | −8.5 | 0.53 | No | No |
| DB13997 | Baloxavir marboxil | 7.3658 | −9.3 | 0.70 | No | No |
| DB15149 | Futibatinib | 7.3568 | −8.1 | 0.54 | Yes | No |
| DB11656 | Rebamipide | 7.3454 | −8.8 | 0.55 | No | No |
| DB14726 | Dabigatran | 7.2764 | −9.5 | 0.54 | No | No |
| DB00796 | Candesartan cilexetil | 7.2675 | −9.3 | 0.59 | No | No |
| DB00293 | Raltitrexed 1 | 6.8772 | −8.4 | 1 | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talubo, N.D.D.; Dela Cruz, E.W.B.; Fowler, P.M.P.T.; Tsai, P.-W.; Tayo, L.L. QSAR-Based Drug Repurposing and RNA-Seq Metabolic Networks Highlight Treatment Opportunities for Hepatocellular Carcinoma Through Pyrimidine Starvation. Cancers 2025, 17, 903. https://doi.org/10.3390/cancers17050903
Talubo NDD, Dela Cruz EWB, Fowler PMPT, Tsai P-W, Tayo LL. QSAR-Based Drug Repurposing and RNA-Seq Metabolic Networks Highlight Treatment Opportunities for Hepatocellular Carcinoma Through Pyrimidine Starvation. Cancers. 2025; 17(5):903. https://doi.org/10.3390/cancers17050903
Chicago/Turabian StyleTalubo, Nicholas Dale D., Emery Wayne B. Dela Cruz, Peter Matthew Paul T. Fowler, Po-Wei Tsai, and Lemmuel L. Tayo. 2025. "QSAR-Based Drug Repurposing and RNA-Seq Metabolic Networks Highlight Treatment Opportunities for Hepatocellular Carcinoma Through Pyrimidine Starvation" Cancers 17, no. 5: 903. https://doi.org/10.3390/cancers17050903
APA StyleTalubo, N. D. D., Dela Cruz, E. W. B., Fowler, P. M. P. T., Tsai, P.-W., & Tayo, L. L. (2025). QSAR-Based Drug Repurposing and RNA-Seq Metabolic Networks Highlight Treatment Opportunities for Hepatocellular Carcinoma Through Pyrimidine Starvation. Cancers, 17(5), 903. https://doi.org/10.3390/cancers17050903