
Granulocyte-Macrophage Colony Stimulating Factor Receptor Contributes to Plexiform Neurofibroma Initiation
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Mouse Dissection and Tumor Number/Size Measurements
2.3. Immunofluorescence (IF) and Immunohistochemistry (IHC)
2.4. Tumor/DRG Processing and Flow Cytometry Analysis
2.5. Mouse Cytokine Array Analysis
2.6. Electron Microscopy
2.7. Statistical Analysis
3. Results
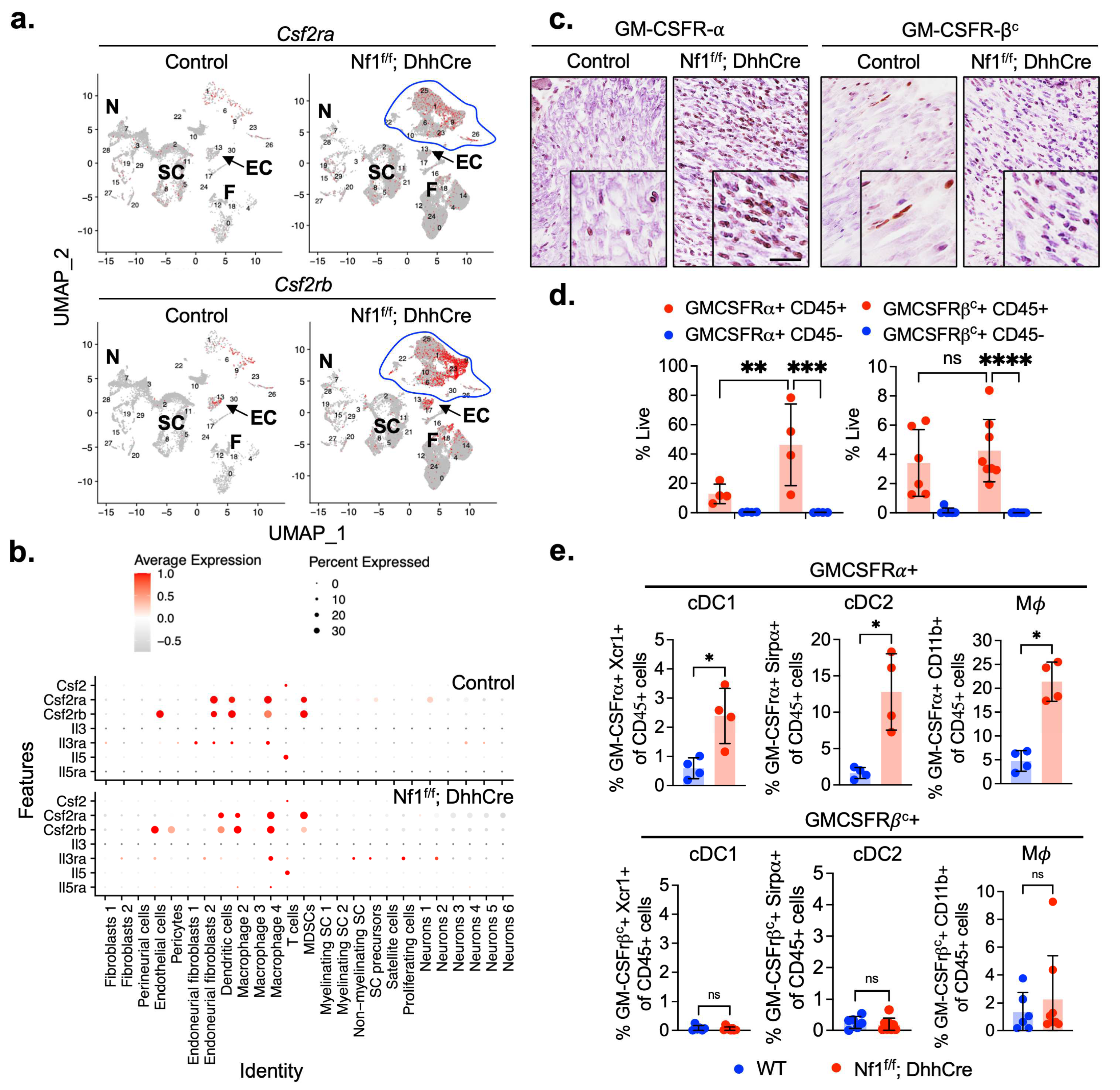
3.1. GM-CSFR-α and GM-CSFR-βc Are Expressed by PNF Macrophages and Dendritic Cells
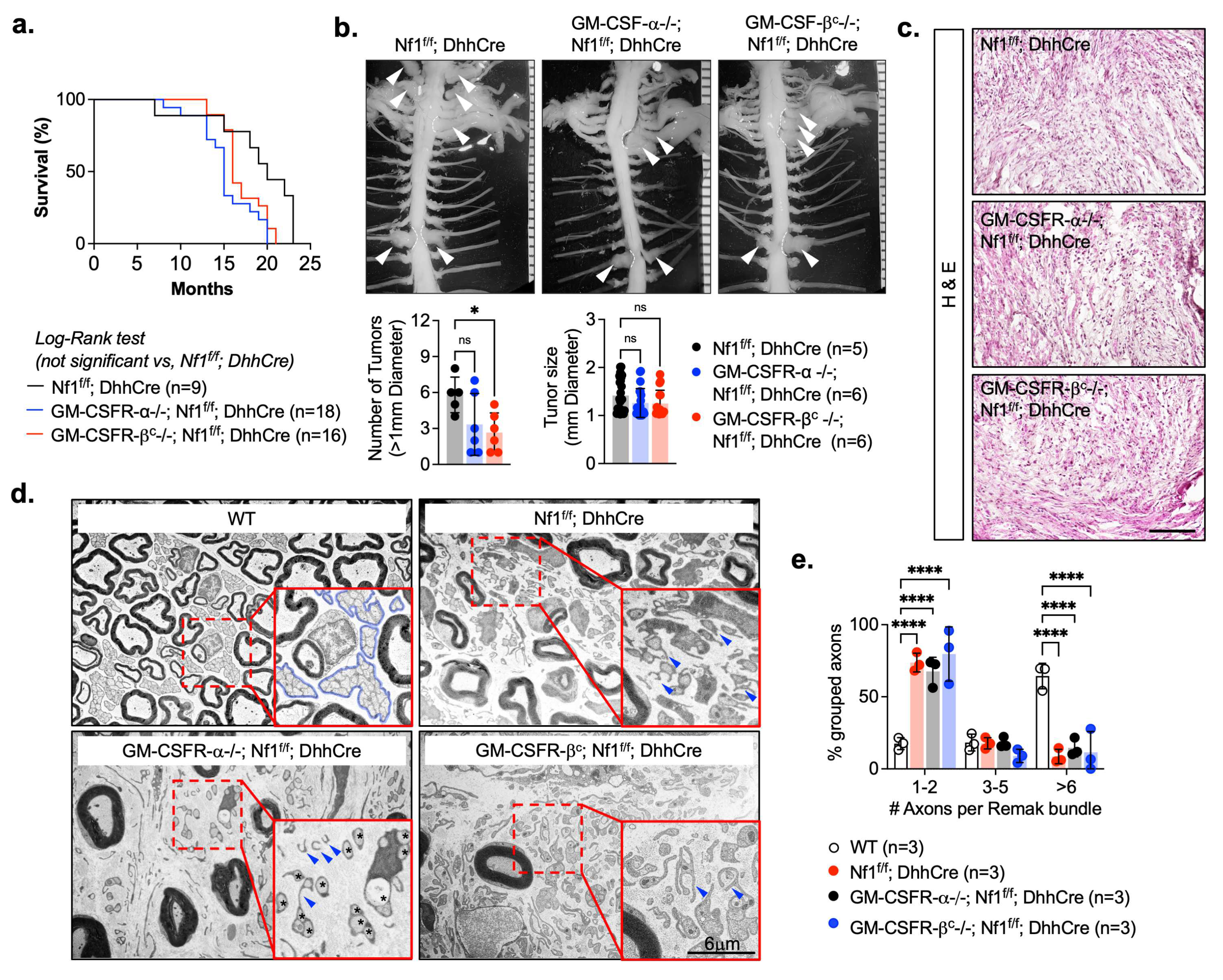
3.2. Csfr2b/Gmcsfr/βc Loss Reduces Neurofibroma Numbers in Nf1f/f; DhhCre Mice
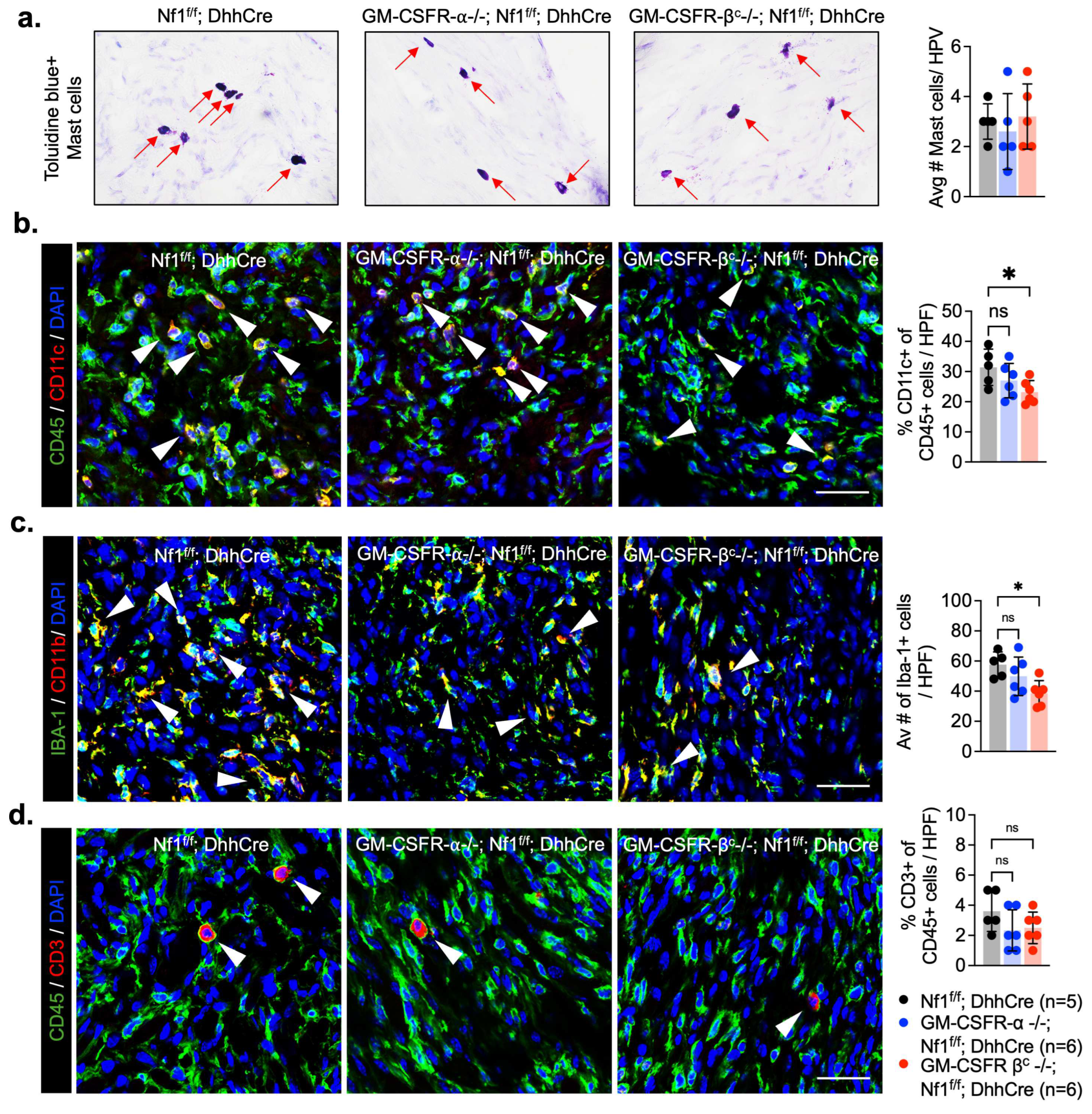
3.3. Deletion of GM-CSFR-βc Reduces Myeloid Cell Infiltrates in PNF
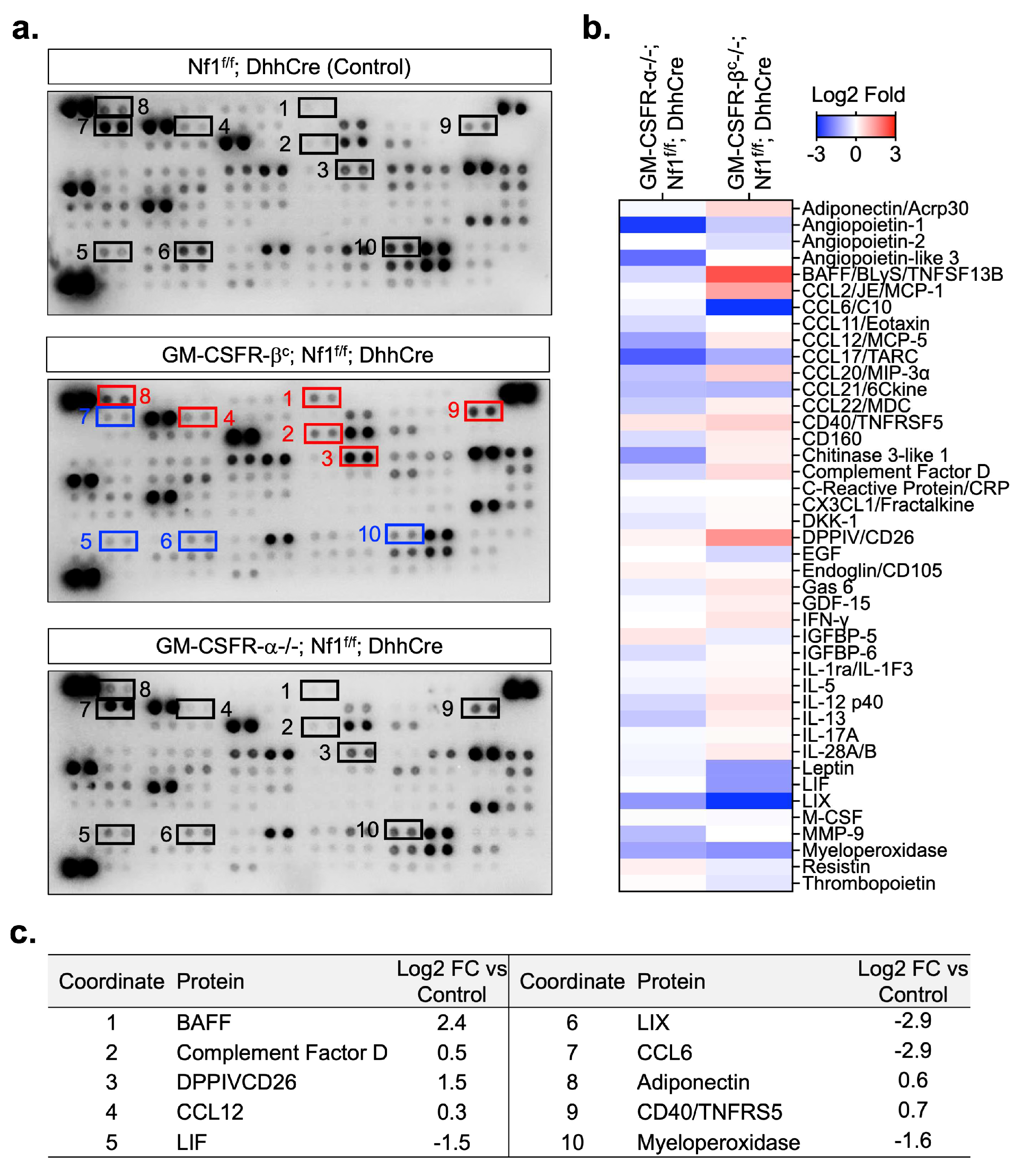
3.4. Loss of GM-CSF Receptors Alters the PNF Inflammatory Proteome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mirza, S.; Walker, A.; Chen, J.; Murphy, J.M.; Young, I.G. The Ig-like domain of human GM-CSF receptor alpha plays a critical role in cytokine binding and receptor activation. Biochem. J. 2010, 426, 307–317. [Google Scholar] [CrossRef]
- Burgess, A.W.; Camakaris, J.; Metcalf, D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J. Biol. Chem. 1977, 252, 1998–2003. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Na, Y.R.; Jung, D.; Gu, G.J.; Seok, S.H. GM-CSF Grown Bone Marrow Derived Cells Are Composed of Phenotypically Different Dendritic Cells and Macrophages. Mol. Cells 2016, 39, 734–741. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Stanley, E.R.; Burgess, A.W.; Shadduck, R.K. Stimulation of macrophage plasminogen activator activity by colony-stimulating factors. J. Cell Physiol. 1980, 103, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D. The colony-stimulating factors and cancer. Nat. Rev. Cancer 2010, 10, 425–434. [Google Scholar] [CrossRef]
- Kumar, A.; Khani, A.T.; Ortiz, A.S.; Swaminathan, S. GM-CSF: A Double-Edged Sword in Cancer Immunotherapy. Front. Immunol. 2022, 13, 901277. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.M.; Neuberg, D.; Friedenberg, W.; Bennett, J.M.; Paietta, E.; Makary, A.Z.; Liesveld, J.L.; Abboud, C.N.; Dewald, G.; Hayes, F.A.; et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: A trial by the Eastern Cooperative Oncology Group. Blood 2004, 103, 479–485. [Google Scholar] [CrossRef]
- Schmeler, K.M.; Vadhan-Raj, S.; Ramirez, P.T.; Apte, S.M.; Cohen, L.; Bassett, R.L.; Iyer, R.B.; Wolf, J.K.; Levenback, C.L.; Gershenson, D.M.; et al. A phase II study of GM-CSF and rIFN-gamma1b plus carboplatin for the treatment of recurrent, platinum-sensitive ovarian, fallopian tube and primary peritoneal cancer. Gynecol. Oncol. 2009, 113, 210–215. [Google Scholar] [CrossRef]
- Correale, P.; Botta, C.; Rotundo, M.S.; Guglielmo, A.; Conca, R.; Licchetta, A.; Pastina, P.; Bestoso, E.; Ciliberto, D.; Cusi, M.G.; et al. Gemcitabine, oxaliplatin, levofolinate, 5-fluorouracil, granulocyte-macrophage colony-stimulating factor, and interleukin-2 (GOLFIG) versus FOLFOX chemotherapy in metastatic colorectal cancer patients: The GOLFIG-2 multicentric open-label randomized phase III trial. J. Immunother. 2014, 37, 26–35. [Google Scholar] [PubMed]
- Correale, P.; Tagliaferri, P.; Fioravanti, A.; Del Vecchio, M.T.; Remondo, C.; Montagnani, F.; Rotundo, M.S.; Ginanneschi, C.; Martellucci, I.; Francini, E.; et al. Immunity feedback and clinical outcome in colon cancer patients undergoing chemoimmunotherapy with gemcitabine + FOLFOX followed by subcutaneous granulocyte macrophage colony-stimulating factor and aldesleukin (GOLFIG-1 Trial). Clin. Cancer Res. 2008, 14, 4192–4199. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Di Magliano, M.P.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, X.; Cheng, X.; Cui, T.; Zhuo, Y.; Ma, W.; Zhao, X.; Zhao, P.; Liu, X.; Feng, W. Granulocyte-macrophage colony-stimulating factor increases tumor growth and angiogenesis directly by promoting endothelial cell function and indirectly by enhancing the mobilization and recruitment of proangiogenic granulocytes. Tumour Biol. 2017, 39, 1010428317692232. [Google Scholar] [CrossRef]
- Anastasaki, C.; Orozco, P.; Gutmann, D.H. RAS and beyond: The many faces of the neurofibromatosis type 1 protein. Dis. Model. Mech. 2022, 15, dmm049362. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rangwala, F.; Fulkerson, P.C.; Ling, B.; Reed, E.; Cox, A.D.; Kamholz, J.; Ratner, N. Role of TC21/R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene 2004, 23, 368–378. [Google Scholar] [CrossRef]
- Kim, H.A.; Rosenbaum, T.; Marchionni, M.A.; Ratner, N.; Declue, J.E. Schwann-Cells from Neurofibromin Deficient Mice Exhibit Activation of P21(Ras), Inhibition of Cell-Proliferation and Morphological-Changes. Oncogene 1995, 11, 325–335. [Google Scholar]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Sherman, L.S.; Atit, R.; Rosenbaum, T.; Cox, A.D.; Ratner, N. Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J. Biol. Chem. 2000, 275, 30740–30745. [Google Scholar] [CrossRef]
- Shannon, K.M.; O’Connell, P.; Martin, G.A.; Paderanga, D.; Olson, K.; Dinndorf, P.; McCormick, F. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N. Engl. J. Med. 1994, 330, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kong, N.; Zhu, Y.; Lauchle, J.O.; Aiyigari, A.; Braun, B.S.; Wang, E.; Kogan, S.C.; Le Beau, M.M.; Parada, L.; et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 2004, 103, 4243–4250. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.A.; O’Marcaigh, A.; Wardak, Z.; Zhang, Y.Y.; Dranoff, G.; Jacks, T.; Clapp, D.W.; Shannon, K.M. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol. Cell 2000, 5, 189–195. [Google Scholar] [CrossRef]
- Bollag, G.; Clapp, D.W.; Shih, S.; Adler, F.; Zhang, Y.Y.; Thompson, P.; Lange, B.J.; Freedman, M.H.; McCormick, F.; Jacks, T.; et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat. Genet. 1996, 12, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.A.; Yang, F.C.; Travers, J.B.; Wenning, M.J.; Hiatt, K.; New, S.; Hood, A.; Shannon, K.; Williams, D.A.; Clapp, D.W. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J. Exp. Med. 2000, 191, 181–188. [Google Scholar] [CrossRef]
- Kim, A.; Morgan, K.; Hasz, D.E.; Wiesner, S.M.; Lauchle, J.O.; Geurts, J.L.; Diers, M.D.; Le, D.T.; Kogan, S.C.; Parada, L.F.; et al. Beta common receptor inactivation attenuates myeloproliferative disease in Nf1 mutant mice. Blood 2007, 109, 1687–1691. [Google Scholar] [CrossRef]
- Largaespada, D.A.; Brannan, C.I.; Jenkins, N.A.; Copeland, N.G. Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat. Genet. 1996, 12, 137–143. [Google Scholar] [CrossRef]
- Serra, E.; Rosenbaum, T.; Winner, U.; Aledo, R.; Ars, E.; Estivill, X.; Lenard, H.G.; Lazaro, C. Schwann cells harbor the somatic NF1 mutation in neurofibromas: Evidence of two different Schwann cell subpopulations. Hum. Mol. Genet. 2000, 9, 3055–3064. [Google Scholar] [CrossRef]
- Wu, J.; Williams, J.P.; Rizvi, T.A.; Kordich, J.J.; Witte, D.; Meijer, D.; Stemmer-Rachamimov, A.O.; Cancelas, J.A.; Ratner, N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 2008, 13, 105–116. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Patel, A.J.; Liao, C.P.; Wang, Y.; Le, L.Q. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer Cell 2014, 26, 695–706. [Google Scholar] [CrossRef]
- Farschtschi, S.; Park, S.J.; Sawitzki, B.; Oh, S.J.; Kluwe, L.; Mautner, V.F.; Kurtz, A. Effector T cell subclasses associate with tumor burden in neurofibromatosis type 1 patients. Cancer Immunol. Immunother. 2016, 65, 1113–1121. [Google Scholar] [CrossRef]
- Haworth, K.B.; Arnold, M.A.; Pierson, C.R.; Choi, K.; Yeager, N.D.; Ratner, N.; Roberts, R.D.; Finlay, J.L.; Cripe, T.P. Immune profiling of NF1-associated tumors reveals histologic subtype distinctions and heterogeneity: Implications for immunotherapy. Oncotarget 2017, 8, 82037–82048. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; McKay, R.M.; Le, L.Q. Tumorigenesis in neurofibromatosis type 1: Role of the microenvironment. Oncogene 2021, 40, 5781–5787. [Google Scholar] [CrossRef] [PubMed]
- Kershner, L.J.; Choi, K.; Wu, J.; Zhang, X.; Perrino, M.; Salomonis, N.; Shern, J.F.; Ratner, N. Multiple Nf1 Schwann cell populations reprogram the plexiform neurofibroma tumor microenvironment. JCI Insight 2022, 7, e154513. [Google Scholar] [CrossRef]
- Liao, C.P.; Booker, R.C.; Brosseau, J.P.; Chen, Z.; Mo, J.; Tchegnon, E.; Wang, Y.; Clapp, D.W.; Le, L.Q. Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J. Clin. Investig. 2018, 128, 2848–2861. [Google Scholar] [CrossRef]
- White, E.E.; Rhodes, S.D. The NF1+/- Immune Microenvironment: Dueling Roles in Neurofibroma Development and Malignant Transformation. Cancers 2024, 16, 994. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Komurov, K.; Fletcher, J.S.; Jousma, E.; Cancelas, J.A.; Wu, J.; Ratner, N. An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Sci. Rep. 2017, 7, 43315. [Google Scholar]
- Rizvi, T.A.; Huang, Y.; Sidani, A.; Atit, R.; Largaespada, D.A.; Boissy, R.E.; Ratner, N. A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve. J. Neurosci. 2002, 22, 9831–9840. [Google Scholar] [CrossRef]
- Ribeiro, S.; Napoli, I.; White, I.J.; Parrinello, S.; Flanagan, A.M.; Suter, U.; Parada, L.F.; Lloyd, A.C. Injury Signals Cooperate with Nf1 Loss to Relieve the Tumor-Suppressive Environment of Adult Peripheral Nerve. Cell Rep. 2013, 5, 126–136. [Google Scholar] [CrossRef]
- Robb, L.; Drinkwater, C.C.; Metcalf, D.; Li, R.; Kontgen, F.; Nicola, N.A.; Begley, C.G. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc. Natl. Acad. Sci. USA 1995, 92, 9565–9569. [Google Scholar] [CrossRef]
- Shima, K.; Arumugam, P.; Sallese, A.; Horio, Y.; Ma, Y.; Trapnell, C.; Wessendarp, M.; Chalk, C.; McCarthy, C.; Carey, B.C.; et al. A murine model of hereditary pulmonary alveolar proteinosis caused by homozygous Csf2ra gene disruption. Am. J. Physiol. Lung Cell Mol. Physiol. 2022, 322, L438–L448. [Google Scholar] [CrossRef] [PubMed]
- Pundavela, J.; Dinglasan, S.A.; Touvron, M.; Hummel, S.A.; Hu, L.; Rizvi, T.A.; Choi, K.; Hildeman, D.A.; Ratner, N. Stimulator of interferon gene facilitates recruitment of effector CD8 T cells that drive neurofibromatosis type 1 nerve tumor initiation and maintenance. Sci. Adv. 2024, 10, eado6342. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.S.; Wu, J.; Jessen, W.J.; Pundavela, J.; Miller, J.A.; Dombi, E.; Kim, M.O.; Rizvi, T.A.; Chetal, K.; Salomonis, N.; et al. Cxcr3-expressing leukocytes are necessary for neurofibroma formation in mice. JCI Insight 2019, 4, e98601. [Google Scholar] [CrossRef]
- Nishinakamura, R.; Nakayama, N.; Hirabayashi, Y.; Inoue, T.; Aud, D.; McNeil, T.; Azuma, S.; Yoshida, S.; Toyoda, Y.; Arai, K.; et al. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor-deficient mice are normal. Immunity 1995, 2, 211–222. [Google Scholar] [CrossRef]
- Rosas, M.; Gordon, S.; Taylor, P.R. Characterisation of the expression and function of the GM-CSF receptor alpha-chain in mice. Eur. J. Immunol. 2007, 37, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chang, L.; Patel, N.; Yang, J.; Lowe, L.; Burns, D.K.; Zhu, Y. Induction of abnormal proliferation by nonmyelinating schwann cells triggers neurofibroma formation. Cancer Cell 2008, 13, 117–128. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Pilch, K.S.; van der Lans, M.; Fazal, S.V.; Benito, C.; Wagstaff, L.J.; Mirsky, R.; Jessen, K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J. Neurosci. 2017, 37, 9086–9099. [Google Scholar] [CrossRef]
- Erlandson, R.A.; Woodruff, J.M. Peripheral nerve sheath tumors: An electron microscopic study of 43 cases. Cancer 1982, 49, 273–287. [Google Scholar] [CrossRef]
- Hirose, T.; Sano, T.; Hizawa, K. Ultrastructural localization of S-100 protein in neurofibroma. Acta Neuropathol. 1986, 69, 103–110. [Google Scholar] [CrossRef]
- Jousma, E.; Rizvi, T.A.; Wu, J.; Janhofer, D.; Dombi, E.; Dunn, R.S.; Kim, M.O.; Masters, A.R.; Jones, D.R.; Cripe, T.P.; et al. Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of Neurofibromatosis type 1. Pediatr. Blood Cancer 2015, 62, 1709–1716. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Caveney, N.A.; Rodriguez, G.E.; Pollmann, C.; Meyer, T.; Borowska, M.T.; Wilson, S.C.; Wang, N.; Xiang, X.; Householder, K.D.; Tao, P.; et al. Structure of the interleukin-5 receptor complex exemplifies the organizing principle of common beta cytokine signaling. Mol. Cell 2024, 84, 1995–2005.e7. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, K. Interleukin-5 and IL-5 receptor in health and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 463–485. [Google Scholar] [CrossRef]
- Perrino, M.R.; Ahmari, N.; Hall, A.; Jackson, M.; Na, Y.; Pundavela, J.; Szabo, S.; Woodruff, T.M.; Dombi, E.; Kim, M.O.; et al. C5aR plus MEK inhibition durably targets the tumor milieu and reveals tumor cell phagocytosis. Life Sci. Alliance 2024, 7, e202302229. [Google Scholar] [CrossRef]
- Gitto, S.B.; Beardsley, J.M.; Nakkina, S.P.; Oyer, J.L.; Cline, K.A.; Litherland, S.A.; Copik, A.J.; Khaled, A.S.; Fanaian, N.; Arnoletti, J.P.; et al. Identification of a novel IL-5 signaling pathway in chronic pancreatitis and crosstalk with pancreatic tumor cells. Cell Commun. Signal 2020, 18, 95. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pundavela, J.; Hall, A.; Dinglasan, S.A.; Choi, K.; Rizvi, T.A.; Trapnell, B.C.; Wu, J.; Ratner, N. Granulocyte-Macrophage Colony Stimulating Factor Receptor Contributes to Plexiform Neurofibroma Initiation. Cancers 2025, 17, 905. https://doi.org/10.3390/cancers17050905
Pundavela J, Hall A, Dinglasan SA, Choi K, Rizvi TA, Trapnell BC, Wu J, Ratner N. Granulocyte-Macrophage Colony Stimulating Factor Receptor Contributes to Plexiform Neurofibroma Initiation. Cancers. 2025; 17(5):905. https://doi.org/10.3390/cancers17050905
Chicago/Turabian StylePundavela, Jay, Ashley Hall, Samantha Anne Dinglasan, Kwangmin Choi, Tilat A. Rizvi, Bruce C. Trapnell, Jianqiang Wu, and Nancy Ratner. 2025. "Granulocyte-Macrophage Colony Stimulating Factor Receptor Contributes to Plexiform Neurofibroma Initiation" Cancers 17, no. 5: 905. https://doi.org/10.3390/cancers17050905
APA StylePundavela, J., Hall, A., Dinglasan, S. A., Choi, K., Rizvi, T. A., Trapnell, B. C., Wu, J., & Ratner, N. (2025). Granulocyte-Macrophage Colony Stimulating Factor Receptor Contributes to Plexiform Neurofibroma Initiation. Cancers, 17(5), 905. https://doi.org/10.3390/cancers17050905