

Targeting Immune Checkpoint Inhibitors for Non-Small-Cell Lung Cancer: Beyond PD-1/PD-L1 Monoclonal Antibodies


Simple Summary
Abstract
1. Introduction
2. CD8 T Cell Exhaustion
3. CTLA-4 Targeting
3.1. Ipilimumab
3.2. Tremelimumab
3.3. Botensilimab
3.4. Cadonilimab (AK-104)
3.5. Erfonrilimab (KN-046)
3.6. Volrustomig (MEDI5752)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aCTLA-4 mAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aCTLA-4 | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Ipilimumab | human IgG1κ | CM-9LA | III | Metastatic, 1st line | 719 | Nivo (aPD-1) +ipi + CT vs. CT | OS in overall population |
| [71,72] |
| CM-227 | III | Metastatic, 1st line | 1189 | Nivo + ipi vs. nivo vs. CT | OS in PD-L1 ≥ 1% of nivo + ipi vs. CT |
| [63,64,65,66,67,68,69,70,71,72,73] | ||
| Tremelimumab | human IgG2a | POSEIDON | III | Metastatic, 1st line | 1013 | Treme + durva (aPD-L1) + CT vs. durva + CT vs. CT | PFS and OS of durva + CT vs. CT |
| [76] |
| Botensilimab | Fc-enhanced human IgG1 | Chand et al. | Ia/b | Metastatic, refractory | 3 with NSCLC | Boten +/− balsti (aPD-1) | Safety |
| [80] |
| aCTLA-4 bsAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aCTLA-4 | Ref |
| Cadonilimab (AK-104) | CTLA-4/PD-1 | AK104-202 | Ib/II | Metastatic, Co A: 2nd line, ICI-naïve Co B: 2nd line, primary resistance to CT-ICI Co C: 2nd line, secondary resistance to CT-ICI | 30 (Co A) 7 (Co B) 16 (Co C) | Cadon | ORR |
| [83] |
| Erfonrilimab (KN-046) | CTLA-4/PD-1 | Zhao et al. | II | Metastatic, 1st line | 87 | Erfon + CT | ORR, DoR |
| [86] |
| Volrustomig (MEDI5752) | CTLA-4/PD-1 | Ahn et al., Spigel et al. | Ib/II | Metastatic, 1st line | 139 | Volru 1500 mg + CT vs. volru 750 mg + CT vs. pembro (aPD-1) + CT | ORR |
| [87,88] |
4. TIGIT Targeting
4.1. Tiragolumab
4.2. Domvanalimab
4.3. Vibostolimab
4.4. Rilvegostomig (AZD2936)
| aTIGIT mAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aTIGIT | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Tiragolumab | Human IgG1κ | CITYSCAPE | II | Metastatic, 1st line, PD-L1 ≥ 1% | 135 | Tirago + atezo (aPD-L1) vs. pcb + atezo | ORR, PFS |
| [104] |
| SKYSCRAPER-01 | III | Metastatic, 1st line, PD-L1 > 50% | 534 | Tirago + atezo vs. pcb + atezo | PFS, OS |
| [106] | ||
| SKYSCRAPER-06 | III | Metastatic, 1st line | 542 | Tirago + atezo + CT vs. pembro + CT | PFS, OS |
| [107] | ||
| Domvanalimab | Fc-silent humanized IgG1 | ARC-7 | II | Metastatic, 1st line, PD-L1 ≥50% | 151 | Domva + zimbe (aPD-1) + etrumadenant vs. domva + zimbe vs. zimbe | ORR, PFS |
| [108] |
| ARC-10 | II | Metastatic, 1st line, PD-L1 ≥50% | 98 | Domva + zimbe vs. zimbe vs. CT | PFS |
| [109,110] | ||
| Vibostolimab | Human IgG1 | KEYVIBE-002 | II | Metastatic, 2nd line | 225 | MK-7684A (vibo + pembro [aPD-1]) + docetaxel vsMK-7684A (vibo + pembro) vs. pcb + docetaxel | PFS |
| [114] |
| aTIGIT bsAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aTIGIT Bispecific | Reference |
| Rilvegostomig | TIGIT/PD-1 | ARTEMIDE-01 | I/II | Metastatic, PD-L1 ≥ 1%, 2nd line, previous ICI (part A, B) | 83 | Rilve 750 or 1500 mg | Safety |
| [117,118] |
| Metastatic, PD-L1 ≥ 1%, ≥2nd line, ICI-naïve (part C, D) | 96 | Rilve 750 or 1500 mg | ORR |
| [119] |
5. LAG-3 Targeting
5.1. Relatlimab (BMS-986016)
5.2. Ieramilimab (LAG-525)
5.3. Favezelimab (MK-4280)
5.4. Fianlimab (REGN3767)
| aLAG-3 mAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aLAG-3 | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Relatlimab (BMS-986016) | Human IgG4 | NEOPredict | II | Early, resectable, stage IB–IIA (single-level N2, UICC 8th edition) | 60 | Nivo (aPD-1) + relavs. nivo | Feasibility of surgery within 43 days |
| [45] |
| RELATIVITY-104 | II | Metastatic, 1st line | 309 | Nivo + rela + CT vs. nivo + CT | ORR |
| [137] | ||
| Ieramilimab (LAG-525) | Humanized IgG4 | Lin et al. | II | Metastatic, ≥2nd line | 42 with NSCLC | Iera + sparta (aPD-1) | ORR |
| [139] |
| Favezelimab (MK-4280) | Humanized IgG4 | KEYNOTE-495/KeyImPaCT | II | Metastatic, 1st line | 243 | Pembro (aPD-1) +lenvatinib vs. pembro + quavon (aCTLA-4) vs. pembro + fave 800 mg | ORR |
| [141,142] |
| aLAG-3 bsAb | Type | Study | Phase | Setting | N | Intervention | 1st endpoint | Results with aLAG-3 | Ref |
| FS118 | LAG-3/PD-L1 | Yap et al. | I | Metastatic, ≥2nd line | 9 with NSCLC | FS118 | Safety |
| [150] |
| Tebotelimab (MGD013) | LAG-3-/PD-1 | Luke et al. | I | Metastatic, ≥2nd line | 35 with NSCLC | Tebo | Safety |
| [152,153] |
6. TIM-3 Targeting
6.1. Cobolimab (TSR-022/GSK4069889)
6.2. Sabatolimab MGB453
6.3. LY3321367
| aTIM-3 mAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aTIM-3 | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Cobolimab (TSR-022/GSK4069889) | Humanized IgG4 | AMBER | I | Metastatic, ≥2nd line after aPD-1/PD-L1 | 84 | Cobo + dostar (aPD-1) | ORR |
| [189,190] |
| Sabatolimab (MGB453) | Humanized IgG4 | Mach et al. | I | Metastatic, ≥2nd line after aPD-1/PD-L1 | 17 with NSCLC | Saba + sparta (aPD-1) | ORR |
| [192] |
| LY3321367 | Humanized IgG1λ, Fc-null | Harding et al. | I | Metastatic, ≥2nd line | 65 with NSCLC | LY3321367 +/−LY300054 (aPD-L1) | Safety, RP2D |
| [193] |
| aTIM-3 bsAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aLAG-3 | Ref |
| Sabestomig (AZD7789) | TIM-3-/PD-1 | Besse et al. | I/IIa | Metastatic, ≥2nd line, ICI-pretreated | 39 | Sabe | Safety |
| [195] |
| LY3415244 | TIM-3/PD-L1 | J1C-MC-JZDA | I | Metastatic, ≥2nd line | 2 with NSCLC | LY3415244 | Safety, RP2D |
| [196] |
7. NKG2A Targeting
Monalizumab
| aNKG2A mAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aLAG-3 | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Monalizumab | Humanized IgG4 | NeoCOAST | II | Resectable, stage IA3-IIIA, neoadjuvant | 83 | Mona + durva (aPD-1) vs. ole (aCD73) + durva vs. danvatirsen + durva vs. durva | MPR |
| [219] |
| COAST | II | Unresectable, stage III, after chemoradiation | 189 | Mona + durva vs. ole + durva vs. durva | ORR |
| [220,221] | ||
| Patel et al. | I/II | Metastatic, 2nd or 3rd line, ICI-naive | 20 with NSCLC | Mona + durva | Safety |
| [223] | ||
| PIONeeR | Ib/IIA | Metastatic, 2nd or 3rd line, after aPD-(L)1 | 114 | Mona + durva vs. ole + durva vs. ceralasertib + durva vs. savolitinib + durva vs. docetaxel | 12 w DCR |
| [224] |
8. Targeting the CD39/CD73/Adenosine Pathway
8.1. CD39 Targeting
IPH5201
8.2. CD73 Targeting
Oleclumab
8.3. Adenosine Receptor Inhibitors
8.3.1. Etrumadenant
8.3.2. Taminadenant
| aCD73 mAb | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with aCD73 | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Oleclumab | Human IgG1λ | NeoCOAST | II | Resectable, stage IA3-IIIA, neoadjuvant | 83 | Mona (aNKG2A) + durva (aPD-L1) vs. ole + durva vs. danvatirsen + durva vs. durva | MPR |
| [219] |
| COAST | II | Unresectable, stage III, after chemoradiation | 189 | Mona + durva vs. ole + durva vs. durva | ORR |
| [220,221] | ||
| PIONeeR | Ib/IIA | Metastatic, 2nd or 3rd line, after aPD-(L)1 | 114 | Mona + durva vs. ole + durva vs. ceralasertib + durva vs. savolitinib + durva vs. docetaxel | 12 w DCR |
| [224] | ||
| Adenosine R I | Type | Study | Phase | Setting | N | Intervention | 1st Endpoint | Results with Adenosine R I | Ref |
| Etrumadenant | Selective dual antagonist of A2a and A2b | ARC-7 | II | Metastatic, 1st line, PD-L1 ≥50% | 151 | Domva (aTIGIT) + zimbe (aPD-1) + etrumadenant vs. domva + zimbe vs. zimbe | ORR, PFS |
| [108] |
| Taminadenant | A2a antagonist | Chiappori et al. | I/Ib | Metastatic, ≥2nd line | 50 | Taminadenant vs. taminadenant + sparta (aPD-1) | Safety |
| [243] |
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hendriks, L.E.L.; Remon, J.; Faivre-Finn, C.; Garassino, M.C.; Heymach, J.V.; Kerr, K.M.; Tan, D.S.W.; Veronesi, G.; Reck, M. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2024, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.L.; Fitzgerald, B.G.; Paz-Ares, L.; Cappuzzo, F.; Jänne, P.A.; Peters, S.; Hirsch, F.R. New promises and challenges in the treatment of advanced non-small-cell lung cancer. Lancet 2024, 404, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Non-oncogene-addicted metastatic non-small-cell lung cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 358–376. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in advanced Nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in metastatic non–small-cell lung cancer. N. Engl. J. Med 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Garassino, M.C.; Gadgeel, S.; Speranza, G.; Felip, E.; Esteban, E.; Dómine, M.; Hochmair, M.J.; Powell, S.F.; Bischoff, H.G.; Peled, N.; et al. Pembrolizumab plus Pemetrexed and Platinum in Nonsquamous non–small-cell lung cancer: 5-year outcomes from the phase 3 KEYNOTE-189 study. J. Clin. Oncol. 2023, 41, 1992–1998. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti–PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Human Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Chamoto, K.; Yaguchi, T.; Tajima, M.; Honjo, T. Insights from a 30-year journey: Function, regulation and therapeutic modulation of PD1. Nat. Rev. Immunol 2023, 23, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T Cell Exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Baessler, A.; Vignali, D.A.A. T Cell exhaustion. Annu. Rev. Immunol. 2024, 42, 179–206. [Google Scholar] [CrossRef]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; Von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Yao, C.; Sun, H.W.; Lacey, N.E.; Ji, Y.; Moseman, E.A.; Shih, H.Y.; Heuston, E.F.; Kirby, M.; Anderson, S.; Cheng, J.; et al. Single-cell RNA-Seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 2019, 20, 890–901. [Google Scholar] [CrossRef]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.L.; Hirsch, F.R. PD-L1 expression in lung cancer. J. Thorac. Oncol. 2016, 11, 964–975. [Google Scholar] [CrossRef]
- Greillier, L.; Tomasini, P.; Barlesi, F. The clinical utility of tumor mutational burden in non-small cell lung cancer. Transl. Lung Cancer Res. 2018, 7, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R.; et al. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022, 8, 1160. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-cell–inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Bibeau, F.; Greillier, L.; Fumet, J.D.; Ilie, A.; Monville, F.; Laugé, C.; Catteau, A.; Boquet, I.; Majdi, A.; et al. Immunoscore immune checkpoint using spatial quantitative analysis of CD8 and PD-L1 markers is predictive of the efficacy of anti- PD1/PD-L1 immunotherapy in non-small cell lung cancer. Ebiomedicine 2023, 92, 104633. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Zhao, X.; Shan, Q.; Xue, H.H. TCF1 in T cell immunity: A broadened frontier. Nat. Rev. Immunol. 2022, 22, 147–157. [Google Scholar] [CrossRef]
- Bordon, Y. TOX for tired T cells. Nat. Rev. Immunol. 2019, 19, 476. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef]
- Belk, J.A.; Daniel, B.; Satpathy, A.T. Epigenetic regulation of T cell exhaustion. Nat. Immunol. 2022, 23, 848–860. [Google Scholar] [CrossRef]
- Giles, J.R.; Ngiow, S.F.; Manne, S.; Baxter, A.E.; Khan, O.; Wang, P.; Staupe, R.; Abdel-Hakeem, M.S.; Huang, H.; Mathew, D.; et al. Shared and distinct biological circuits in effector, memory and exhausted CD8+ T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat. Immunol. 2022, 23, 1600–1613. [Google Scholar] [CrossRef]
- Andrews, L.P.; Butler, S.C.; Cui, J.; Cillo, A.R.; Cardello, C.; Liu, C.; Brunazzi, E.A.; Baessler, A.; Xie, B.; Kunning, S.R.; et al. LAG-3 and PD-1 synergize on CD8+ T cells to drive t cell exhaustion and hinder autocrine IFN-γ-dependent anti-tumor immunity. Cell 2024, 187, 4355–4372.e22. [Google Scholar] [CrossRef]
- Cillo, A.R.; Cardello, C.; Shan, F.; Karapetyan, L.; Kunning, S.; Sander, C.; Rush, E.; Karunamurthy, A.; Massa, R.C.; Rohatgi, A.; et al. Blockade of LAG-3 and PD-1 leads to co-expression of cytotoxic and exhaustion gene modules in CD8+ T cells to promote antitumor immunity. Cell 2024, 187, 4373–4388.e15. [Google Scholar] [CrossRef]
- Hofmann, M.; Thimme, R.; Schamel, W.W. PD-1 and LAG-3: Synergistic fostering of T cell exhaustion. Sig. Transduct. Target Ther. 2024, 9, 291. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; Manne, S.; Huang, Y.J.; Azar, T.; Chen, Z.; Mathew, D.; Chen, Q.; Khan, O.; Wu, J.E.; Alcalde, V.; et al. LAG-3 sustains TOX expression and regulates the CD94/NKG2-Qa-1b axis to govern exhausted CD8 T Cell NK receptor expression and cytotoxicity. Cell 2024, 187, 4336–4354.e19. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Morgensztern, D.; Chaudhry, A.; Iannotti, N.; Acevedo, A.; Balaburski, G.; Balogh, A.; Peters, S. 1359TiP RELATIVITY-104: First-Line Relatlimab (RELA) + Nivolumab (NIVO) with chemotherapy vs nivo with chemotherapy in stage IV or recurrent Non-Small Cell Lung Cancer (NSCLC): A phase II, randomized, double-blind study. Ann. Oncol. 2021, 32, S1030. [Google Scholar] [CrossRef]
- Schuler, M.; Cuppens, K.; Plönes, T.; Wiesweg, M.; Du Pont, B.; Hegedus, B.; Köster, J.; Mairinger, F.; Darwiche, K.; Paschen, A.; et al. Neoadjuvant nivolumab with or without relatlimab in resectable non-small-cell lung cancer: A randomized phase 2 trial. Nat. Med. 2024, 30, 1602–1611. [Google Scholar] [CrossRef]
- Augustin, R.C.; Bao, R.; Luke, J.J. Targeting Cbl-b in cancer immunotherapy. J. Immunother. Cancer 2023, 11, e006007. [Google Scholar] [CrossRef]
- Chocarro, L.; Blanco, E.; Fernandez-Rubio, L.; Garnica, M.; Zuazo, M.; Garcia, M.J.; Bocanegra, A.; Echaide, M.; Johnston, C.; Edwards, C.J.; et al. PD-1/LAG-3 Co-signaling profiling uncovers CBL ubiquitin ligases as key immunotherapy targets. EMBO Mol. Med. 2024, 16, 1791–1816. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dai, E.; Li, Y.; Han, G.; Pei, G.; Ingram, D.R.; Thakkar, K.; Qin, J.J.; Dang, M.; Le, X.; et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat. Med. 2023, 29, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Globig, A.M.; Zhao, S.; Roginsky, J.; Maltez, V.I.; Guiza, J.; Avina-Ochoa, N.; Heeg, M.; Araujo Hoffmann, F.; Chaudhary, O.; Wang, J.; et al. The Β1-adrenergic receptor links sympathetic nerves to T cell exhaustion. Nature 2023, 622, 383–392. [Google Scholar] [CrossRef]
- Baek, A.E. Stress exhausts T cells. Sci. Signal. 2023, 16, eadl0724. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; Hou, Y.; Lin, Y.; Zhao, H.; Shi, Y.; Chen, K.; Nian, C.; Tang, J.; Pan, L.; et al. Osr2 functions as a biomechanical checkpoint to aggravate CD8+ T cell exhaustion in tumor. Cell 2024, 187, 3409–3426.e24. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.D.; Le Gros, G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol. Cell Biol. 1999, 77, 1–10. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in regulatory T cells for cancer immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Walker, L.S.K.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Na.t Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef]
- Lax, B.M.; Palmeri, J.R.; Lutz, E.A.; Sheen, A.; Stinson, J.A.; Duhamel, L.; Santollani, L.; Kennedy, A.; Rothschilds, A.M.; Spranger, S.; et al. Both intratumoral regulatory T cell depletion and CTLA-4 antagonism are required for maximum efficacy of anti-CTLA-4 antibodies. Proc. Natl. Acad. Sci. USA 2023, 120, e2300895120. [Google Scholar] [CrossRef]
- Tang, F.; Du, X.; Liu, M.; Zheng, P.; Liu, Y. Anti-CTLA-4 antibodies in cancer immunotherapy: Selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. 2018, 8, 30. [Google Scholar] [CrossRef]
- Sato, Y.; Casson, C.N.; Matsuda, A.; Kim, J.I.; Shi, J.Q.; Iwasaki, S.; Chen, S.; Modrell, B.; Chan, C.; Tavares, D.; et al. Fc-independent functions of anti-CTLA-4 antibodies contribute to anti-tumor efficacy. Cancer Immunol. Immunother. 2022, 71, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; De La Mora Jimenez, E.; et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Melero, I.; Yau, T.; Kang, Y.K.; Kim, T.Y.; Santoro, A.; Sangro, B.; Kudo, M.; Hou, M.M.; Matilla, A.; Tovoli, F.; et al. Nivolumab plus ipilimumab combination therapy in patients with advanced hepatocellular carcinoma previously treated with sorafenib: 5-year results from CheckMate 040. Ann. Oncol. 2024, 35, 537–548. [Google Scholar] [CrossRef]
- Andre, T.; Elez, E.; Van Cutsem, E.; Jensen, L.H.; Bennouna, J.; Mendez, G.; Schenker, M.; De La Fouchardiere, C.; Limon, M.L.; Yoshino, T.; et al. Nivolumab plus ipilimumab in microsatellite-instability–high metastatic colorectal cancer. N. Engl. J. Med. 2024, 391, 2014–2026. [Google Scholar] [CrossRef]
- Hossain, M.A. A Comprehensive review of immune checkpoint inhibitors for cancer treatment. Int. Immunopharmacol. 2024, 143, 113365. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, P.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. Final, 10-year outcomes with nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2025, 392, 11–22. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Ciuleanu, T.E.; Schenker, M.; Bordenave, S.; Cobo, M.; Juan-Vidal, O.; Reinmuth, N.; Richardet, E.; Felip, E.; Menezes, J.; et al. Five-year outcomes with first-line nivolumab plus ipilimumab with 2 cycles of chemotherapy versus 4 cycles of chemotherapy alone in patients with metastatic non-small cell lung cancer in the randomized CheckMate 9LA trial. Eur. J. Cancer 2024, 211, 114296. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lee, J.S.; Ciuleanu, T.E.; Bernabe Caro, R.; Nishio, M.; Urban, L.; Audigier-Valette, C.; Lupinacci, L.; Sangha, R.; Pluzanski, A.; et al. Five-year survival outcomes with nivolumab plus ipilimumab versus chemotherapy as first-line treatment for metastatic non–small-cell lung cancer in CheckMate 227. J. Clin. Oncol. 2023, 41, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Johnson, M.L.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Kim, S.W.; Ursol, G.; Hussein, M.; Lim, F.L.; et al. Durvalumab with or without tremelimumab in combination with chemotherapy as first-line therapy for metastatic non–small-cell lung cancer: The phase III POSEIDON study. J. Clin. Oncol. 2023, 41, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Dougan, S.K.; Dougan, M. Immune mechanisms of toxicity from checkpoint inhibitors. Trends Cancer 2023, 9, 543–553. [Google Scholar] [CrossRef]
- Lebbé, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of two dosing regimens for nivolumab in combination with ipilimumab in patients with advanced melanoma: Results from the phase IIIb/IV CheckMate 511 trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef]
- O’Day, S.; Khoueiry, A.E.; Ramamurthy, C.; Bullock, A.; Shapiro, I.; Serina; Han, H.; Namagerdi, L.O.; Kaleta, R.; Wijatyk, A.; et al. 398 AGEN1181, an Fc engineered anti-CTLA-4 Antibody, Demonstrates Clinical Activity, Alone or in Combination with Balstilimab (Anti-PD-1), and Broadens the Therapeutic Potential of CTLA-4 Therapy. J. Immuno Ther. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Chand, D.; Savitsky, D.A.; Krishnan, S.; Mednick, G.; Delepine, C.; Garcia-Broncano, P.; Soh, K.T.; Wu, W.; Wilkens, M.K.; Udartseva, O.; et al. Botensilimab, an Fc-enhanced anti–CTLA-4 antibody, is effective against tumors poorly responsive to conventional immunotherapy. Cancer Discov. 2024, 14, 2407–2429. [Google Scholar] [CrossRef]
- Bullock, A.J.; Schlechter, B.L.; Fakih, M.G.; Tsimberidou, A.M.; Grossman, J.E.; Gordon, M.S.; Wilky, B.A.; Pimentel, A.; Mahadevan, D.; Balmanoukian, A.S.; et al. Botensilimab plus balstilimab in relapsed/refractory microsatellite stable metastatic colorectal cancer: A phase 1 trial. Nat. Med. 2024, 30, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Huang, Z.; Zhong, T.; Zhang, P.; Wang, Z.M.; Xia, M.; Li, B. Cadonilimab, a tetravalent PD-1/CTLA-4 bispecific antibody with trans-binding and enhanced target binding avidity. Mabs 2023, 15, 2180794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, Y.; Fan, Y.; Zhou, J.; Yang, N.; Yu, Q.; Zhuang, W.; Song, W.; Wang, Z.M.; Li, B.; et al. A multicenter, open-label phase Ib/II study of cadonilimab (anti PD-1 and CTLA-4 bispecific antibody) monotherapy in previously treated advanced non–small-cell lung cancer (AK104-202 Study). Lung Cancer 2023, 184, 107355. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Su, C.E.P. 11D.01 A phase II study of cadonilimab plus chemotherapy as first-line treatment for PD-L1-negative advanced non-small cell lung cancer: LungCadX. J. Thorac. Oncol. 2024, 19, S614. [Google Scholar] [CrossRef]
- Ma, Y.; Xue, J.; Zhao, Y.; Zhang, Y.; Huang, Y.; Yang, Y.; Fang, W.; Guo, Y.; Li, Q.; Ge, X.; et al. Phase I trial of KN046, a novel bispecific antibody targeting PD-L1 and CTLA-4 in patients with advanced solid tumors. J. Immunother. Cancer 2023, 11, e006654. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, G.; Li, X.; Wu, J.; Chang, B.; Hu, S.; Yang, S.; Xu, T.; Liu, Y.; Wang, N.; et al. KN046, a bispecific antibody against PD-L1 and CTLA-4, plus chemotherapy as first-line treatment for metastatic NSCLC: A multicenter phase 2 trial. Cell Rep. Med. 2024, 5, 101470. [Google Scholar] [CrossRef]
- Ahn, M.J.; Kim, S.W.; Costa, E.C.; Rodríguez, L.M.; Oliveira, J.; Insa Molla, M.A.; Majem, M.; Costa, L.; Su, W.C.; Lee, K.H.; et al. LBA56 MEDI5752 or Pembrolizumab (P) plus Carboplatin/Pemetrexed (CP) in treatment-Naïve (1L) non-small cell lung cancer (NSCLC): A phase Ib/II trial. Ann. Oncol. 2022, 33, S1423. [Google Scholar] [CrossRef]
- Spigel, D.R.; Ahn, M.J.; Majem, M.; Medina Rodríguez, L.; Lee, K.H.; Carcereny, E.; Hernández, A.A.; Insa, A.; Cho, E.K.; Besse, B.; et al. OA11.04 volrustomig + platinum doublet chemotherapy (CTx) in first-line non-small cell lung cancer (NSCLC): Phase 1b trial update. J. Thorac. Oncol. 2024, 19, S33–S34. [Google Scholar] [CrossRef]
- Johnson, M.L.; Arriola, E.; Kato, T.; Girard, N.; Gadgeel, S.M.; Wang, J.; Li, X.; Lowery, C.; Krug, L.M.; Ahn, M.J. eVOLVE-Lung02: A phase 3 study of first-line (1L) volrustomig plus chemotherapy (CT) versus pembrolizumab plus CT in metastatic non-small-cell lung cancer (mNSCLC) with PD-L1 tumor cell (TC) expression < 50%. J. Clin. Oncol. 2024, 42, TPS8652. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Bañas, H.; Casas-Avilés, I.; Durán, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: Novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef]
- Molfetta, R.; Zitti, B.; Lecce, M.; Milito, N.D.; Stabile, H.; Fionda, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R. CD155: A multi-functional molecule in tumor progression. Int. J. Mol. Sci. 2020, 21, 922. [Google Scholar] [CrossRef]
- Preillon, J.; Cuende, J.; Rabolli, V.; Garnero, L.; Mercier, M.; Wald, N.; Pappalardo, A.; Denies, S.; Jamart, D.; Michaux, A.C.; et al. Restoration of T-cell effector function, depletion of tregs, and direct killing of tumor cells: The multiple mechanisms of action of a-TIGIT antagonist antibodies. Mol. Cancer Ther. 2021, 20, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon—γ production of natural killer cells via β—Arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8 + T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Lepletier, A.; Madore, J.; O’Donnell, J.S.; Johnston, R.L.; Li, X.Y.; McDonald, E.; Ahern, E.; Kuchel, A.; Eastgate, M.; Pearson, S.-A.; et al. Tumor CD155 expression is associated with resistance to anti-PD1 immunotherapy in metastatic melanoma. Clin. Cancer Res. 2020, 26, 3671–3681. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, J.; Chen, Y.; Cui, J.; Lei, Y.; Cui, Y.; Jiang, N.; Jiang, W.; Chen, L.; Chen, Y.; et al. Combined evaluation of the expression status of CD155 and TIGIT plays an important role in the prognosis of LUAD (lung adenocarcinoma). Int. Immunopharmacol. 2020, 80, 106198. [Google Scholar] [CrossRef]
- Wen, J.; Mao, X.; Cheng, Q.; Liu, Z.; Liu, F. A pan-cancer analysis revealing the role of TIGIT in tumor microenvironment. Sci. Rep. 2021, 11, 22502. [Google Scholar] [CrossRef] [PubMed]
- Waight, J.D.; Chand, D.; Dietrich, S.; Gombos, R.; Horn, T.; Gonzalez, A.M.; Manrique, M.; Swiech, L.; Morin, B.; Brittsan, C.; et al. Selective FcγR Co-engagement on APCs modulates the activity of therapeutic antibodies targeting T cell antigens. Cancer Cell 2018, 33, 1033–1047.e5. [Google Scholar] [CrossRef]
- Sivick Gauthier, K.E.; Piovesan, D.; Groot, A.E.D.; Anderson, A.E.; Ray, R.D.; Serwas, N.K.; Cho, S.; Patnaik, A.; Foster, P.; Mitchell, C.G.; et al. 475 Fc-Silent Anti-TIGIT Antibodies Potentiate Anti-Tumor Immunity without Depleting Regulatory T Cells. J. ImmunoTherapy Cancer 2023, 11, A534. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Guan, X.; Hu, R.; Choi, Y.; Srivats, S.; Nabet, B.Y.; Silva, J.; McGinnis, L.; Hendricks, R.; Nutsch, K.; Banta, K.L.; et al. Anti-TIGIT antibody improves PD-L1 blockade through myeloid and treg cells. Nature 2024, 627, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Roche Reports Update on Phase III SKYSCRAPER-01 Study Results. Available online: https://www.roche.com/media/releases/med-cor-2024-11-26 (accessed on 26 November 2024).
- Socinski, M.A.; Rodriguez Abreu, D.; Lee, D.H.; Cappuzzo, F.; Nishio, M.; Lovly, C.M.; Ozyilkan, O.; Li, Q.; Johnson, M.L.; Garon, E.B.; et al. LBA2 SKYSCRAPER-06: Efficacy and safety of tiragolumab plus atezolizumab plus chemotherapy (Tira + Atezo + Chemo) vs pembrolizumab plus chemotherapy (Pembro + Chemo) in patients (Pts) with advanced non-squamous non-small cell lung cancer (NSq NSCLC). Immuno-Oncol. Technol. 2024, 24, 101025. [Google Scholar] [CrossRef]
- Johnson, M.L.; Fox, W.; Lee, Y.G.; Lee, K.H.; Ahn, H.K.; Kim, Y.C.; Lee, K.Y.; Lee, J.S.; He, X.; Park, C.; et al. ARC-7: Randomized phase 2 study of domvanalimab + zimberelimab ± etrumadenant versus zimberelimab in first-line, metastatic, PD-L1-high non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2022, 40, 397600. [Google Scholar] [CrossRef]
- Naidoo, J.; Peters, S.; Runglodvatana, Y.; Li, J.Y.C.; Fong, C.H.; Ho, G.F.; How, S.H.; Juengsamarn, J.; Todd, T.; Marina, N.; et al. 1461 phase 2 randomized study of domvanalimab combined with zimberelimab in front-line, PD-(L)1 high, locally advanced or metastatic non-small cell lung cancer (NSCLC): Results from ARC-10 part 1. J. ImmunoTherapy Cancer 2024, 12. [Google Scholar] [CrossRef]
- Arcus Biosciences Announces That Domvanalimab Plus Zimberelimab Improved Overall Survival in ARC-10, a Randomized Study in Patients with PD-L1-High Non-Small Cell Lung Cancer. Available online: https://investors.arcusbio.com/investors-and-media/press-releases/press-release-details/2024/Arcus-Biosciences-Announces-that-Domvanalimab-Plus-Zimberelimab-Improved-Overall-Survival-in-ARC-10-a-Randomized-Study-in-Patients-with-PD-L1-High-Non-Small-Cell-Lung-Cancer/default.aspx (accessed on 5 November 2024).
- Rodriguez-Abreu, D.; Gray, J.E.; Ahn, M.J.; Johnson, M.L.; Yu, X.; Chen, X.; Phuong, P.H.; Kim, J.; Reck, M. STAR-121: A phase 3, randomized study of Domvanalimab (DOM) and Zimberelimab (ZIM) in combination with chemotherapy vs pembrolizumab (Pembro) and chemotherapy in patients with untreated metastatic non-small cell lung cancer (mNSCLC) with no actionable gene alterations. J. Clin. Oncol. 2023, 41, TPS9141. [Google Scholar] [CrossRef]
- Spira, A.I.; Chiu, J.; Wang, C.C.; Zer, A.; Conibear, J.; Phuong, P.H.; Park, J.K.; Seto, A.; Zhang, J.; Cho, B.C. VELOCITY-lung: A phase (Ph) 2 study evaluating safety and efficacy of domvanalimab (Dom) + Zimberelimab (Zim) + Sacituzumab Govitecan (SG), or Etrumadenant (Etruma) + Dom + Zim, or Etruma + Zim in patients (Pts) with treatment-naïve metastatic non-small cell lung cancer (mNSCLC). J. Clin. Oncol. 2023, 41, TPS9155. [Google Scholar] [CrossRef]
- Niu, J.; Maurice-Dror, C.; Lee, D.H.; Kim, D.W.; Nagrial, A.; Voskoboynik, M.; Chung, H.C.; Mileham, K.; Vaishampayan, U.; Rasco, D.; et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer☆. Ann. Oncol. 2022, 33, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Peled, N.; Mazieres, J.; Kowalski, D.M.; Lam, W.S.; Hochmair, M.J.; Majem, M.; Kim, S.H.; Blanco, A.C.; Cuppens, K.; Casarini, I.A.; et al. 121P MK-7684A (Vibostolimab [Vibo] plus Pembrolizumab [Pembro] coformulation) with/without Docetaxel in metastatic NSCLC after platinum-chemotherapy (Chemo) and immunotherapy. Immuno. Oncol. Technol. 2023, 20, 100593. [Google Scholar] [CrossRef]
- Cho, B.C.; Juergens, R.A.; Cheng, Y.; De Castro, G.; Erman, M.; Bauman, J.R.; Takahashi, T.; Schwarzenberger, P.; Li, C.; Pietanza, M.C.; et al. Abstract CT561: KeyVibe-003: Randomized, double-blind, phase 3 study of first-line pembrolizumab with and without vibostolimab (Anti-TIGIT) in patients with PD-L1-positive metastatic NSCLC. Cancer Res. 2022, 82, CT561. [Google Scholar] [CrossRef]
- Garassino, M.C.; Felip, E.; Awad, M.M.; Wang, W.; Kim, S.J.; Pietanza, M.C.; Rodriguez-Abreu, D. A randomized, double-blind, phase 3 trial of MK-7684A plus chemotherapy versus pembrolizumab plus chemotherapy as first-line therapy for metastatic non–small cell lung cancer (NSCLC): KeyVibe-007. J. Clin. Oncol. 2022, 40, TPS9160. [Google Scholar] [CrossRef]
- Rohrberg, K.S.; Brandão, M.; Castanon Alvarez, E.; Felip, E.; Gort, E.H.; Hiltermann, T.J.J.N.; Izumi, H.; Kim, D.W.; Kim, S.W.; Paz-Ares, L.G.; et al. Safety, Pharmacokinetics (PK), Pharmacodynamics (PD) and preliminary efficacy of AZD2936, a bispecific antibody targeting PD-1 and TIGIT, in checkpoint inhibitor (CPI)-experienced advanced/metastatic non-small-cell lung cancer (NSCLC): First report of ARTEMIDE-01. J. Clin. Oncol. 2023, 41, 9050. [Google Scholar] [CrossRef]
- Brandão, M.; Hiltermann, J.T.J.N.; Wauters, E.; Solomon, B.J.; Castanon Alvarez, E.; Felip, E.; Gort, E.H.; Izumi, H.; Kim, D.W.; Paz-Ares, L.; et al. 1446P preliminary efficacy and safety of rilvegostomig (AZD2936), a bispecific antibody targeting PD-1 and TIGIT, in checkpoint inhibitor (CPI)-pretreated advanced/metastatic non-small-cell lung cancer (NSCLC): ARTEMIDE-01. Ann. Oncol. 2023, 34, S822–S823. [Google Scholar] [CrossRef]
- Hiltermann, T.J.N.; Izumi, H.; Cho, B.C.; Cunha, S.; Danchaivijitr, P.; Felip, E.; Ho, G.F.; Leventakos, K.; Li, Y.; Sugawara, S.; et al. OA11.03 efficacy and safety of Rilvegostomig, an anti-PD-1/TIGIT bispecific, for CPI-naïve metastatic NSCLC with PD-L1 1-49% or ≥50%. J. Thorac. Oncol. 2024, 19, S33. [Google Scholar] [CrossRef]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Guy, C.; Mitrea, D.M.; Chou, P.C.; Temirov, J.; Vignali, K.M.; Liu, X.; Zhang, H.; Kriwacki, R.; Bruchez, M.P.; Watkins, S.C.; et al. LAG3 associates with TCR–CD3 complexes and suppresses signaling by driving co-receptor–Lck dissociation. Nat. Immunol. 2022, 23, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-Cell responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Triebel, F. LAG-3: A regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 2003, 24, 619–622. [Google Scholar] [CrossRef]
- Andrews, L.P.; Somasundaram, A.; Moskovitz, J.M.; Szymczak-Workman, A.L.; Liu, C.; Cillo, A.R.; Lin, H.; Normolle, D.P.; Moynihan, K.D.; Taniuchi, I.; et al. Resistance to PD1 blockade in the absence of metalloprotease-mediated LAG3 shedding. Sci. Immunol. 2020, 5, eabc2728. [Google Scholar] [CrossRef]
- Liang, B.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; Flores, M.; Li, N.; Schweighoffer, E.; Greenberg, S.; et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 2008, 180, 5916–5926. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Zhang, Q.; Beres, A.; Vignali, D. Regulatory T cells use LAG-3 as the “brake” in autoimmune diabetes but the “accelerator” in tumor environment (IRC4P.487). J. Immunol. 2014, 192, 60.14. [Google Scholar] [CrossRef]
- Thaker, Y.R.; Andrews, L.P.; Workman, C.J.; Vignali, D.A.A.; Sharpe, A.H. Treg-specific LAG3 deletion reveals a key role for LAG3 in regulatory T Cells to inhibit CNS autoimmunity. J. Immunol. 2018, 200 (Suppl. S1), 101.7. [Google Scholar] [CrossRef]
- Kim, D.; Kim, G.; Yu, R.; Lee, J.; Kim, S.; Gleason, M.R.; Qiu, K.; Montauti, E.; Wang, L.L.; Fang, D.; et al. Inhibitory co-receptor Lag3 supports Foxp3+ regulatory T cell function by restraining Myc-dependent metabolic programming. Immunity 2024, 57, 2634–2650.e5. [Google Scholar] [CrossRef]
- Dykema, A.G.; Zhang, J.; Cheung, L.S.; Connor, S.; Zhang, B.; Zeng, Z.; Cherry, C.M.; Li, T.; Caushi, J.X.; Nishimoto, M.; et al. Lung tumor–infiltrating treg have divergent transcriptional profiles and function linked to checkpoint blockade response. Sci. Immunol. 2023, 8, eadg1487. [Google Scholar] [CrossRef]
- Su, J.; Fu, Y.; Cui, Z.; Abidin, Z.; Yuan, J.; Zhang, X.; Li, R.; Zhao, C. Relatlimab: A novel drug targeting immune checkpoint LAG-3 in melanoma therapy. Front. Pharmacol. 2024, 14, 1349081. [Google Scholar] [CrossRef] [PubMed]
- Detterbeck, F.C. The eighth edition TNM stage classification for lung cancer: What does it mean on main street? J. Thorac. Cardiovasc. Surg. 2018, 155, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Girard, N.; Burotto, M.; Paz-Ares, L.G.; Reck, M.; Schenker, M.; Lingua, A.; Orlandi, F.J.; Naidoo, J.; Beardlsey, E.K.; Velcheti, V.; et al. LBA53 nivolumab (NIVO) plus relatlimab with platinum-doublet chemotherapy (PDCT) vs NIVO + PDCT as first-line (1L) treatment (Tx) for stage IV or recurrent NSCLC: Results from the randomized phase II RELATIVITY-104 study. Ann. Oncol. 2024, 35, S1243–S1244. [Google Scholar] [CrossRef]
- Schöffski, P.; Tan, D.S.W.; Martín, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer. 2022, 10, e003776. [Google Scholar] [CrossRef]
- Lin, C.C.; Garralda, E.; Schöffski, P.; Hong, D.S.; Siu, L.L.; Martin, M.; Maur, M.; Hui, R.; Soo, R.A.; Chiu, J.; et al. A phase 2, multicenter, open-label study of anti-LAG-3 ieramilimab in combination with anti-PD-1 spartalizumab in patients with advanced solid malignancies. OncoImmunology 2024, 13, 2290787. [Google Scholar] [CrossRef]
- Mishra, A.K.; Shahid, S.; Karade, S.S.; Agnihotri, P.; Kolesnikov, A.; Hasan, S.S.; Mariuzza, R.A. CryoEM structure of a therapeutic antibody (Favezelimab) bound to human LAG3 determined using a bivalent fab as fiducial marker. Structure 2023, 31, 1149–1157.e3. [Google Scholar] [CrossRef]
- Gutierrez, M.; Lam, W.S.; Hellmann, M.; Gubens, M.; Aggarwal, C.; Weng Tan, D.S.; Felip, E.; Chiu, J.W.; Lee, J.S.; Yang, J.C.-H.; et al. 457 KEYNOTE-495/KeyImPaCT: Interim Analysis of a Randomized, Biomarker-Directed, Phase 2 Trial of Pembrolizumab-Based Combination Therapy for Non–Small Cell Lung Cancer (NSCLC). J. ImmunoTherapy Cancer 2021, 9. [Google Scholar] [CrossRef]
- Gutierrez, M.; Lam, W.S.; Hellmann, M.D.; Gubens, M.A.; Aggarwal, C.; Tan, D.S.W.; Felip, E.; Chiu, J.W.Y.; Lee, J.S.; Yang, J.C.H.; et al. Biomarker-directed, pembrolizumab-based combination therapy in non-small cell lung cancer: Phase 2 KEYNOTE-495/KeyImPaCT trial interim results. Nat. Med. 2023, 29, 1718–1727. [Google Scholar] [CrossRef]
- Faulkner, N.E.; Melkadze, T.; Nair, S.; Gabrail, N.; Kunta, G.; Ibrahim, E.; Dreisbach, L.; Brungs, D.; Chikhladze, N.; Gogishvili, M.; et al. A phase 2/3 study of fianlimab plus cemiplimab versus cemiplimab in patients with advanced non-small cell lung cancer with tumors expressing PD-L1 ≥50%. J. Clin. Oncol. 2024, 42, TPS8663. [Google Scholar] [CrossRef]
- Gabrail, N.; Gogishvili, M.; Makharadze, T.; Chikhladze, N.; Faulkner, N.E.; Nair, S.; Ibrahim, E.; Brungs, D.; Dreisbach, L.; Kunta, G.; et al. A phase 2/3 study of fianlimab, cemiplimab, plus chemotherapy versus cemiplimab plus chemotherapy in first-line treatment of advanced non-small cell lung cancer. J. Clin. Oncol. 2024, 42, TPS8660. [Google Scholar] [CrossRef]
- Johnson, M.L.; Patel, M.R.; Ulahannan, S.V.; Hansen, A.; George, B.; Chu, Q.S.C.; Elgadi, M.; Ge, M.; Duffy, C.; Graeser, R.; et al. Phase I study of BI 754111 (Anti-LAG-3) plus BI 754091(Anti-PD-1) in patients (Pts) with advanced solid cancers, followed by expansion in pts with microsatellite stable metastatic colorectal cancer (mCRC), anti-PD-(L)1-pretreated non-small cell lung cancer (NSCLC) and other solid tumors. Ann. Oncol. 2018, 29, viii441. [Google Scholar] [CrossRef]
- Johnson, M.L.; Patel, M.R.; Cherry, M.; Kang, Y.K.; Yamaguchi, K.; Oh, D.Y.; Hussein, M.A.; Kitano, S.; Kondo, S.; Hansen, A.R.; et al. Safety of BI 754111, an anti-LAG-3 monoclonal antibody (mAb), in combination with BI 754091, an anti-PD-1 mAb, in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3063. [Google Scholar] [CrossRef]
- Zettl, M.; Wurm, M.; Schaaf, O.; Mostböck, S.; Tirapu, I.; Apfler, I.; Lorenz, I.C.; Frego, L.; Kenny, C.; Thibodeau, M.; et al. Combination of two novel blocking antibodies, anti-PD-1 antibody ezabenlimab (BI 754091) and anti-LAG-3 antibody BI 754111, leads to increased immune cell responses. OncoImmunology 2022, 11, 2080328. [Google Scholar] [CrossRef]
- Ghosh, S.; Sharma, G.; Travers, J.; Kumar, S.; Choi, J.; Jun, H.T.; Kehry, M.; Ramaswamy, S.; Jenkins, D. TSR-033, a novel therapeutic antibody targeting LAG-3, enhances T-cell function and the activity of PD-1 blockade in vitro and in vivo. Mol. Cancer Ther. 2019, 18, 632–641. [Google Scholar] [CrossRef]
- Grandal, M.M.; Melander, M.C.; Bhatia, V.K.; Gjetting, T.; Lindsted, T.; Fröhlich, C.; Lantto, J.; Horak, I.D.; Kragh, M.; Kofoed, K.; et al. Abstract 5626: Preclinical characterization of Sym022, a novel anti-LAG3 antibody. Cancer Res. 2018, 78, 5626. [Google Scholar] [CrossRef]
- Yap, T.A.; LoRusso, P.M.; Wong, D.J.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Holz, J.B.; Grabowska, U.; Gradinaru, C.; Leung, K.M.; Marshall, S.; et al. A phase 1 first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1 in patients with advanced cancer and PD-L1 resistance. Clin. Cancer Res. 2023, 29, 888–898. [Google Scholar] [CrossRef]
- Rohrberg, K.S.; Garralda, E.; Calvo, E.; Moreno Garcia, V.; Guidi, M.; Kraus, D.G.; McIntyre, C.; Kao, H.; Codarri Deak, L.; Michielin, F.; et al. 745P clinical activity, safety, and PK/PD from the first in human study (NP41300) of RO7247669, a PD1-LAG3 bispecific antibody. Ann. Oncol. 2022, 33, S884–S885. [Google Scholar] [CrossRef]
- Luke, J.J.; Patel, M.R.; Blumenschein, G.R.; Hamilton, E.; Chmielowski, B.; Ulahannan, S.V.; Connolly, R.M.; Santa-Maria, C.A.; Wang, J.; Bahadur, S.W.; et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: A phase 1 trial. Nat. Med. 2023, 29, 2814–2824. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific antibodies: From research to clinical application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Dixon, K.O.; Lahore, G.F.; Kuchroo, V.K. Beyond T cell exhaustion: TIM-3 regulation of myeloid cells. Sci. Immunol. 2024, 9, eadf2223. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory t cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Morita, T.; Annan, D.A.; Horlad, H.; Ohnishi, K.; Yamada, S.; Nakayama, T.; Kitada, S.; Suzu, S.; Kinoshita, I.; et al. The coordinated actions of TIM-3 on cancer and myeloid cells in the regulation of tumorigenicity and clinical prognosis in clear cell renal cell carcinomas. Cancer Immunol. Res. 2015, 3, 999–1007. [Google Scholar] [CrossRef]
- Dixon, K.O.; Tabaka, M.; Schramm, M.A.; Xiao, S.; Tang, R.; Dionne, D.; Anderson, A.C.; Rozenblatt-Rosen, O.; Regev, A.; Kuchroo, V.K. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 2021, 595, 101–106. [Google Scholar] [CrossRef]
- Phong, B.L.; Avery, L.; Sumpter, T.L.; Gorman, J.V.; Watkins, S.C.; Colgan, J.D.; Kane, L.P. Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J. Exp. Med. 2015, 212, 2289–2304. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand Galectin-9 negatively regulates t helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Ma, X.; Ma, Y.; Du, Y.; Feng, J. A New emerging target in cancer immunotherapy: Galectin-9 (LGALS9). Genes Dis. 2023, 10, 2366–2382. [Google Scholar] [CrossRef]
- Elahi, S.; Niki, T.; Hirashima, M.; Horton, H. Galectin-9 binding to Tim-3 renders activated human CD4+ T cells less susceptible to HIV-1 infection. Blood 2012, 119, 4192–4204. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, A.; Bigam, S.; Elahi, S. Galectin-9 promotes natural killer cells activity via interaction with CD44. Front. Immunol. 2023, 14, 1131379. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yoon, C.H.; Gandhi, A.; Hanley, T.; Castrillon, C.; Kondo, Y.; Lin, X.; Kim, W.; Yang, C.; Driouchi, A.; et al. High-dimensional mapping of human CEACAM1 expression on immune cells and association with melanoma drug resistance. Commun. Med. 2024, 4, 128. [Google Scholar] [CrossRef]
- Takeuchi, A.; Yokoyama, S.; Nakamori, M.; Nakamura, M.; Ojima, T.; Yamaguchi, S.; Mitani, Y.; Shively, J.E.; Yamaue, H. Loss of CEACAM1 is associated with poor prognosis and peritoneal dissemination of patients with gastric cancer. Sci. Rep. 2019, 9, 12702. [Google Scholar] [CrossRef]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.L.E.; Lee, H.H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T Cell/transmembrane, Ig, and Mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef]
- Van De Weyer, P.S.; Muehlfeit, M.; Klose, C.; Bonventre, J.V.; Walz, G.; Kuehn, E.W. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand Galectin-9. Biochem. Biophys. Res. Commun. 2006, 351, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Schraven, B.; Veillette, A. PAG-associated fynt regulates calcium signaling and promotes anergy in T lymphocytes. Mol. Cell. Biol. 2007, 27, 1960–1973. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef]
- Clayton, K.L.; Haaland, M.S.; Douglas-Vail, M.B.; Mujib, S.; Chew, G.M.; Ndhlovu, L.C.; Ostrowski, M.A. T cell Ig and mucin domain–containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J. Immunol. 2014, 192, 782–791. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed]
- De Mingo Pulido, Á.; Hänggi, K.; Celias, D.P.; Gardner, A.; Li, J.; Batista-Bittencourt, B.; Mohamed, E.; Trillo-Tinoco, J.; Osunmakinde, O.; Peña, R.; et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 2021, 54, 1154–1167.e7. [Google Scholar] [CrossRef]
- Redmond, W.L.; Sherman, L.A. Peripheral tolerance of CD8 T lymphocytes. Immunity 2005, 22, 275–284. [Google Scholar] [CrossRef]
- Luckashenak, N.; Schroeder, S.; Endt, K.; Schmidt, D.; Mahnke, K.; Bachmann, M.F.; Marconi, P.; Deeg, C.A.; Brocker, T. Constitutive crosspresentation of tissue antigens by dendritic cells controls CD8+ T cell tolerance in vivo. Immunity 2008, 28, 521–532. [Google Scholar] [CrossRef]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Y.; Zhang, J.P.; Liang, J.; Li, L.; Zheng, L. Tim-3 expression defines regulatory T cells in human tumors. PLoS ONE 2013, 8, e58006. [Google Scholar] [CrossRef]
- Banerjee, H.; Nieves-Rosado, H.; Kulkarni, A.; Murter, B.; McGrath, K.V.; Chandran, U.R.; Chang, A.; Szymczak-Workman, A.L.; Vujanovic, L.; Delgoffe, G.M.; et al. Expression of Tim-3 drives phenotypic and functional changes in treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep. 2021, 36, 109699. [Google Scholar] [CrossRef]
- Sakuishi, K.; Ngiow, S.F.; Sullivan, J.M.; Teng, M.W.L.; Kuchroo, V.K.; Smyth, M.J.; Anderson, A.C. TIM3+ FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. OncoImmunology 2013, 2, e23849. [Google Scholar] [CrossRef]
- Zhu, C.; Dixon, K.O.; Newcomer, K.; Gu, G.; Xiao, S.; Zaghouani, S.; Schramm, M.A.; Wang, C.; Zhang, H.; Goto, K.; et al. Tim-3 adaptor protein Bat3 is a molecular checkpoint of T cell terminal differentiation and exhaustion. Sci. Adv. 2021, 7, eabd2710. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Falchook, G.S.; Ribas, A.; Davar, D.; Eroglu, Z.; Wang, J.S.; Luke, J.J.; Hamilton, E.P.; Di Pace, B.; Wang, T.; Ghosh, S.; et al. Phase 1 trial of TIM-3 inhibitor cobolimab monotherapy and in combination with PD-1 inhibitors nivolumab or dostarlimab (AMBER). J. Clin. Oncol. 2022, 40, 2504. [Google Scholar] [CrossRef]
- Davar, D.; Eroglu, Z.; Milhem, M.; Becerra, C.; Gutierrez, M.; Ribas, A.; Pace, B.D.; Wang, T.; Zhang, H.; Ghosh, S.; et al. 596 AMBER, part 2B: A phase 1 study of cobolimab plus dostarlimab in patients with advanced/metastatic non-small cell lung cancer (NSCLC) previously treated with anti-PD(L)-1 therapy. J. ImmunoTherapy Cancer 2023, 11. [Google Scholar] [CrossRef]
- Kim, H.R.; Gridelli, C.; Kapur, D.; Tufman, A.; Felip, E.; Velcheti, V.; Kim, Y.J.; Goetze, T.O.; Lopez, P.G.; Corre, R.; et al. P1.11-01 cobolimab with dostarlimab and docetaxel in patients with advanced non-small cell lung cancer (NSCLC): COSTAR lung. J. Thorac. Oncol. 2022, 17, S109–S110. [Google Scholar] [CrossRef]
- Mach, N.; Curigliano, G.; Santoro, A.; Kim, D.W.; Tai, D.W.M.; Hodi, S.; Wilgenhof, S.; Doi, T.; Longmire, T.; Sun, H.; et al. Phase (Ph) II study of MBG453 + Spartalizumab in patients (Pts) with non-small cell lung cancer (NSCLC) and melanoma pretreated with Anti–PD-1/L1 therapy. Ann. Oncol. 2019, 30, v491–v492. [Google Scholar] [CrossRef]
- Harding, J.J.; Moreno, V.; Bang, Y.J.; Hong, M.H.; Patnaik, A.; Trigo, J.; Szpurka, A.M.; Yamamoto, N.; Doi, T.; Fu, S.; et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: A phase Ia/b Study of LY3321367 with or without an anti-PD-L1 antibody. Clin. Cancer Res. 2021, 27, 2168–2178. [Google Scholar] [CrossRef]
- Waight, J.; Iyer, P.; Breous-Nystrom, E.; Riordan, C.; Findeis, M.; Underwood, D.; Connolly, J.; Sanicola-Nadel, M.; Nastri, H.; Scherle, P.; et al. Abstract 3825: INCAGN02390, a novel antagonist antibody that targets the co-inhibitory receptor TIM-3. Cancer Res. 2018, 78, 3825. [Google Scholar] [CrossRef]
- Besse, B.; Italiano, A.; Cousin, S.; Ruiter, G.; Felip, E.; Castanon Alvarez, E.; Ramalingam, S.S.; Rolfo, C.D.; Spigel, D.R.; Andrew, D.; et al. 1313MO Safety and preliminary efficacy of AZD7789, a bispecific antibody targeting PD-1 and TIM-3, in patients (Pts) with stage IIIB–IV non-small-cell lung cancer (NSCLC) with previous anti-PD-(L)1 therapy. Ann. Oncol. 2023, 34, S755. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Bivi, N.; Calderon, B.; Shimizu, T.; Delafontaine, B.; Liu, Z.T.; Szpurka, A.M.; Copeland, V.; Hodi, F.S.; Rottey, S.; et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin. Cancer Res. 2021, 27, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Antonia, S.J. The NKG2A immune checkpoint—A new direction in cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 277–278. [Google Scholar] [CrossRef]
- Cazzetta, V.; Depierreux, D.; Colucci, F.; Mikulak, J.; Mavilio, D. NKG2A immune checkpoint in Vδ2 T Cells: Emerging application in cancer immunotherapy. Cancers 2023, 15, 1264. [Google Scholar] [CrossRef]
- Raulet, D.H.; Vance, R.E.; McMahon, C.W. Regulation of the natural killer cell receptor repertoire. Annu. Rev. Immunol. 2001, 19, 291–330. [Google Scholar] [CrossRef]
- Kabat, J.; Borrego, F.; Brooks, A.; Coligan, J.E. Role that each NKG2A immunoreceptor tyrosine-based inhibitory motif plays in mediating the human CD94/NKG2A inhibitory signal. J. Immunol. 2002, 169, 1948–1958. [Google Scholar] [CrossRef]
- Lanier, L.L. NK CELL RECOGNITION. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Gemelli, M.; Noonan, D.M.; Carlini, V.; Pelosi, G.; Barberis, M.; Ricotta, R.; Albini, A. Overcoming resistance to checkpoint inhibitors: Natural killer cells in non-small cell lung cancer. Front. Oncol 2022, 12, 886440. [Google Scholar] [CrossRef] [PubMed]
- Kyrysyuk, O.; Wucherpfennig, K.W. Designing cancer immunotherapies that engage T cells and NK cells. Annu. Rev. Immunol. 2023, 41, 17–38. [Google Scholar] [CrossRef]
- Viant, C.; Fenis, A.; Chicanne, G.; Payrastre, B.; Ugolini, S.; Vivier, E. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat. Commun. 2014, 5, 5108. [Google Scholar] [CrossRef] [PubMed]
- Niogret, C.; Miah, S.M.S.; Rota, G.; Fonta, N.P.; Wang, H.; Held, W.; Birchmeier, W.; Sexl, V.; Yang, W.; Vivier, E.; et al. Shp-2 is critical for ERK and metabolic engagement downstream of IL-15 receptor in NK cells. Nat. Commun. 2019, 10, 1444. [Google Scholar] [CrossRef]
- Yazdi, M.T.; Van Riet, S.; Van Schadewijk, A.; Fiocco, M.; Van Hall, T.; Taube, C.; Hiemstra, P.S.; Van Der Burg, S.H. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget 2016, 7, 3477–3488. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.H.; Li, S.; Khodadadi-Jamayran, A.; Jen, J.; Han, H.; Guidry, K.; Chen, T.; Hao, Y.; Fedele, C.; Zebala, J.A.; et al. Combined inhibition of SHP2 and CXCR1/2 promotes antitumor T-cell response in NSCLC. Cancer Discov. 2022, 12, 47–61. [Google Scholar] [CrossRef]
- Borst, L.; Sluijter, M.; Sturm, G.; Charoentong, P.; Santegoets, S.J.; Van Gulijk, M.; Van Elsas, M.J.; Groeneveldt, C.; Van Montfoort, N.; Finotello, F.; et al. NKG2A is a late immune checkpoint on CD8 T cells and marks repeated stimulation and cell division. Intl J. Cancer 2022, 150, 688–704. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.J.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity 1998, 8, 693–701. [Google Scholar] [CrossRef]
- Vales-Gomez, M. Kinetics and Peptide Dependency of the Binding of the Inhibitory NK Receptor CD94/NKG2-A and the Activating Receptor CD94/NKG2-C to HLA-E. EMBO J. 1999, 18, 4250–4260. [Google Scholar] [CrossRef]
- Kaiser, B.K.; Barahmand-pour, F.; Paulsene, W.; Medley, S.; Geraghty, D.E.; Strong, R.K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J. Immunol. 2005, 174, 2878–2884. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, N.; Wieten, L.; Popeijus, H.E.; Voorter, C.E.M.; Tilanus, M.G.J. HLA-E regulates NKG2C+ natural killer cell function through presentation of a restricted peptide repertoire. Hum. Immunol. 2015, 76, 578–586. [Google Scholar] [CrossRef]
- Siemaszko, J.; Marzec-Przyszlak, A.; Bogunia-Kubik, K. Activating NKG2C receptor: Functional characteristics and current strategies in clinical applications. Arch. Immunol. Ther. Exp. 2023, 71, 9. [Google Scholar] [CrossRef] [PubMed]
- Battin, C.; Kaufmann, G.; Leitner, J.; Tobias, J.; Wiedermann, U.; Rölle, A.; Meyer, M.; Momburg, F.; Steinberger, P. NKG2A -checkpoint Inhibition and its blockade critically depends on peptides presented by its ligand HLA-E. Immunology 2022, 166, 507–521. [Google Scholar] [CrossRef]
- Nuttin, L.; Latour, C.; Revy, S.; Aucagne, R.; Lalle, G.; Ghiringhelli, F.; Limagne, E. Abstract 3864: Role of NK/TAM cross-talk on T-CD8+ recruitment and efficacy of chemoimmunotherapy and MEKi combination in “cold” tumors. Cancer Res. 2024, 84, 3864. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; Kar, G.; Spicer, J.D.; García-Campelo, R.; Weder, W.; Daniel, D.B.; Spigel, D.R.; Hussein, M.; Mazieres, J.; Oliveira, J.; et al. Neoadjuvant durvalumab alone or combined with novel immuno-oncology agents in resectable lung cancer: The phase II NeoCOAST platform trial. Cancer Discov. 2023, 13, 2394–2411. [Google Scholar] [CrossRef]
- Herbst, R.S.; Majem, M.; Barlesi, F.; Carcereny, E.; Chu, Q.; Monnet, I.; Sanchez-Hernandez, A.; Dakhil, S.; Camidge, D.R.; Winzer, L.; et al. COAST: An open-label, phase II, multidrug platform study of durvalumab alone or in combination with oleclumab or monalizumab in patients with unresectable, stage III non–small-cell lung cancer. J. Clin. Oncol. 2022, 40, 3383–3393. [Google Scholar] [CrossRef]
- Aggarwal, C.; Martinez-Marti, A.; Majem, M.; Barlesi, F.; Carcereny, E.; Chu, Q.S.; Monnet, I.; Sanchez, A.; Dakhil, S.R.; Camidge, D.R.; et al. Updated results from COAST, a phase 2 study of Durvalumab (D) ± Oleclumab (O) or Monalizumab (M) in patients (Pts) with stage III unresectable non-small cell lung cancer (uNSCLC). J. Clin. Oncol. 2024, 42, 8046. [Google Scholar] [CrossRef]
- Barlesi, F.; Cho, B.C.; Goldberg, S.B.; Yoh, K.; Zimmer Gelatti, A.C.; Mann, H.; Gopinathan, A.; Bielecka, Z.F.; Newton, M.; Aggarwal, C. PACIFIC-9: Phase III trial of Durvalumab + Oleclumab or Monalizumab in unresectable stage III non-small-cell lung cancer. Future Oncol. 2024, 20, 2137–2147. [Google Scholar] [CrossRef]
- Patel, S.P.; Alonso-Gordoa, T.; Banerjee, S.; Wang, D.; Naidoo, J.; Standifer, N.E.; Palmer, D.C.; Cheng, L.Y.; Kourtesis, P.; Ascierto, M.L.; et al. Phase 1/2 study of monalizumab plus durvalumab in patients with advanced solid tumors. J Immunother Cancer 2024, 12, e007340. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Cropet, C.; Jeanson, A.; Pérol, M.; Mazieres, J.; Fontaine, C.; Rabeau, A.; Meurer, M.; Greillier, L.; Le Ray, M.; et al. LBA8 Precision immuno-oncology for advanced non-small cell lung cancer (NSCLC) patients with PD-(L)1 inhibitors resistance (PIONeeR): A phase Ib/IIa clinical trial targeting identified resistance pathways. Ann. Oncol. 2024, 35, S1242. [Google Scholar] [CrossRef]
- Melander, M.; Laugel, B. Abstract LB220: S095029: A novel clinical-stage Fc-silenced NKG2A-blocking antibody with best-in-class potential. Cancer Res. 2023, 83, LB220. [Google Scholar] [CrossRef]
- Spreafico, A.; Naing, A.; Barve, M.; Patnaik, A.; Ianopulos, X.; Drean, P.; Hemon, A.; Kantari, C.C.; He, P.; Askoxylakis, V.; et al. 1495 A Phase 1 Multicenter Study of the Safety and Efficacy of the NKG2A Targeting Antibody S095029 as Single Agent and in Combination with Anti-PD1 in Patients with Advanced Malignancies. J. ImmunoTherapy Cancer 2024, 12. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef]
- Kaplinsky, N.; Williams, K.; Watkins, D.; Adams, M.; Stanbery, L.; Nemunaitis, J. Regulatory role of CD39 and CD73 in tumor immunity. Future Oncol. 2024, 20, 1367–1380. [Google Scholar] [CrossRef]
- Resta, R.; Yamashita, Y.; Thompson, L.F. Ecto-enzyme and Signaling Functions of Lymphocyte CD 7 3. Immunological Reviews 1998, 161, 95–109. [Google Scholar] [CrossRef]
- Liu, D.; Zhao, J.; Li, L.; Wang, J.; Wang, C.; Wu, Y.; Huang, Y.; Xing, D.; Chen, W. CD73: Agent development potential and its application in diabetes and atherosclerosis. Front. Immunol. 2024, 15, 1515875. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD 39 and CD 73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef]
- Imbimbo, M.; Hollebecque, A.; Italiano, A.; McKean, M.; Macarulla, T.; Castanon Alvarez, E.; Carneiro, B.A.; Mager, R.; Barnhart, V.; Murtomaki, E.; et al. 188P IPH5201 as monotherapy or in combination with durvalumab (D) in advanced solid tumours. Immuno. Oncol. Technol. 2022, 16, 100300. [Google Scholar] [CrossRef]
- Barlesi, F.; Mercier, O.; Łowczak, A.; Mandziuk, S.; Kuzdzal, J.; Koutras, A.; Dasgupta, A.; Leyco, A.J.; Andre, P.; Mager, R.; et al. 1290TiP a phase II multicenter, open label, non-randomized study of neoadjuvant and adjuvant treatment with IPH5201 and durvalumab in patients with resectable, early-stage (II to IIIA) non-small cell lung cancer (MATISSE). Ann. Oncol. 2023, 34, S744. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Cha, Y.; George, B.; Grewal, J.S.; Merchan, J.R.; Mckean, M.; Soares, H.P.; Cruz, T.D.; Moesta, A.K.; Jahn, T.M.; et al. 659P combination treatment with TTX-030, a first-in-class anti-CD39 antibody, in patients with advanced pancreatic cancer. Ann. Oncol. 2024, 35, S519–S520. [Google Scholar] [CrossRef]
- Thompson, L.F.; Ruedi, J.M.; Glass, A.; Low, M.G.; Lucas, A.H. Antibodies to 5’-nucleotidase (CD73), a glycosyl-phosphatidylinositol-anchored protein, cause human peripheral blood T cells to proliferate. J. Immunol. 1989, 143, 1815–1821. [Google Scholar] [CrossRef]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Invest. 2011, 121, 2371–2382. [Google Scholar] [CrossRef]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.P.; Möller, A.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef]
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.A.; Haibe-Kains, B.; Karn, T.; et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015, 75, 4494–4503. [Google Scholar] [CrossRef]
- Allard, B.; Turcotte, M.; Spring, K.; Pommey, S.; Royal, I.; Stagg, J. Anti-CD73 therapy impairs tumor angiogenesis. Int. J. Cancer 2014, 134, 1466–1473. [Google Scholar] [CrossRef]
- Bendell, J.; LoRusso, P.; Overman, M.; Noonan, A.M.; Kim, D.-W.; Strickler, J.H.; Kim, S.W.; Clarke, S.; George, T.J.; Grimison, P.S.; et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunol. Immunother 2023, 72, 2443–2458. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. international union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Creelan, B.; Tanvetyanon, T.; Gray, J.E.; Haura, E.B.; Thapa, R.; Barlow, M.L.; Chen, Z.; Chen, D.T.; Beg, A.A.; et al. Phase I study of taminadenant (PBF509/NIR178), an adenosine 2A receptor antagonist, with or without spartalizumab (PDR001), in patients with advanced non–small cell lung cancer. Clin. Cancer Res. 2022, 28, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
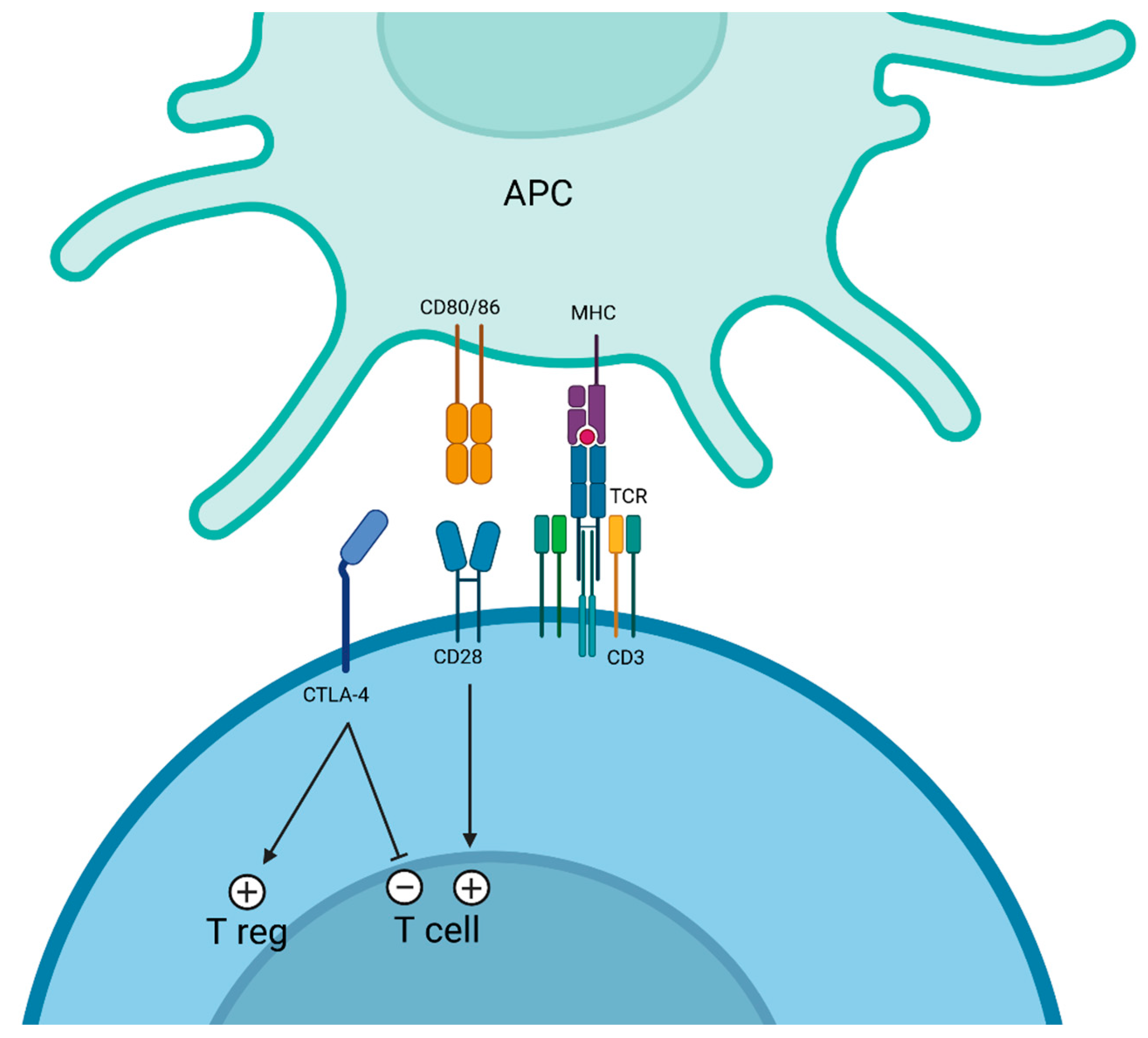
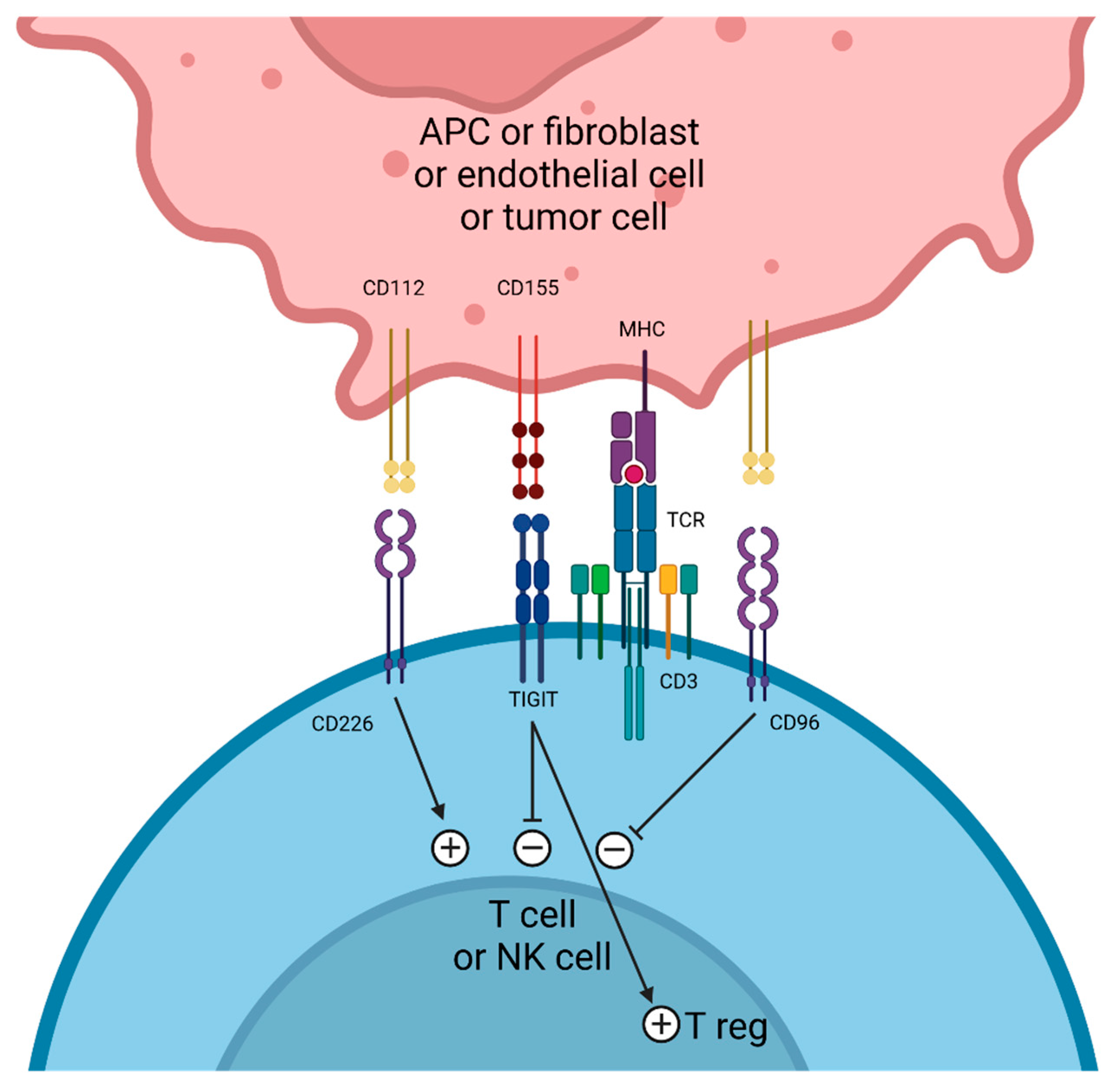

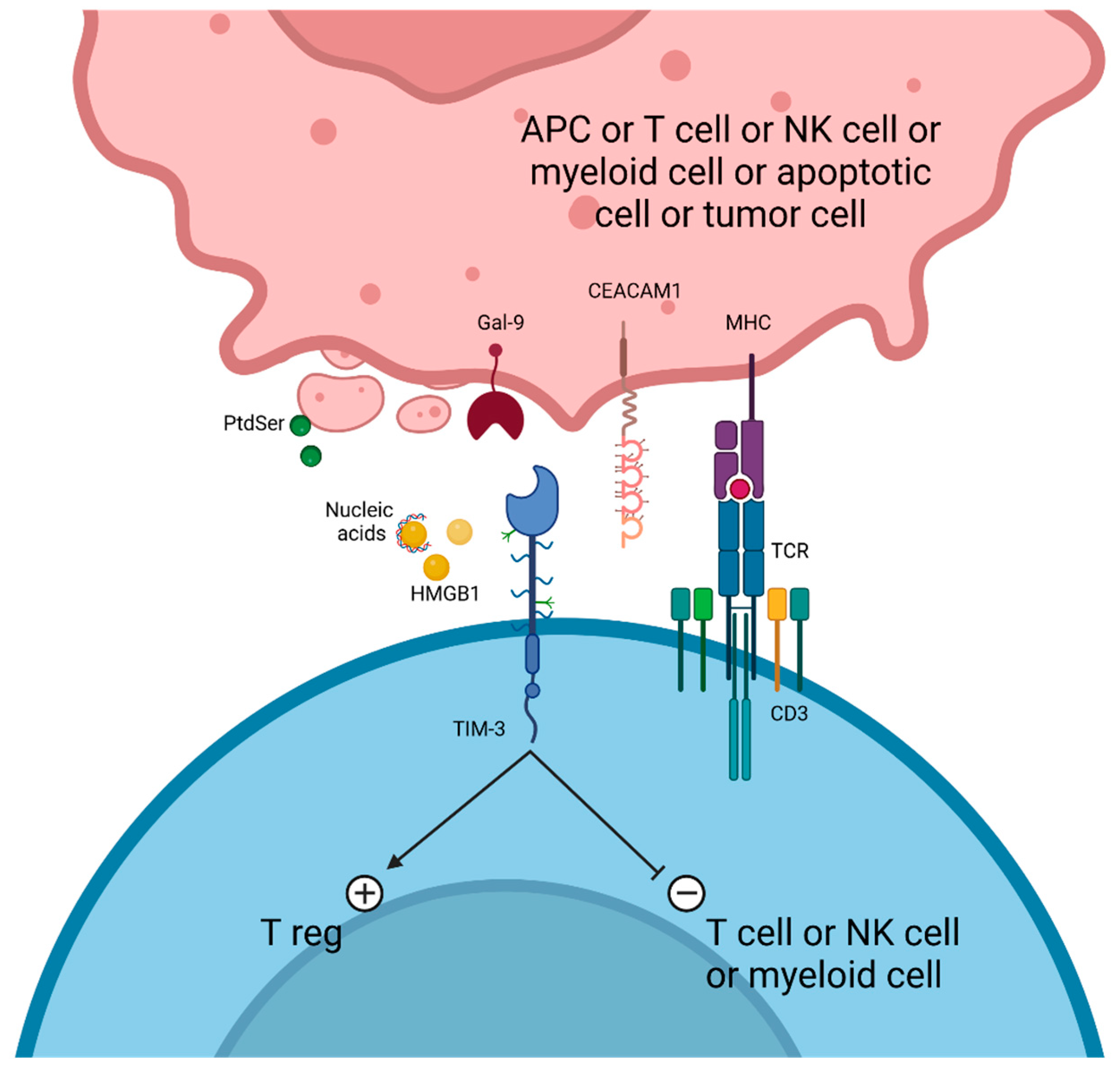
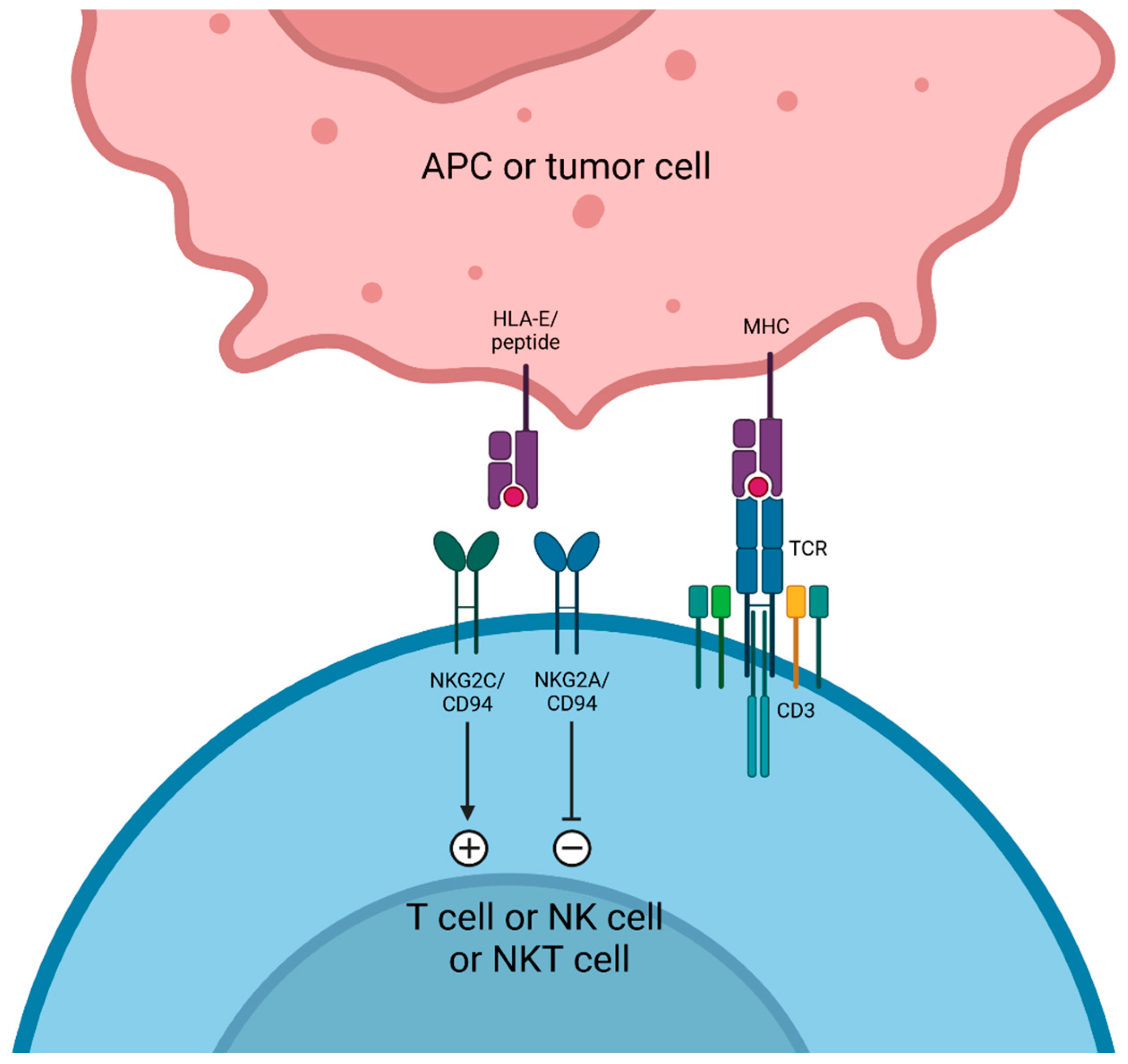
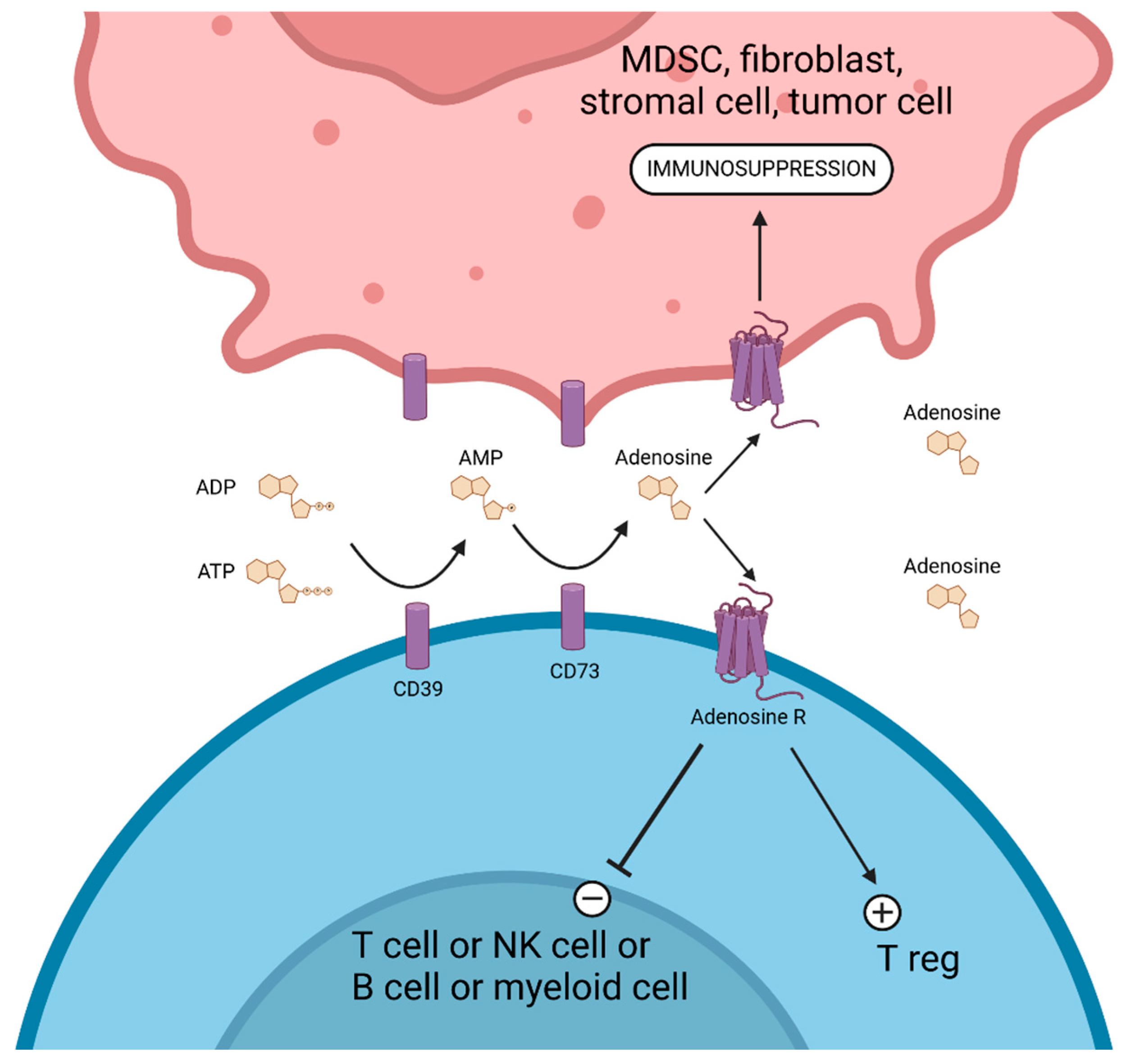
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roussot, N.; Kaderbhai, C.; Ghiringhelli, F. Targeting Immune Checkpoint Inhibitors for Non-Small-Cell Lung Cancer: Beyond PD-1/PD-L1 Monoclonal Antibodies. Cancers 2025, 17, 906. https://doi.org/10.3390/cancers17050906
Roussot N, Kaderbhai C, Ghiringhelli F. Targeting Immune Checkpoint Inhibitors for Non-Small-Cell Lung Cancer: Beyond PD-1/PD-L1 Monoclonal Antibodies. Cancers. 2025; 17(5):906. https://doi.org/10.3390/cancers17050906
Chicago/Turabian StyleRoussot, Nicolas, Courèche Kaderbhai, and François Ghiringhelli. 2025. "Targeting Immune Checkpoint Inhibitors for Non-Small-Cell Lung Cancer: Beyond PD-1/PD-L1 Monoclonal Antibodies" Cancers 17, no. 5: 906. https://doi.org/10.3390/cancers17050906
APA StyleRoussot, N., Kaderbhai, C., & Ghiringhelli, F. (2025). Targeting Immune Checkpoint Inhibitors for Non-Small-Cell Lung Cancer: Beyond PD-1/PD-L1 Monoclonal Antibodies. Cancers, 17(5), 906. https://doi.org/10.3390/cancers17050906