
Translational Advances in Oncogene and Tumor-Suppressor Gene Research

 , , , ,
, , , ,  , , , , and
, , , , and 
Simple Summary
Abstract
1. Introduction
2. Molecular Mechanisms
2.1. Tumor-Suppressor Gene Inactivation and Oncogene Activation
2.2. Key Signaling Pathways Involved
2.3. Epigenetic Regulation
3. Challenges and Controversies
3.1. Tumor Heterogeneity
3.2. Drug-Resistance Mechanisms
3.3. The “Gray” Area Between Oncogenes and Tumor-Suppressor Genes
4. Clinical Implications
4.1. Diagnostic Approaches
4.2. Therapeutic Strategies
4.3. Personalized Medicine Applications
5. Emerging Technologies and Future Perspectives
5.1. Single-Cell Sequencing
5.2. CRISPR-Cas9 Screening
5.3. Artificial Intelligence and Machine-Learning Applications
5.4. Liquid Biopsy
5.5. Tumor Microenvironment, Cancer Vaccines, and Oncolytic Viruses
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-Binding Cassette |
| ADC | Antibody–Drug Conjugate |
| AFH | Angiomatoid Fibrous Histiocytoma |
| AI | Artificial Intelligence |
| AKT | Protein Kinase B |
| ALL | Acute Lymphoblastic Leukemia |
| ALK | Anaplastic Lymphoma Kinase |
| ALV | Avian Leukosis Virus |
| AML | Acute Myeloid Leukemia |
| APC | Adenomatous Polyposis Coli |
| AS-PCR | Allele-Specific Polymerase Chain Reaction |
| BAD | Bcl-2-Associated Death Promoter |
| BCG | Bacillus Calmette–Guerin |
| BCRP | Breast Cancer Resistance Protein |
| Bcl-2 | B-cell Lymphoma-2 |
| bp | Base Pairs |
| BRCA1 | Breast Cancer Gene 1 |
| CAF | Cancer-Associated Fibroblasts |
| CAD | Computer-Aided Diagnoses |
| CAR | Chimeric Antigen Receptor |
| Cas9 | CRISPR-associated Endonuclease 9 |
| Cas9n | Cas9 Mutant Nickase |
| CCNE | Cyclin E |
| CDK | Cyclin-Dependent Kinase |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| cDNA | Complementary DNA |
| c-MYC | Cellular Myelocytomatosis Oncogene |
| CKI | Cyclin-Dependent Kinase Inhibitors |
| CNN | Convolutional Neural Network |
| CML | Chronic Myelogenous Leukemia |
| CNV | Copy Number Variations |
| CRS | Cytokine Release Syndrome |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CTNNB1 | Catenin-Beta-1 |
| CTCs | Circulating Tumor Cells |
| ctDNA | Circulating Tumor DNA |
| DAMP | Damage-Associated Molecular Patterns |
| DC | Dendritic Cell |
| DEL | Double-Expression Lymphoma |
| DLBCL | Large B-cell Lymphoma |
| DNMT | DNA Methyltransferase |
| DVL | Disheveled Protein |
| ECM | Extracellular Matrix |
| EGF | Epidermal Growth Factor |
| EFGR | Epidermal Growth Factor |
| ELCAP | Early Lung Cancer Action Program |
| EMT | Epithelial to Mesenchymal Transition |
| ERK | Extracellular Signal-Regulated Kinase |
| EWSR1 | EWS RNA-Binding Protein 1 |
| EVs | Extracellular Vesicles |
| FACS | Fluorescence-Activated Cell Sorting |
| FASTK | Fas-Activated Serine/Threonine Kinase |
| FDA | U.S. Food and Drug Administration |
| FFPE | Formalin-Fixed Paraffin Embedded |
| FL | Follicular Lymphoma |
| FOXO | Forkhead Box O |
| GAC | Gastric Adenocarcinoma |
| GADD45 | Growth Arrest and DNA Damage-Inducible 45 |
| GEF | Guanine Nucleotide Exchange Factors |
| GEJ | Gastroesophageal Junction Adenocarcinoma |
| GIST | Gastrointestinal Stromal Tumor |
| GOF | Gain of Function |
| GSK-3β | Glycogen Synthase Kinase-3β |
| H2AW | H2A.W Histone |
| H3K27me3 | Trimethylation of Histone H3 at Lysine 27 |
| HCC | Hepatocellular Carcinoma |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HL | Hodgkin Lymphoma |
| HPV | Human Papillomavirus |
| HR+ | Hormone Receptor Positive (Estrogen and Progesterone) |
| IARC | International Agency for Research on Cancer |
| EEF1D | Eukaryotic Translation Elongation Factor 1 Delta |
| IFNγ | Interferon gamma |
| IGF-1 | Insulin-Like Growth Factor-1 |
| IGFALS | Insulin-Like Growth Factor Binding Protein Acid Labile Subunit |
| IL-2 | Interleukin 2 |
| kbp | Kilobase Pairs |
| LIDC | Lung Image Database Consortium |
| LOF | Loss of Function |
| LOH | Loss of Heterozygosity |
| MAPK | Mitogen-Activated Protein Kinase |
| mbp | Megabase Pairs |
| MCL | Mantle Cell Lymphoma |
| mCRPC | Metastatic Castration-Resistant Prostate Cancer |
| MDM2 | Murine Double Minute 2 |
| MDMX | Murine Double Minute X |
| MEK | Mitogen-Activated Protein Kinase Kinase |
| ML | Machine Learning |
| MMEJ | Microhomology-Mediated End Joining |
| miRNA | MicroRNAs |
| MLH1 | MutL Protein Homolog 1 |
| MM | Multiple Myeloma |
| MMPs | Matrix Metalloproteinases |
| MRP | Multidrug Resistance-Associated Proteins |
| MRD | Minimal Residual Disease |
| mtDNA | Mitochondrial DNA |
| mTORC2 | mTOR Complex 2 |
| NGS | Next-Generation Sequencing |
| NK | Natural Killer |
| NMIBC | Non-Muscle-Invasive Bladder Cancer |
| NSCLC | Non-Small Cell Lung Cancer |
| PARP1 | Poly (ADP-ribose) Polymerase 1 |
| PI3K | Phosphoinositide 3-Kinase |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| PLAT | Tissue Type Plasminogen Activator |
| PDK1 | 3-Phosphoinositide-Dependent Protein Kinase 1 |
| POLQ | DNA Polymerase Theta |
| POLR3K | RNA Polymerase III Subunit K |
| PP1 | Protein Phosphatase |
| PRC2 | Polycomb Repressive Complex 2 |
| PTCL | Peripheral T-Cell Lymphoma |
| PTEN | Phosphate and Tensin Homolog |
| PTPRN | Protein Tyrosine Phosphatase Receptor Type N |
| p21 | CDKN1A |
| pNET | Primitive Neuro-Ectodermal Tumor |
| RCC | Renal Cell Carcinoma |
| RIT | Radioimmunotherapy |
| ROS | Reactive Oxygen Species |
| RSV | Rous Sarcoma Virus |
| RTK | Receptor Tyrosine Kinases |
| sgRNA | Single Guide RNA |
| SSB | Single-Stranded Breaks |
| SNP | Single-Nucleotide Polymorphism |
| SNV | Single Nucleotide Variants |
| TAM | Tumor-Associated Macrophage |
| TAA | Tumor-Associated Antigen |
| TCGA | The Cancer Genome Atlas |
| TGFβ1 | Transforming Growth Factor Beta 1 |
| TNBC | Triple-Negative Breast Cancer |
| TNF | Tumor Necrosis Factor |
| TSA | Tumor-Specific Antigen |
| TS | Targeted Sequencing |
| TME | Tumor Microenvironment |
| T-VEC | Talimogene Laherparepvec |
| VAF | Variant Allele Frequency |
| VCAN | Versican |
| VEGF | Vascular Endothelial Growth Factor |
| WES | Whole Exome Sequencing |
| WGS | Whole Genome Sequencing |
| WHO | World Health Organization |
References
- Roy, P.; Saikia, B. Cancer and Cure: A Critical Analysis. Indian J. Cancer 2016, 53, 441. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Cancer Factsheets. Available online: https://gco.iarc.fr/today/en/fact-sheets-cancers (accessed on 5 January 2025).
- Gilbertson, R.J. Mapping Cancer Origins. Cell 2011, 145, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA Mutations in Human Cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Kim, M.; Mahmood, M.; Reznik, E.; Gammage, P.A. Mitochondrial DNA Is a Major Source of Driver Mutations in Cancer. Trends Cancer 2022, 8, 1046–1059. [Google Scholar] [CrossRef]
- Porta-Pardo, E.; Valencia, A.; Godzik, A. Understanding Oncogenicity of Cancer Driver Genes and Mutations in the Cancer Genomics Era. FEBS Lett. 2020, 594, 4233–4246. [Google Scholar] [CrossRef]
- Lechner, J.F.; Tesfaigzi, J.; Gerwin, B.I. Oncogenes and Tumor-Suppressor Genes in Mesothelioma—A Synopsis. Environ. Health Perspect. 1997, 105, 1061–1067. [Google Scholar] [CrossRef]
- Dakal, T.C.; Dhabhai, B.; Pant, A.; Moar, K.; Chaudhary, K.; Yadav, V.; Ranga, V.; Sharma, N.K.; Kumar, A.; Maurya, P.K.; et al. Oncogenes and Tumor Suppressor Genes: Functions and Roles in Cancers. MedComm 2024, 5, e582. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of Heterozygosity of Essential Genes Represents a Widespread Class of Potential Cancer Vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef]
- Fluiter, K.; Housman, D.; ten Asbroek, A.L.M.A.; Baas, F. Killing Cancer by Targeting Genes That Cancer Cells Have Lost: Allele-Specific Inhibition, a Novel Approach to the Treatment of Genetic Disorders. Cell. Mol. Life Sci. 2003, 60, 834–843. [Google Scholar] [CrossRef]
- Torry, D.S.; Cooper, G.M. Proto-Oncogenes in Development and Cancer. Am. J. Reprod. Immunol. 1991, 25, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Hijiya, N.; Gewirtz, A.M. Oncogenes, Protooncogenes, and Tumor Suppressor Genes in Acute Myelogenous Leukemia. J. Pediatr. Hematol. Oncol. 1995, 17, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kopelovich, L.; Shea-Herbert, B. Heritable One-Hit Events Defining Cancer Prevention? Cell Cycle 2013, 12, 2553–2557. [Google Scholar] [CrossRef]
- Park, S.; Supek, F.; Lehner, B. Higher Order Genetic Interactions Switch Cancer Genes from Two-Hit to One-Hit Drivers. Nat. Commun. 2021, 12, 7051. [Google Scholar] [CrossRef] [PubMed]
- Simatou, A.; Simatos, G.; Goulielmaki, M.; Spandidos, D.A.; Baliou, S.; Zoumpourlis, V. Historical Retrospective of the SRC Oncogene and New Perspectives (Review). Mol. Clin. Oncol. 2020, 13, 21. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-Suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef]
- Roy, S.; Bondada, M.S.; Zhang, Y.; Moffat, K.; Nair, V.; Yao, Y. Proviral ALV-LTR Sequence Is Essential for Continued Proliferation of the ALV-Transformed B Cell Line. Int. J. Mol. Sci. 2022, 23, 11263. [Google Scholar] [CrossRef]
- Morsberger, L.; Pallavajjala, A.; Long, P.; Hardy, M.; Park, R.; Parish, R.; Nozari, A.; Zou, Y.S. HER2 Amplification by Next-Generation Sequencing to Identify HER2-Positive Invasive Breast Cancer with Negative HER2 Immunohistochemistry. Cancer Cell Int. 2022, 22, 350. [Google Scholar] [CrossRef]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef]
- Gariglio, P. (Ed.) Oncogenes and Tumor Suppressor Genes. In Molecular Oncology: Principles and Recent Advances; Bentham Science Publishers: Sharjah, United Arab Emirates, 2012; pp. 64–82. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Quinlan, M.P.; Settleman, J. Isoform-Specific Ras Functions in Development and Cancer. Future Oncol. 2009, 5, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Hutter, S.; Bolin, S.; Weishaupt, H.; Swartling, F. Modeling and Targeting MYC Genes in Childhood Brain Tumors. Genes 2017, 8, 107. [Google Scholar] [CrossRef]
- Ong, J.Y.; Torres, J.Z. Dissecting the Mechanisms of Cell Division. J. Biol. Chem. 2019, 294, 11382–11390. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Friedman, P.N.; Marshak, D.R.; Prives, C.; Beach, D. Human P53 Is Phosphorylated by P60-Cdc2 and Cyclin B-Cdc2. Proc. Natl. Acad. Sci. USA 1990, 87, 4766–4770. [Google Scholar] [CrossRef]
- Canavese, M.; Santo, L.; Raje, N. Cyclin Dependent Kinases in Cancer. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development-Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic Analysis of CDK and Cyclin Proteins in Premetazoan Lineages. BMC Evol. Biol. 2014, 14, 10. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Dong, P.; Gassler, N.; Taheri, M.; Baniahmad, A.; Dilmaghani, N.A. A Review on the Role of Cyclin Dependent Kinases in Cancers. Cancer Cell Int. 2022, 22, 325. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The History and Future of Targeting Cyclin-Dependent Kinases in Cancer Therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To Cycle or Not to Cycle: A Critical Decision in Cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Topacio, B.R.; Zatulovskiy, E.; Cristea, S.; Xie, S.; Tambo, C.S.; Rubin, S.M.; Sage, J.; Kõivomägi, M.; Skotheim, J.M. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol. Cell 2019, 74, 758–770.e4. [Google Scholar] [CrossRef] [PubMed]
- Pavletich, N.P. Mechanisms of Cyclin-Dependent Kinase Regulation: Structures of Cdks, Their Cyclin Activators, and Cip and INK4 Inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, R.; Teixeira, L.K. Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front. Cell Dev. Biol. 2021, 9, 774845. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Adams, P.D. Regulation of the Retinoblastoma Tumor Suppressor Protein by Cyclin/Cdks. Biochim. Biophys. Acta-Rev. Cancer 2001, 1471, M123–M133. [Google Scholar] [CrossRef]
- Rubin, S.M.; Sage, J.; Skotheim, J.M. Integrating Old and New Paradigms of G1/S Control. Mol. Cell 2020, 80, 183–192. [Google Scholar] [CrossRef]
- Tamrakar, S. Role of PRB Dephosphorylation in Cell Cycle Regulation. Front. Biosci. 2000, 5, d121. [Google Scholar] [CrossRef]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in Cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef]
- Zhang, M.; Kim, S.; Yang, H.W. Non-Canonical Pathway for Rb Inactivation and External Signaling Coordinate Cell-Cycle Entry without CDK4/6 Activity. Nat. Commun. 2023, 14, 7847. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor Suppressor P53: Biology, Signaling Pathways, and Therapeutic Targeting. Biochim. Biophys. Acta-Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting P53 Pathways: Mechanisms, Structures and Advances in Therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Stevaux, O.; Dyson, N.J. A Revised Picture of the E2F Transcriptional Network and RB Function. Curr. Opin. Cell Biol. 2002, 14, 684–691. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling Mechanisms of P53-Mediated Tumour Suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Hou, H.; Sun, D.; Zhang, X. The Role of MDM2 Amplification and Overexpression in Therapeutic Resistance of Malignant Tumors. Cancer Cell Int. 2019, 19, 216. [Google Scholar] [CrossRef]
- Ma, M.; Hua, S.; Min, X.; Wang, L.; Li, J.; Wu, P.; Liang, H.; Zhang, B.; Chen, X.; Xiang, S. P53 Positively Regulates the Proliferation of Hepatic Progenitor Cells Promoted by Laminin-521. Signal Transduct. Target. Ther. 2022, 7, 290. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. P53 Has a Direct Apoptogenic Role at the Mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Vaddavalli, P.L.; Schumacher, B. The P53 Network: Cellular and Systemic DNA Damage Responses in Cancer and Aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Sinha, A.; Zou, Y.; Patel, A.S.; Yoo, S.; Jiang, F.; Sato, T.; Kong, R.; Watanabe, H.; Zhu, J.; Massion, P.P.; et al. Early-Stage Lung Adenocarcinoma MDM2 Genomic Amplification Predicts Clinical Outcome and Response to Targeted Therapy. Cancers 2022, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Monleon, A.; Kryh Öberg, H.; Gaarder, J.; Berbegall, A.P.; Javanmardi, N.; Djos, A.; Ussowicz, M.; Taschner-Mandl, S.; Ambros, I.M.; Øra, I.; et al. Amplification of CDK4 and MDM2: A Detailed Study of a High-Risk Neuroblastoma Subgroup. Sci. Rep. 2022, 12, 12420. [Google Scholar] [CrossRef]
- Padilla-Nash, H.M.; McNeil, N.E.; Yi, M.; Nguyen, Q.-T.; Hu, Y.; Wangsa, D.; Mack, D.L.; Hummon, A.B.; Case, C.; Cardin, E.; et al. Aneuploidy, Oncogene Amplification and Epithelial to Mesenchymal Transition Define Spontaneous Transformation of Murine Epithelial Cells. Carcinogenesis 2013, 34, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. Signaling Pathways of the Insulin-like Growth Factor Binding Proteins. Endocr. Rev. 2023, 44, 753–778. [Google Scholar] [CrossRef]
- Bianco, R.; Caputo, R.; Caputo, R.; Damiano, V.; De Placido, S.; Ficorella, C.; Agrawal, S.; Bianco, A.R.; Ciardiello, F.; Tortora, G. Combined Targeting of Epidermal Growth Factor Receptor and MDM2 by Gefitinib and Antisense MDM2 Cooperatively Inhibit Hormone-Independent Prostate Cancer. Clin. Cancer Res. 2004, 10, 4858–4864. [Google Scholar] [CrossRef]
- Laroche, A.; Chaire, V.; Algeo, M.-P.; Karanian, M.; Fourneaux, B.; Italiano, A. MDM2 Antagonists Synergize with PI3K/MTOR Inhibition in Well-Differentiated/Dedifferentiated Liposarcomas. Oncotarget 2017, 8, 53968–53977. [Google Scholar] [CrossRef] [PubMed]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 Phosphorylation by Akt Regulates the P53 Response to Oxidative Stress to Promote Cell Proliferation and Tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. Ubiquitin-Independent P53 Proteasomal Degradation. Cell Death Differ. 2010, 17, 103–108. [Google Scholar] [CrossRef]
- Lossaint, G.; Horvat, A.; Gire, V.; Bačević, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulić, V. Reciprocal Regulation of P21 and Chk1 Controls the Cyclin D1-RB Pathway to Mediate Senescence Onset after G2 Arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef]
- Seong, H.-A.; Ha, H. Thr55 Phosphorylation of P21 by MPK38/MELK Ameliorates Defects in Glucose, Lipid, and Energy Metabolism in Diet-Induced Obese Mice. Cell Death Dis. 2019, 10, 380. [Google Scholar] [CrossRef]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral Role of Noxa in P53-Mediated Apoptotic Response. Genes Dev. 2003, 17, 2233–2238. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by P53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thomas, H.R.; Li, Z.; Yeo, N.C.; Scott, H.E.; Dang, N.; Hossain, M.I.; Andrabi, S.A.; Parant, J.M. Puma, Noxa, P53, and P63 Differentially Mediate Stress Pathway Induced Apoptosis. Cell Death Dis. 2021, 12, 659. [Google Scholar] [CrossRef]
- Basu, A. The Relationship between BcI2, Bax and P53: Consequences for Cell Cycle Progression and Cell Death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Zhu, C.; Wang, K. A Review of Current Evidence about LncRNA MEG3: A Tumor Suppressor in Multiple Cancers. Front. Cell Dev. Biol. 2022, 10, 997633. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Nandan, M.O.; Yang, V.W. An Update on the Biology of RAS/RAF Mutations in Colorectal Cancer. Curr. Color. Cancer Rep. 2011, 7, 113–120. [Google Scholar] [CrossRef]
- Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Clark, G.J.; Der, C.J. Increasing Complexity of Ras Signaling. Oncogene 1998, 17, 1395–1413. [Google Scholar] [CrossRef]
- Osaka, N.; Hirota, Y.; Ito, D.; Ikeda, Y.; Kamata, R.; Fujii, Y.; Chirasani, V.R.; Campbell, S.L.; Takeuchi, K.; Senda, T.; et al. Divergent Mechanisms Activating RAS and Small GTPases Through Post-Translational Modification. Front. Mol. Biosci. 2021, 8, 707439. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK Pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Srdanović, S.; Wolter, M.; Trinh, C.H.; Ottmann, C.; Warriner, S.L.; Wilson, A.J. Understanding the Interaction of 14-3-3 Proteins with h DMX and h DM2: A Structural and Biophysical Study. FEBS J. 2022, 289, 5341–5358. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, G.-D.; Shi, Z.; Qi, L.-L.; Zhou, L.-Y.; Fu, Z.-X. The Ras/Raf/MEK/ERK Signaling Pathway and Its Role in the Occurrence and Development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radzlejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A Family of Protein-Serine/Threonine Kinases That Are Activated and Tyrosine Phosphorylated in Response to Insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted Therapies: Is the Undruggable Drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Wardell, C.; Poznański, P.; Pierzchała, M.; Taniguchi, H. Oncogenic Mutation BRAF V600E Changes Phenotypic Behavior of THLE-2 Liver Cells through Alteration of Gene Expression. Int. J. Mol. Sci. 2022, 23, 1548. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The Role of Microenvironment in Tumor Angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The Role of VEGF in Cancer-Induced Angiogenesis and Research Progress of Drugs Targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Habrowska-Górczyńska, D.E.; Kozieł, M.J.; Urbanek, K.A.; Kowalska, K.; Piastowska-Ciesielska, A.W. FOXO3a/PI3K/Akt Pathway Participates in the ROS- Induced Apoptosis Triggered by α-ZEL and β-ZEL. Sci. Rep. 2024, 14, 13281. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/MTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted Role of MTOR (Mammalian Target of Rapamycin) Signaling Pathway in Human Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. MTOR Signaling Pathway and MTOR Inhibitors in Cancer: Progress and Challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Yang, L.; Miao, L.; Liang, F.; Huang, H.; Teng, X.; Li, S.; Nuriddinov, J.; Selzer, M.E.; Hu, Y. The MTORC1 Effectors S6K1 and 4E-BP Play Different Roles in CNS Axon Regeneration. Nat. Commun. 2014, 5, 5416. [Google Scholar] [CrossRef]
- Mukherjee, R.; Vanaja, K.G.; Boyer, J.A.; Gadal, S.; Solomon, H.; Chandarlapaty, S.; Levchenko, A.; Rosen, N. Regulation of PTEN Translation by PI3K Signaling Maintains Pathway Homeostasis. Mol. Cell 2021, 81, 708–723.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, T.; Li, B.; Rong, Y.; Verkhratsky, A.; Peng, L. Crosstalk Between MAPK/ERK and PI3K/AKT Signal Pathways During Brain Ischemia/Reperfusion. ASN Neuro 2015, 7, 1759091415602463. [Google Scholar] [CrossRef]
- Dai, J.; Bercury, K.K.; Macklin, W.B. Interaction of MTOR and Erk1/2 Signaling to Regulate Oligodendrocyte Differentiation. Glia 2014, 62, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.A.; Khan, M.; Monica, V.; Kar, D. The Molecular Basis of Wnt/β-Catenin Signaling Pathways in Neurodegenerative Diseases. Int. J. Cell Biol. 2023, 2023, 9296092. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 Receptors for Wnt/β-Catenin Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt Signaling in the Nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Schlosshauer, P.W.; Pirog, E.C.; Levine, R.L.; Ellenson, L.H. Mutational Analysis of the CTNNB1 and APC Genes in Uterine Endometrioid Carcinoma. Mod. Pathol. 2000, 13, 1066–1071. [Google Scholar] [CrossRef]
- Sehgal, P.; Lanauze, C.; Wang, X.; Hayer, K.E.; Torres-Diz, M.; Leu, N.A.; Sela, Y.; Stanger, B.Z.; Lengner, C.J.; Thomas-Tikhonenko, A. MYC Hyperactivates Wnt Signaling in APC/CTNNB1-Mutated Colorectal Cancer Cells through MiR-92a–Dependent Repression of DKK3. Mol. Cancer Res. 2021, 19, 2003–2014. [Google Scholar] [CrossRef]
- Xue, W.; Yang, L.; Chen, C.; Ashrafizadeh, M.; Tian, Y.; Sun, R. Wnt/β-Catenin-Driven EMT Regulation in Human Cancers. Cell. Mol. Life Sci. 2024, 81, 79. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, L.; Hu, C.; Liang, S.; Fei, X.; Yan, N.; Zhang, Y.; Zhang, F. WNT Pathway Inhibitor Pyrvinium Pamoate Inhibits the Self-Renewal and Metastasis of Breast Cancer Stem Cells. Int. J. Oncol. 2016, 48, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Daisy Precilla, S.; Biswas, I.; Kuduvalli, S.S.; Anitha, T.S. Crosstalk between PI3K/AKT/MTOR and WNT/β-Catenin Signaling in GBM—Could Combination Therapy Checkmate the Collusion? Cell. Signal. 2022, 95, 110350. [Google Scholar] [CrossRef]
- Fleming-de-Moraes, C.D.; Rocha, M.R.; Tessmann, J.W.; de Araujo, W.M.; Morgado-Diaz, J.A. Crosstalk between PI3K/Akt and Wnt/β-Catenin Pathways Promote Colorectal Cancer Progression Regardless of Mutational Status. Cancer Biol. Ther. 2022, 23, 1–13. [Google Scholar] [CrossRef]
- Langhammer, T.-S.; Roolf, C.; Krohn, S.; Kretzschmar, C.; Huebner, R.; Rolfs, A.; Freund, M.; Junghanss, C. PI3K/Akt Signaling Interacts with Wnt/β-Catenin Signaling But Does Not Induce an Accumulation of β-Catenin in the Nucleus of Acute Lymphoblastic Leukemia Cell Lines. Blood 2013, 122, 4886. [Google Scholar] [CrossRef]
- Koujah, L.; Madavaraju, K.; Agelidis, A.M.; Patil, C.D.; Shukla, D. Heparanase-Induced Activation of AKT Stabilizes β-Catenin and Modulates Wnt/β-Catenin Signaling during Herpes Simplex Virus 1 Infection. MBio 2021, 12, e02792-21. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of β-Catenin by AKT Promotes β-Catenin Transcriptional Activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Ro, E.J.; Choi, K.-Y. Interaction between Wnt/β-Catenin and RAS-ERK Pathways and an Anti-Cancer Strategy via Degradations of β-Catenin and RAS by Targeting the Wnt/β-Catenin Pathway. npj Precis. Oncol. 2018, 2, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Pizzute, T.; Pei, M. A Review of Crosstalk between MAPK and Wnt Signals and Its Impact on Cartilage Regeneration. Cell Tissue Res. 2014, 358, 633–649. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Fundamental Role of Epigenetic Events in Cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Sonar, S.; Nyahatkar, S.; Kalele, K.; Adhikari, M.D. Role of DNA Methylation in Cancer Development and Its Clinical Applications. Clin. Transl. Discov. 2024, 4, e279. [Google Scholar] [CrossRef]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. In DNA Methyltransferases-Role and Function; Springer: Berlin/Heidelberg, Germany, 2016; pp. 151–172. [Google Scholar] [CrossRef]
- Stojchevski, R.; Velichkovikj, S.; Arsov, T. Genetic and Epigenetic Basis of Obesity-Induced Inflammation and Diabetes. In Obesity, Diabetes and Inflammation: Molecular Mechanisms and Clinical Management; Springer: Cham, Switzerland, 2023; pp. 129–146. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA Methylation Profiles in Cancer Diagnosis and Therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA Hypermethylation in Disease: Mechanisms and Clinical Relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef]
- Niv, Y. Microsatellite Instability and MLH1 Promoter Hypermethylation in Colorectal Cancer. World J. Gastroenterol. 2007, 13, 1867. [Google Scholar] [CrossRef]
- Oubaddou, Y.; Oukabli, M.; Fenniche, S.; Elktaibi, A.; Elochi, M.R.; Al Bouzidi, A.; Qmichou, Z.; Dakka, N.; Diorio, C.; Richter, A.; et al. BRCA1 Promoter Hypermethylation in Malignant Breast Tumors and in the Histologically Normal Adjacent Tissues to the Tumors: Exploring Its Potential as a Biomarker and Its Clinical Significance in a Translational Approach. Genes 2023, 14, 1680. [Google Scholar] [CrossRef]
- Tramontano, A.; Boffo, F.L.; Russo, G.; De Rosa, M.; Iodice, I.; Pezone, A. Methylation of the Suppressor Gene P16INK4a: Mechanism and Consequences. Biomolecules 2020, 10, 446. [Google Scholar] [CrossRef]
- Besselink, N.; Keijer, J.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E. The Genome-Wide Mutational Consequences of DNA Hypomethylation. Sci. Rep. 2023, 13, 6874. [Google Scholar] [CrossRef]
- Choi, J.-Y.; James, S.R.; Link, P.A.; McCann, S.E.; Hong, C.-C.; Davis, W.; Nesline, M.K.; Ambrosone, C.B.; Karpf, A.R. Association between Global DNA Hypomethylation in Leukocytes and Risk of Breast Cancer. Carcinogenesis 2009, 30, 1889–1897. [Google Scholar] [CrossRef]
- Tripathi, K.; Goel, A.; Singhai, A.; Garg, M. Promoter Hypomethylation as Potential Confounder of Ras Gene Overexpression and Their Clinical Significance in Subsets of Urothelial Carcinoma of Bladder. Mol. Biol. Rep. 2021, 48, 2183–2199. [Google Scholar] [CrossRef]
- Ehrlich, M. Dna Hypomethylation In Cancer Cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.S.; Loriot, A.; Diacofotaki, A.; Van Tongelen, A.; De Smet, C. Transcriptional Overlap Links DNA Hypomethylation with DNA Hypermethylation at Adjacent Promoters in Cancer. Sci. Rep. 2021, 11, 17346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic Modifications of Histones in Cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell. Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; von Bülow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone Modifications and Their Role in Epigenetics of Atopy and Allergic Diseases. Allergy Asthma Clin. Immunol. 2018, 14, 39. [Google Scholar] [CrossRef]
- Arif, K.M.T.; Elliott, E.K.; Haupt, L.M.; Griffiths, L.R. Regulatory Mechanisms of Epigenetic MiRNA Relationships in Human Cancer and Potential as Therapeutic Targets. Cancers 2020, 12, 2922. [Google Scholar] [CrossRef]
- Reynolds, P.A.; Sigaroudinia, M.; Zardo, G.; Wilson, M.B.; Benton, G.M.; Miller, C.J.; Hong, C.; Fridlyand, J.; Costello, J.F.; Tlsty, T.D. Tumor Suppressor P16INK4A Regulates Polycomb-Mediated DNA Hypermethylation in Human Mammary Epithelial Cells. J. Biol. Chem. 2006, 281, 24790–24802. [Google Scholar] [CrossRef]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef] [PubMed]
- Albero, R.; Enjuanes, A.; Demajo, S.; Castellano, G.; Pinyol, M.; García, N.; Capdevila, C.; Clot, G.; Suárez-Cisneros, H.; Shimada, M.; et al. Cyclin D1 Overexpression Induces Global Transcriptional Downregulation in Lymphoid Neoplasms. J. Clin. Investig. 2018, 128, 4132–4147. [Google Scholar] [CrossRef] [PubMed]
- Braile, M.; Luciano, N.; Carlomagno, D.; Salvatore, G.; Orlandella, F.M. Insight into the Role of the MiR-584 Family in Human Cancers. Int. J. Mol. Sci. 2024, 25, 7448. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Torres, A.; Torres, K.; Pesci, A.; Ceccaroni, M.; Paszkowski, T.; Cassandrini, P.; Zamboni, G.; Maciejewski, R. Deregulation of MiR-100, MiR-99a and MiR-199b in Tissues and Plasma Coexists with Increased Expression of MTOR Kinase in Endometrioid Endometrial Carcinoma. BMC Cancer 2012, 12, 369. [Google Scholar] [CrossRef]
- Fan, D.; Ren, B.; Yang, X.; Liu, J.; Zhang, Z. Upregulation of MiR-501-5p Activates the Wnt/β-Catenin Signaling Pathway and Enhances Stem Cell-like Phenotype in Gastric Cancer. J. Exp. Clin. Cancer Res. 2016, 35, 177. [Google Scholar] [CrossRef]
- Zhang, C.; Kang, C.; Wang, P.; Cao, Y.; Lv, Z.; Yu, S.; Wang, G.; Zhang, A.; Jia, Z.; Han, L.; et al. MicroRNA-221 and -222 Regulate Radiation Sensitivity by Targeting the PTEN Pathway. Int. J. Radiat. Oncol. 2011, 80, 240–248. [Google Scholar] [CrossRef]
- Ma, Y.; Shen, N.; Wicha, M.S.; Luo, M. The Roles of the Let-7 Family of MicroRNAs in the Regulation of Cancer Stemness. Cells 2021, 10, 2415. [Google Scholar] [CrossRef]
- Zhu, L.; Jiang, M.; Wang, H.; Sun, H.; Zhu, J.; Zhao, W.; Fang, Q.; Yu, J.; Chen, P.; Wu, S.; et al. A Narrative Review of Tumor Heterogeneity and Challenges to Tumor Drug Therapy. Ann. Transl. Med. 2021, 9, 1351. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Liu, J.; Dang, H.; Wang, X.W. The Significance of Intertumor and Intratumor Heterogeneity in Liver Cancer. Exp. Mol. Med. 2018, 50, e416. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.-L.; et al. The Patterns and Dynamics of Genomic Instability in Metastatic Pancreatic Cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-D.; Sun, Z.; Zhu, Z.-B.; Liu, X.; Chen, J.-Z.; Hao, L.; Zhu, J.-F.; Pang, K.; Wu, D.; Dong, Y.; et al. Integrated Single-Cell and Spatial Transcriptomic Profiling Reveals Higher Intratumour Heterogeneity and Epithelial-Fibroblast Interactions in Recurrent Bladder Cancer. Clin. Transl. Med. 2023, 13, e1338. [Google Scholar] [CrossRef]
- Morrissy, A.S.; Cavalli, F.M.G.; Remke, M.; Ramaswamy, V.; Shih, D.J.H.; Holgado, B.L.; Farooq, H.; Donovan, L.K.; Garzia, L.; Agnihotri, S.; et al. Spatial Heterogeneity in Medulloblastoma. Nat. Genet. 2017, 49, 780–788. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-Cancer Analysis of the Extent and Consequences of Intratumor Heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Fu, F.; Nowak, M.A.; Bonhoeffer, S. Spatial Heterogeneity in Drug Concentrations Can Facilitate the Emergence of Resistance to Cancer Therapy. PLoS Comput. Biol. 2015, 11, e1004142. [Google Scholar] [CrossRef]
- Zhou, S.; Zheng, J.; Zhai, W.; Chen, Y. Spatio-Temporal Heterogeneity in Cancer Evolution and Tumor Microenvironment of Renal Cell Carcinoma with Tumor Thrombus. Cancer Lett. 2023, 572, 216350. [Google Scholar] [CrossRef]
- de Oliveira, D.; Dall’Oglio, M.F.; Reis, S.T.; Zerati, M.; Souza, I.C.; Leite, K.R.; Srougi, M. Chromosome 9p Deletions Are an Independent Predictor of Tumor Progression Following Nephrectomy in Patients with Localized Clear Cell Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 601–606. [Google Scholar] [CrossRef]
- Narimatsu, T.; Matsuura, K.; Nakada, C.; Tsukamoto, Y.; Hijiya, N.; Kai, T.; Inoue, T.; Uchida, T.; Nomura, T.; Sato, F.; et al. Downregulation of NDUFB6 Due to 9p24.1-p13.3 Loss Is Implicated in Metastatic Clear Cell Renal Cell Carcinoma. Cancer Med. 2015, 4, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, B.D.; Leslie, E.M. Efflux Transporters in Anti-Cancer Drug Resistance: Molecular and Functional Identification and Characterization of Multidrug Resistance Proteins (MRPs/ABCCs). In Drug Efflux Pumps in Cancer Resistance Pathways: From Molecular Recognition and Characterization to Possible Inhibition Strategies in Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 31–65. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Ashique, S.; Bhowmick, M.; Pal, R.; Khatoon, H.; Kumar, P.; Sharma, H.; Garg, A.; Kumar, S.; Das, U. Multi Drug Resistance in Colorectal Cancer-Approaches to Overcome, Advancements and Future Success. Adv. Cancer Biol.-Metastasis 2024, 10, 100114. [Google Scholar] [CrossRef]
- Wallbillich, N.J.; Lu, H. Role of C-Myc in Lung Cancer: Progress, Challenges, and Prospects. Chin. Med. J. Pulm. Crit. Care Med. 2023, 1, 129–138. [Google Scholar] [CrossRef]
- Al-Abdulla, R.; Perez-Silva, L.; Lozano, E.; Macias, R.I.R.; Herraez, E.; Abad, M.; Segues, N.; Bujanda, L.; Briz, O.; Marin, J.J.G. Sensitizing Gastric Adenocarcinoma to Chemotherapy by Pharmacological Manipulation of Drug Transporters. Biochem. Pharmacol. 2020, 171, 113682. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the Breast Cancer Resistance Protein (BCRP/ABCG2) in Drug Transport—An Update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Uceda-Castro, R.; Margarido, A.S.; Song, J.-Y.; de Gooijer, M.C.; Messal, H.A.; Chambers, C.R.; Nobis, M.; Çitirikkaya, C.H.; Hahn, K.; Seinstra, D.; et al. BCRP Drives Intrinsic Chemoresistance in Chemotherapy-Naïve Breast Cancer Brain Metastasis. Sci. Adv. 2023, 9, eabp9530. [Google Scholar] [CrossRef]
- Belan, O.; Sebald, M.; Adamowicz, M.; Anand, R.; Vancevska, A.; Neves, J.; Grinkevich, V.; Hewitt, G.; Segura-Bayona, S.; Bellelli, R.; et al. POLQ Seals Post-Replicative SsDNA Gaps to Maintain Genome Stability in BRCA-Deficient Cancer Cells. Mol. Cell 2022, 82, 4664–4680.e9. [Google Scholar] [CrossRef]
- Lemée, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.-J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.-L.; Leduc, C.; et al. DNA Polymerase θ up-Regulation Is Associated with Poor Survival in Breast Cancer, Perturbs DNA Replication, and Promotes Genetic Instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef]
- Higgins, G.S.; Harris, A.L.; Prevo, R.; Helleday, T.; McKenna, W.G.; Buffa, F.M. Overexpression of POLQ Confers a Poor Prognosis in Early Breast Cancer Patients. Oncotarget 2010, 1, 175–184. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Rojo, F.; García-Parra, J.; Zazo, S.; Tusquets, I.; Ferrer-Lozano, J.; Menendez, S.; Eroles, P.; Chamizo, C.; Servitja, S.; Ramírez-Merino, N.; et al. Nuclear PARP-1 Protein Overexpression Is Associated with Poor Overall Survival in Early Breast Cancer. Ann. Oncol. 2012, 23, 1156–1164. [Google Scholar] [CrossRef]
- Gilabert, M.; Launay, S.; Ginestier, C.; Bertucci, F.; Audebert, S.; Pophillat, M.; Toiron, Y.; Baudelet, E.; Finetti, P.; Noguchi, T.; et al. Poly(ADP-Ribose) Polymerase 1 (PARP1) Overexpression in Human Breast Cancer Stem Cells and Resistance to Olaparib. PLoS ONE 2014, 9, e104302. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, A.; Partipilo, G.; De Summa, S.; Giotta, F.; Simone, G.; Mangia, A. Nuclear PARP1 Expression and Its Prognostic Significance in Breast Cancer Patients. Tumor Biol. 2016, 37, 6143–6153. [Google Scholar] [CrossRef]
- Wang, F.; Gouttia, O.G.; Wang, L.; Peng, A. PARP1 Upregulation in Recurrent Oral Cancer and Treatment Resistance. Front. Cell Dev. Biol. 2022, 9, 804962. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Oh, G.; Wang, A.; Wang, L.; Li, J.; Werba, G.; Weissinger, D.; Zhao, E.; Dhara, S.; Hernandez, R.E.; Ackermann, A.; et al. POLQ Inhibition Elicits an Immune Response in Homologous Recombination–Deficient Pancreatic Adenocarcinoma via CGAS/STING Signaling. J. Clin. Investig. 2023, 133, e165934. [Google Scholar] [CrossRef]
- Liu, P.; Hao, L.; Liu, M.; Hu, S. Glutathione-Responsive and -Exhausting Metal Nanomedicines for Robust Synergistic Cancer Therapy. Front. Bioeng. Biotechnol. 2023, 11, 1161472. [Google Scholar] [CrossRef]
- Niu, B.; Liao, K.; Zhou, Y.; Wen, T.; Quan, G.; Pan, X.; Wu, C. Application of Glutathione Depletion in Cancer Therapy: Enhanced ROS-Based Therapy, Ferroptosis, and Chemotherapy. Biomaterials 2021, 277, 121110. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, S. Modulating Intracellular Oxidative Stress via Engineered Nanotherapeutics. J. Control. Release 2020, 319, 333–343. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras Copy Number Defines Metabolic Reprogramming and Therapeutic Susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a Novel Pharmacological Mechanism of Anti-Cancer Drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Talty, R.; Jain, A.; Cai, Y.; Zheng, J.; Shen, X.; Muca, E.; Paty, P.B.; Bosenberg, M.W.; Khan, S.A.; et al. Discovery of Decreased Ferroptosis in Male Colorectal Cancer Patients with KRAS Mutations. Redox Biol. 2023, 62, 102699. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef]
- Datta, N.; Chakraborty, S.; Basu, M.; Ghosh, M.K. Tumor Suppressors Having Oncogenic Functions: The Double Agents. Cells 2020, 10, 46. [Google Scholar] [CrossRef]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant P53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The Gut Microbiome Switches Mutant P53 from Tumour-Suppressive to Oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the Control of Apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of Function Between Tumor Prevention and Carcinogenesis. JNCI J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Qin, D. Next-Generation Sequencing and Its Clinical Application. Cancer Biol. Med. 2019, 16, 4–10. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-Generation Sequencing Technologies: An Overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of Applications of High-Throughput Sequencing in Personalized Medicine: Barriers and Facilitators of Future Progress in Research and Clinical Application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Beedanagari, S.; John, K. Next Generation Sequencing. In Encyclopedia of Toxicology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 501–503. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Choo, Z.-N.; Behr, J.M.; Deshpande, A.; Hadi, K.; Yao, X.; Tian, H.; Takai, K.; Zakusilo, G.; Rosiene, J.; Da Cruz Paula, A.; et al. Most Large Structural Variants in Cancer Genomes Can Be Detected without Long Reads. Nat. Genet. 2023, 55, 2139–2148. [Google Scholar] [CrossRef]
- Bagger, F.O.; Borgwardt, L.; Jespersen, A.S.; Hansen, A.R.; Bertelsen, B.; Kodama, M.; Nielsen, F.C. Whole Genome Sequencing in Clinical Practice. BMC Med. Genom. 2024, 17, 39. [Google Scholar] [CrossRef]
- Souche, E.; Beltran, S.; Brosens, E.; Belmont, J.W.; Fossum, M.; Riess, O.; Gilissen, C.; Ardeshirdavani, A.; Houge, G.; van Gijn, M.; et al. Recommendations for Whole Genome Sequencing in Diagnostics for Rare Diseases. Eur. J. Hum. Genet. 2022, 30, 1017–1021. [Google Scholar] [CrossRef]
- The Lancet Oncology. Incorporating Whole-Genome Sequencing into Cancer Care. Lancet Oncol. 2024, 25, 945. [Google Scholar] [CrossRef] [PubMed]
- Kinnersley, B.; Sud, A.; Everall, A.; Cornish, A.J.; Chubb, D.; Culliford, R.; Gruber, A.J.; Lärkeryd, A.; Mitsopoulos, C.; Wedge, D.; et al. Analysis of 10,478 Cancer Genomes Identifies Candidate Driver Genes and Opportunities for Precision Oncology. Nat. Genet. 2024, 56, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.; Anyane-Yeboa, K.; Wynn, J.; Wilson, A.; Truitt Cho, M.; Guzman, E.; Sisson, R.; Egan, C.; Chung, W.K. The Usefulness of Whole-Exome Sequencing in Routine Clinical Practice. Genet. Med. 2014, 16, 922–931. [Google Scholar] [CrossRef]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The Promise of Whole-Exome Sequencing in Medical Genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Modai, S.; Shomron, N. Molecular Risk Factors for Schizophrenia. Trends Mol. Med. 2016, 22, 242–253. [Google Scholar] [CrossRef]
- Ganatra, H.; Tan, J.K.; Simmons, A.; Bigogno, C.M.; Khurana, V.; Ghose, A.; Ghosh, A.; Mahajan, I.; Boussios, S.; Maniam, A.; et al. Applying Whole-Genome and Whole-Exome Sequencing in Breast Cancer: A Review of the Landscape. Breast Cancer 2024, 31, 999–1009. [Google Scholar] [CrossRef]
- Hamdi, Y.; Boujemaa, M.; Ben Rekaya, M.; Ben Hamda, C.; Mighri, N.; El Benna, H.; Mejri, N.; Labidi, S.; Daoud, N.; Naouali, C.; et al. Family Specific Genetic Predisposition to Breast Cancer: Results from Tunisian Whole Exome Sequenced Breast Cancer Cases. J. Transl. Med. 2018, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Arjun Kumar, E.; Palladino, G.; Korfi, K.; Wang, J. Applications and Analysis of Targeted Genomic Sequencing in Cancer Studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Tawana, K.; Wang, J.; Renneville, A.; Bödör, C.; Hills, R.; Loveday, C.; Savic, A.; Van Delft, F.W.; Treleaven, J.; Georgiades, P.; et al. Disease Evolution and Outcomes in Familial AML with Germline CEBPA Mutations. Blood 2015, 126, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-T.; Choi, Y.-L.; Yun, J.W.; Kim, N.K.D.; Kim, S.-Y.; Jeon, H.J.; Nam, J.-Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and Detection of Low-Allele-Fraction Variants in Clinical Cancer Samples. Nat. Commun. 2017, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal Diversification of Primary Breast Cancer Revealed by Multiregion Sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Padma, V.V. An Overview of Targeted Cancer Therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef]
- Shuel, S.L. Targeted Cancer Therapies: Clinical Pearls for Primary Care. Can. Fam. Physician 2022, 68, 515–518. [Google Scholar] [CrossRef]
- Min, H.-Y.; Lee, H.-Y. Molecular Targeted Therapy for Anticancer Treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- PETERS, G.J. From ‘Targeted Therapy’ to Targeted Therapy. Anticancer Res. 2019, 39, 3341–3345. [Google Scholar] [CrossRef]
- Charlton, P.; Spicer, J. Targeted Therapy in Cancer. Medicine 2016, 44, 34–38. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Liu, G.-H.; Chen, T.; Zhang, X.; Ma, X.-L.; Shi, H.-S. Small Molecule Inhibitors Targeting the Cancers. MedComm 2022, 3, e181. [Google Scholar] [CrossRef]
- Hertzman Johansson, C.; Egyhazi Brage, S. BRAF Inhibitors in Cancer Therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour Micro-Environment Elicits Innate Resistance to RAF Inhibitors through HGF Secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Asić, K. Dominant Mechanisms of Primary Resistance Differ from Dominant Mechanisms of Secondary Resistance to Targeted Therapies. Crit. Rev. Oncol. Hematol. 2016, 97, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Balik, K.; Modrakowska, P.; Maj, M.; Kaźmierski, Ł.; Bajek, A. Limitations of Molecularly Targeted Therapy. Med. Res. J. 2019, 4, 99–105. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, N.K.; Dwiwedi, P.; Charan, J.; Kaur, R.; Sidhu, P.; Chugh, V.K. Monoclonal Antibodies: A Review. Curr. Clin. Pharmacol. 2018, 13, 85–99. [Google Scholar] [CrossRef]
- Kumar, M.; Jalota, A.; Sahu, S.K.; Haque, S. Therapeutic Antibodies for the Prevention and Treatment of Cancer. J. Biomed. Sci. 2024, 31, 6. [Google Scholar] [CrossRef]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.-M.; Ravetch, J.V. Type I and Type II Fc Receptors Regulate Innate and Adaptive Immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Wang, T.T.; Dahan, R.; Maamary, J.; Ravetch, J.V. Signaling by Antibodies: Recent Progress. Annu. Rev. Immunol. 2017, 35, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Trastuzumab for Early-Stage, HER2-Positive Breast Cancer: A Meta-Analysis of 13,864 Women in Seven Randomised Trials. Lancet Oncol. 2021, 22, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-Positive Breast Cancer: Advances and Future Directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef]
- Zhou, J.; Ji, Q.; Li, Q. Resistance to Anti-EGFR Therapies in Metastatic Colorectal Cancer: Underlying Mechanisms and Reversal Strategies. J. Exp. Clin. Cancer Res. 2021, 40, 328. [Google Scholar] [CrossRef]
- Kasi, P.M.; Afable, M.G.; Herting, C.; Lukanowski, M.; Jin, Z. Anti-EGFR Antibodies in the Management of Advanced Colorectal Cancer. Oncologist 2023, 28, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Gordeev, A.; Vaal, A.; Puchkova, M.; Smirnova, I.; Doronin, A.; Znobishcheva, A.; Zhmudanova, D.; Aleksandrov, A.; Sukchev, M.; Imyanitov, E.; et al. Preclinical Comparison of Prolgolimab, Pembrolizumab and Nivolumab. Sci. Rep. 2024, 14, 23136. [Google Scholar] [CrossRef]
- McMahon, D.J.; McLaughlin, R.; Naidoo, J. Is Immunotherapy Beneficial in Patients with Oncogene-Addicted Non-Small Cell Lung Cancers? A Narrative Review. Cancers 2024, 16, 527. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Shahid, K.; Khalife, M.; Dabney, R.; Phan, A.T. Immunotherapy and Targeted Therapy—The New Roadmap in Cancer Treatment. Ann. Transl. Med. 2019, 7, 595. [Google Scholar] [CrossRef]
- McLean, L.; Leal, J.L.; Solomon, B.J.; John, T. Immunotherapy in Oncogene Addicted Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2021, 10, 2736–2751. [Google Scholar] [CrossRef]
- Zhang, J.; Vokes, N.; Li, M.; Xu, J.; Bai, H.; Wang, J.; Wang, Z.; Zhang, J. Overcoming EGFR-TKI Resistance by Targeting the Tumor Microenvironment. Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Dailah, H.G.; Hommdi, A.A.; Koriri, M.D.; Algathlan, E.M.; Mohan, S. Potential Role of Immunotherapy and Targeted Therapy in the Treatment of Cancer: A Contemporary Nursing Practice. Heliyon 2024, 10, e24559. [Google Scholar] [CrossRef] [PubMed]
- Otano, I.; Ucero, A.C.; Zugazagoitia, J.; Paz-Ares, L. At the Crossroads of Immunotherapy for Oncogene-Addicted Subsets of NSCLC. Nat. Rev. Clin. Oncol. 2023, 20, 143–159. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in Cancer Treatment: A Review of 15 Years of Clinical Experience and Future Outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Bodet-Milin, C.; Kraeber-Bodéré, F.; Eugène, T.; Guérard, F.; Gaschet, J.; Bailly, C.; Mougin, M.; Bourgeois, M.; Faivre-Chauvet, A.; Chérel, M.; et al. Radioimmunotherapy for Treatment of Acute Leukemia. Semin. Nucl. Med. 2016, 46, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.-K.V.; Press, O.W. Radioimmunotherapy of Human Tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef]
- Abbasi, A.; Dadashpour, M.; Alipourfard, I. Calculation of Radium-223 and Actinium-225 α-Emitter Radiopharmaceuticals Dose Rates in Treatment of Metastatic Castration-Resistant Prostate Cancer. J. Cancer Res. Ther. 2021, 17, 348–352. [Google Scholar] [CrossRef]
- Cremonesi, M.; Ferrari, M.; Grana, C.M.; Vanazzi, A.; Stabin, M.; Bartolomei, M.; Papi, S.; Prisco, G.; Martinelli, G.; Paganelli, G.; et al. High-Dose Radioimmunotherapy with 90Y-Ibritumomab Tiuxetan: Comparative Dosimetric Study for Tailored Treatment. J. Nucl. Med. 2007, 48, 1871–1879. [Google Scholar] [CrossRef]
- Guo, Y.; Parry, J.J.; Laforest, R.; Rogers, B.E.; Anderson, C.J. The Role of P53 in Combination Radioimmunotherapy with 64Cu-DOTA-Cetuximab and Cisplatin in a Mouse Model of Colorectal Cancer. J. Nucl. Med. 2013, 54, 1621–1629. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, J.; Haggstrom, L.R.; Lim, E. Antibody–Drug Conjugates: Ushering in a New Era of Cancer Therapy. Pharmaceutics 2023, 15, 2017. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.; Wang, E.S. Gemtuzumab Ozogamicin for the Treatment of Acute Myeloid Leukemia. Expert Rev. Clin. Pharmacol. 2018, 11, 549–559. [Google Scholar] [CrossRef]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody–Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther.-Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- De Marco, R.C.; Monzo, H.J.; Ojala, P.M. CAR T Cell Therapy: A Versatile Living Drug. Int. J. Mol. Sci. 2023, 24, 6300. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Goyco Vera, D.; Waghela, H.; Nuh, M.; Pan, J.; Lulla, P. Approved CAR-T Therapies Have Reproducible Efficacy and Safety in Clinical Practice. Hum. Vaccines Immunother. 2024, 20, 2378543. [Google Scholar] [CrossRef]
- Keefe, D.M.K.; Bateman, E.H. Potential Successes and Challenges of Targeted Cancer Therapies. JNCI Monogr. 2019, 2019, lgz008. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the Mechanisms and Challenges of Cancer Drug Resistance. Cell Commun. Signal. 2024, 22, 109. [Google Scholar] [CrossRef]
- American Cancer Society. Managing Cancer-Related Side Effects. Available online: https://www.cancer.org/cancer/managing-cancer/side-effects.html (accessed on 5 March 2025).
- Kiss, B.; Borbély, J. Business Risk Mitigation in the Development Process of New Monoclonal Antibody Drug Conjugates for Cancer Treatment. Pharmaceutics 2023, 15, 1761. [Google Scholar] [CrossRef]
- Dropulić, B. CAR-T and Cellular Gene Therapies Are Too Expensive. Nat. Med. 2024, 30, 2714. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Dhiman, V.K.; Pandey, M.; Dhiman, V.K.; Sharma, A.; Pandey, H.; Verma, S.K.; Pandey, R. Personalized Medicine: An Alternative for Cancer Treatment. Cancer Treat. Res. Commun. 2024, 42, 100860. [Google Scholar] [CrossRef]
- Hoeben, A.; Joosten, E.A.J.; van den Beuken-van Everdingen, M.H.J. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2021, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef]
- Vicente, A.M.; Ballensiefen, W.; Jönsson, J.-I. How Personalised Medicine Will Transform Healthcare by 2030: The ICPerMed Vision. J. Transl. Med. 2020, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Saeed, R.F.; Awan, U.A.; Saeed, S.; Mumtaz, S.; Akhtar, N.; Aslam, S. Targeted Therapy and Personalized Medicine. In Therapeutic Approaches in Cancer Treatment; Springer: Cham, Switzerland, 2023; pp. 177–205. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer Chemotherapy and beyond: Current Status, Drug Candidates, Associated Risks and Progress in Targeted Therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Cecchin, E.; Stocco, G. Pharmacogenomics and Personalized Medicine. Genes 2020, 11, 679. [Google Scholar] [CrossRef] [PubMed]
- Sadee, W.; Wang, D.; Hartmann, K.; Toland, A.E. Pharmacogenomics: Driving Personalized Medicine. Pharmacol. Rev. 2023, 75, 789–814. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the MTOR Signaling Network for Cancer Therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef]
- Böhm, R.; Imseng, S.; Jakob, R.P.; Hall, M.N.; Maier, T.; Hiller, S. The Dynamic Mechanism of 4E-BP1 Recognition and Phosphorylation by MTORC1. Mol. Cell 2021, 81, 2403–2416.e5. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid Biopsy in Cancer: Current Status, Challenges and Future Prospects. Signal Transduct. Target. Ther. 2024, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Vegesna, R.; Mukherjee, S.; Kammula, A.V.; Dhruba, S.R.; Wu, W.; Kerr, D.L.; Nair, N.U.; Jones, M.G.; Yosef, N.; et al. PERCEPTION Predicts Patient Response and Resistance to Treatment Using Single-Cell Transcriptomics of Their Tumors. Nat. Cancer 2024, 5, 938–952. [Google Scholar] [CrossRef]
- Corti, C.; Cobanaj, M.; Dee, E.C.; Criscitiello, C.; Tolaney, S.M.; Celi, L.A.; Curigliano, G. Artificial Intelligence in Cancer Research and Precision Medicine: Applications, Limitations and Priorities to Drive Transformation in the Delivery of Equitable and Unbiased Care. Cancer Treat. Rev. 2023, 112, 102498. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of Single-Cell Sequencing in Cancer Research: Progress and Perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Paolillo, C.; Londin, E.; Fortina, P. Single-Cell Genomics. Clin. Chem. 2019, 65, 972–985. [Google Scholar] [CrossRef]
- Zafar, H.; Wang, Y.; Nakhleh, L.; Navin, N.; Chen, K. Monovar: Single-Nucleotide Variant Detection in Single Cells. Nat. Methods 2016, 13, 505–507. [Google Scholar] [CrossRef]
- Frumkin, D.; Wasserstrom, A.; Itzkovitz, S.; Harmelin, A.; Rechavi, G.; Shapiro, E. Amplification of Multiple Genomic Loci from Single Cells Isolated by Laser Micro-Dissection of Tissues. BMC Biotechnol. 2008, 8, 17. [Google Scholar] [CrossRef]
- Keays, K.M.; Owens, G.P.; Ritchie, A.M.; Gilden, D.H.; Burgoon, M.P. Laser Capture Microdissection and Single-Cell RT-PCR without RNA Purification. J. Immunol. Methods 2005, 302, 90–98. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The Technology and Biology of Single-Cell RNA Sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-Length MRNA-Seq from Single-Cell Levels of RNA and Individual Circulating Tumor Cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.-B.; Lönnerberg, P.; Linnarsson, S. Characterization of the Single-Cell Transcriptional Landscape by Highly Multiplex RNA-Seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Q.; Li, P.; Liu, T.; Liu, X.; Zhu, F.; Zhang, L. Single-Cell Transcriptome Sequencing Provides Insight into Multiple Chemotherapy Resistance in a Patient with Refractory DLBCL: A Case Report. Front. Immunol. 2024, 15, 1303310. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-Related Inflammation and Treatment Effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef]
- Liu, M.-K.; Liu, F.; Dai, Y.-T.; Weng, X.-Q.; Cheng, L.-L.; Fan, L.-Q.; Liu, H.; Jiang, L.; Sun, X.-J.; Fang, H.; et al. Case Report: Molecular and Microenvironment Change upon Midostaurin Treatment in Mast Cell Leukemia at Single-Cell Level. Front. Immunol. 2023, 14, 1210909. [Google Scholar] [CrossRef]
- Wang, H.; Boussouar, A.; Mazelin, L.; Tauszig-Delamasure, S.; Sun, Y.; Goldschneider, D.; Paradisi, A.; Mehlen, P. The Proto-Oncogene c-Kit Inhibits Tumor Growth by Behaving as a Dependence Receptor. Mol. Cell 2018, 72, 413–425.e5. [Google Scholar] [CrossRef]
- Ustun, C.; Arock, M.; Kluin-Nelemans, H.C.; Reiter, A.; Sperr, W.R.; George, T.; Horny, H.-P.; Hartmann, K.; Sotlar, K.; Damaj, G.; et al. Advanced Systemic Mastocytosis: From Molecular and Genetic Progress to Clinical Practice. Haematologica 2016, 101, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Debaize, L.; Jakobczyk, H.; Avner, S.; Gaudichon, J.; Rio, A.-G.; Sérandour, A.A.; Dorsheimer, L.; Chalmel, F.; Carroll, J.S.; Zörnig, M.; et al. Interplay between Transcription Regulators RUNX1 and FUBP1 Activates an Enhancer of the Oncogene c-KIT and Amplifies Cell Proliferation. Nucleic Acids Res. 2018, 46, 11214–11228. [Google Scholar] [CrossRef]
- Li, Q.; Lai, Q.; He, C.; Fang, Y.; Yan, Q.; Zhang, Y.; Wang, X.; Gu, C.; Wang, Y.; Ye, L.; et al. RUNX1 Promotes Tumour Metastasis by Activating the Wnt/β-Catenin Signalling Pathway and EMT in Colorectal Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 334. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Mukhopadhyay, A. A Comprehensive Review of Machine Learning Techniques for Multi-Omics Data Integration: Challenges and Applications in Precision Oncology. Brief. Funct. Genom. 2024, 23, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Bunnik, E.M.; Le Roch, K.G. An Introduction to Functional Genomics and Systems Biology. Adv. Wound Care 2013, 2, 490–498. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Dai, Y.; Zhao, Z.; Zhu, J.; Guo, H.; Yang, R. Single-Cell Sequencing to Multi-Omics: Technologies and Applications. Biomark. Res. 2024, 12, 110. [Google Scholar] [CrossRef]
- Chakraborty, S.; Sharma, G.; Karmakar, S.; Banerjee, S. Multi-OMICS Approaches in Cancer Biology: New Era in Cancer Therapy. Biochim. Biophys. Acta-Mol. Basis Dis. 2024, 1870, 167120. [Google Scholar] [CrossRef]
- Cho, S.B. Uncovering Oncogenic Mechanisms of Tumor Suppressor Genes in Breast Cancer Multi-Omics Data. Int. J. Mol. Sci. 2022, 23, 9624. [Google Scholar] [CrossRef]
- Zhao, M.; Zhao, Z. Concordance of Copy Number Loss and Down-Regulation of Tumor Suppressor Genes: A Pan-Cancer Study. BMC Genom. 2016, 17, 532. [Google Scholar] [CrossRef]
- Halaburkova, A.; Cahais, V.; Novoloaca, A.; da Silva Araujo, M.G.; Khoueiry, R.; Ghantous, A.; Herceg, Z. Pan-Cancer Multi-Omics Analysis and Orthogonal Experimental Assessment of Epigenetic Driver Genes. Genome Res. 2020, 30, 1517–1532. [Google Scholar] [CrossRef] [PubMed]
- Schupp, P.G.; Shelton, S.J.; Brody, D.J.; Eliscu, R.; Johnson, B.E.; Mazor, T.; Kelley, K.W.; Potts, M.B.; McDermott, M.W.; Huang, E.J.; et al. Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections. Cancers 2024, 16, 2429. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, Unmet and Neglected Challenges in Multi-Omics Studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-T.; Lu, Y.; Wu, J.; Zhang, C.; Tan, H.-Y.; Bian, Z.-X.; Wang, N.; Feng, Y. CRISPR-Cas9 Library Screening Approach for Anti-Cancer Drug Discovery: Overview and Perspectives. Theranostics 2022, 12, 3329–3344. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics 2021, 15, 353–361. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Vanoli, F.; Song, E.; Dermawan, J.K.; Fishinevich, E.; Sung, P.; Min, S.S.; Xie, Z.; de Traux de Wardin, H.; Hwang, S.; Maki, R.G.; et al. Modeling Extraordinary Response Through Targeting Secondary Alterations in Fusion-Associated Sarcoma. JCO Precis. Oncol. 2024, 8, e2300688. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Vanoli, F.; Herviou, L.; Sung, Y.-S.; Zhang, L.; Singer, S.; Tap, W.D.; Benayed, R.; Bale, T.A.; Benhamida, J.K.; et al. Comprehensive Genomic Profiling of EWSR1/FUS::CREB Translocation-Associated Tumors Uncovers Prognostically Significant Recurrent Genetic Alterations and Methylation-Transcriptional Correlates. Mod. Pathol. 2022, 35, 1055–1065. [Google Scholar] [CrossRef]
- Wang, S.-W.; Gao, C.; Zheng, Y.-M.; Yi, L.; Lu, J.-C.; Huang, X.-Y.; Cai, J.-B.; Zhang, P.-F.; Cui, Y.-H.; Ke, A.-W. Current Applications and Future Perspective of CRISPR/Cas9 Gene Editing in Cancer. Mol. Cancer 2022, 21, 57. [Google Scholar] [CrossRef]
- Zhang, B. CRISPR/Cas Gene Therapy. J. Cell. Physiol. 2021, 236, 2459–2481. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, C.; Wang, Y.; Koivisto, O.; Zhou, J.; Shu, Y.; Zhang, H. Nanotechnology-Based Delivery of CRISPR/Cas9 for Cancer Treatment. Adv. Drug Deliv. Rev. 2021, 176, 113891. [Google Scholar] [CrossRef]
- Huang, S.; Yang, J.; Shen, N.; Xu, Q.; Zhao, Q. Artificial Intelligence in Lung Cancer Diagnosis and Prognosis: Current Application and Future Perspective. Semin. Cancer Biol. 2023, 89, 30–37. [Google Scholar] [CrossRef]
- Chen, Y.; Han, Q.; Huang, Z.; Lyu, M.; Ai, Z.; Liang, Y.; Yan, H.; Wang, M.; Xiang, Z. Value of IVIM in Differential Diagnoses between Benign and Malignant Solitary Lung Nodules and Masses: A Meta-Analysis. Front. Surg. 2022, 9, 817443. [Google Scholar] [CrossRef]
- Wan, Y.-L.; Wu, P.; Huang, P.-C.; Tsay, P.-K.; Pan, K.-T.; Trang, N.; Chuang, W.-Y.; Wu, C.-Y.; Lo, S. The Use of Artificial Intelligence in the Differentiation of Malignant and Benign Lung Nodules on Computed Tomograms Proven by Surgical Pathology. Cancers 2020, 12, 2211. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Yang, D.; Zhu, Y.; Liu, Y.; Hu, J.; Bai, C. 3D Gray Density Coding Feature for Benign-malignant Pulmonary Nodule Classification on Chest CT. Med. Phys. 2021, 48, 7826–7836. [Google Scholar] [CrossRef]
- Mehta, K.; Jain, A.; Mangalagiri, J.; Menon, S.; Nguyen, P.; Chapman, D.R. Lung Nodule Classification Using Biomarkers, Volumetric Radiomics, and 3D CNNs. J. Digit. Imaging 2021, 34, 647–666. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.; Hancock, M.; Clunie, D.; Brochhausen, M.; Bona, J.; Kirby, J.; Freymann, J.; Pieper, S.; Aerts, H.J.W.L.; Kikinis, R.; et al. DICOM Re-encoding of Volumetrically Annotated Lung Imaging Database Consortium (LIDC) Nodules. Med. Phys. 2020, 47, 5953–5965. [Google Scholar] [CrossRef]
- Armato, S.G.; McLennan, G.; Bidaut, L.; McNitt-Gray, M.F.; Meyer, C.R.; Reeves, A.P.; Zhao, B.; Aberle, D.R.; Henschke, C.I.; Hoffman, E.A.; et al. The Lung Image Database Consortium (LIDC) and Image Database Resource Initiative (IDRI): A Completed Reference Database of Lung Nodules on CT Scans. Med. Phys. 2011, 38, 915–931. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and Mutation Prediction from Non–Small Cell Lung Cancer Histopathology Images Using Deep Learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.Y.; Chu, T.N.; Ladi-Seyedian, S.-S. Genomics and Artificial Intelligence. Urol. Clin. N. Am. 2024, 51, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Reed, M.; Zhu, H.; Cheng, Y.; Almeida, J.; Fruhbeck, G.; Ribeiro, R.; Hu, P. Epigenome-Wide DNA Methylation and Transcriptome Profiling of Localized and Locally Advanced Prostate Cancer: Uncovering New Molecular Markers. Genomics 2022, 114, 110474. [Google Scholar] [CrossRef]
- Armakolas, A.; Kotsari, M.; Koskinas, J. Liquid Biopsies, Novel Approaches and Future Directions. Cancers 2023, 15, 1579. [Google Scholar] [CrossRef]
- Connal, S.; Cameron, J.M.; Sala, A.; Brennan, P.M.; Palmer, D.S.; Palmer, J.D.; Perlow, H.; Baker, M.J. Liquid Biopsies: The Future of Cancer Early Detection. J. Transl. Med. 2023, 21, 118. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Siddiqi, S.; Ahmed, S.; Hands, R.; Dorudi, S. Quantification of Cytokeratin 20, Carcinoembryonic Antigen and Guanylyl Cyclase C MRNA Levels in Lymph Nodes May Not Predict Treatment Failure in Colorectal Cancer Patients. Int. J. Cancer 2004, 108, 412–417. [Google Scholar] [CrossRef]
- Mastoraki, S.; Strati, A.; Tzanikou, E.; Chimonidou, M.; Politaki, E.; Voutsina, A.; Psyrri, A.; Georgoulias, V.; Lianidou, E. ESR1 Methylation: A Liquid Biopsy–Based Epigenetic Assay for the Follow-up of Patients with Metastatic Breast Cancer Receiving Endocrine Treatment. Clin. Cancer Res. 2018, 24, 1500–1510. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid Biopsy: Current Technology and Clinical Applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Ma, S.; Zhou, M.; Xu, Y.; Gu, X.; Zou, M.; Abudushalamu, G.; Yao, Y.; Fan, X.; Wu, G. Clinical Application and Detection Techniques of Liquid Biopsy in Gastric Cancer. Mol. Cancer 2023, 22, 7. [Google Scholar] [CrossRef]
- Turning the Tide of Early Cancer Detection. Nat. Med. 2024, 30, 1217. [CrossRef] [PubMed]
- Han, H.S.; Lee, K.-W. Liquid Biopsy: An Emerging Diagnostic, Prognostic, and Predictive Tool in Gastric Cancer. J. Gastric Cancer 2024, 24, 4. [Google Scholar] [CrossRef]
- Fatima, S.; Ma, Y.; Safrachi, A.; Haider, S.; Spring, K.J.; Vafaee, F.; Scott, K.F.; Roberts, T.L.; Becker, T.M.; de Souza, P. Harnessing Liquid Biopsies to Guide Immune Checkpoint Inhibitor Therapy. Cancers 2022, 14, 1669. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Erratum: Clonal Evolution and Resistance to EGFR Blockade in the Blood of Colorectal Cancer Patients. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef]
- Cossu, A.M.; Scrima, M.; Lombardi, A.; Grimaldi, A.; Russo, M.; Ottaiano, A.; Caraglia, M.; Bocchetti, M. Future Directions and Management of Liquid Biopsy in Non-Small Cell Lung Cancer. Explor. Target. Anti-Tumor Ther. 2020, 1, 239–252. [Google Scholar] [CrossRef]
- Canale, M.; Pasini, L.; Bronte, G.; Delmonte, A.; Cravero, P.; Crinò, L.; Ulivi, P. Role of Liquid Biopsy in Oncogene-Addicted Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2019, 8, S265–S279. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Liquid Biopsy and Minimal Residual Disease—Latest Advances and Implications for Cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Johnston, A.D.; Ross, J.P.; Ma, C.; Fung, K.Y.C.; Locke, W.J. Epigenetic Liquid Biopsies for Minimal Residual Disease, What’s around the Corner? Front. Oncol. 2023, 13, 1103797. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Minimal Residual Disease as a Target for Liquid Biopsy in Patients with Solid Tumours. Nat. Rev. Clin. Oncol. 2025, 22, 65–77. [Google Scholar] [CrossRef]
- Alba-Bernal, A.; Lavado-Valenzuela, R.; Domínguez-Recio, M.E.; Jiménez-Rodriguez, B.; Queipo-Ortuño, M.I.; Alba, E.; Comino-Méndez, I. Challenges and Achievements of Liquid Biopsy Technologies Employed in Early Breast Cancer. eBioMedicine 2020, 62, 103100. [Google Scholar] [CrossRef]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid Biopsy: A Step Closer to Transform Diagnosis, Prognosis and Future of Cancer Treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef]
- Yu, S.; Wang, S.; Wang, X.; Xu, X. The Axis of Tumor-Associated Macrophages, Extracellular Matrix Proteins, and Cancer-Associated Fibroblasts in Oncogenesis. Cancer Cell Int. 2024, 24, 335. [Google Scholar] [CrossRef]
- Czekay, R.-P.; Cheon, D.-J.; Samarakoon, R.; Kutz, S.M.; Higgins, P.J. Cancer-Associated Fibroblasts: Mechanisms of Tumor Progression and Novel Therapeutic Targets. Cancers 2022, 14, 1231. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Butti, R.; Panda, V.K.; Malhotra, D.; Das, S.; Mitra, T.; Kapse, P.; Gosavi, S.W.; Kundu, G.C. Modulation of the Tumor Microenvironment and Mechanism of Immunotherapy-Based Drug Resistance in Breast Cancer. Mol. Cancer 2024, 23, 92. [Google Scholar] [CrossRef]
- Kaczmarek, M.; Poznańska, J.; Fechner, F.; Michalska, N.; Paszkowska, S.; Napierała, A.; Mackiewicz, A. Cancer Vaccine Therapeutics: Limitations and Effectiveness—A Literature Review. Cells 2023, 12, 2159. [Google Scholar] [CrossRef]
- Memorial Sloan Kettering Cancer Center. Cancer Vaccines: The Types, How They Work, and Which Cancers They Treat. Available online: https://www.mskcc.org/cancer-care/diagnosis-treatment/cancer-treatments/immunotherapy/cancer-vaccines (accessed on 25 January 2025).
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. MRNA Vaccine for Cancer Immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Pérez-Baños, A.; Gleisner, M.A.; Flores, I.; Pereda, C.; Navarrete, M.; Araya, J.P.; Navarro, G.; Quezada-Monrás, C.; Tittarelli, A.; Salazar-Onfray, F. Whole Tumour Cell-Based Vaccines: Tuning the Instruments to Orchestrate an Optimal Antitumour Immune Response. Br. J. Cancer 2023, 129, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Pandya, A.; Shah, Y.; Kothari, N.; Postwala, H.; Shah, A.; Parekh, P.; Chorawala, M.R. The Future of Cancer Immunotherapy: DNA Vaccines Leading the Way. Med. Oncol. 2023, 40, 200. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, I.; Muralidhar, A.; McNeel, D.G. Vaccines as Treatments for Prostate Cancer. Nat. Rev. Urol. 2023, 20, 544–559. [Google Scholar] [CrossRef]
- Madan, R.A.; Gulley, J.L. Sipuleucel-T: Harbinger of a New Age of Therapeutics for Prostate Cancer. Expert Rev. Vaccines 2011, 10, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Gu, X.; Li, Y.; Wu, Q. Mechanisms of BCG in the Treatment of Bladder Cancer-Current Understanding and the Prospect. Biomed. Pharmacother. 2020, 129, 110393. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Redelman-Sidi, G. BCG in Bladder Cancer Immunotherapy. Cancers 2022, 14, 3073. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Du, J. Human Papillomavirus Vaccines: An Updated Review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef]
- Aggarwal, S.; Agarwal, P.; Singh, A.K. Human Papilloma Virus Vaccines: A Comprehensive Narrative Review. Cancer Treat. Res. Commun. 2023, 37, 100780. [Google Scholar] [CrossRef]
- Flores, J.E.; Thompson, A.J.; Ryan, M.; Howell, J. The Global Impact of Hepatitis B Vaccination on Hepatocellular Carcinoma. Vaccines 2022, 10, 793. [Google Scholar] [CrossRef]
- Cao, M.; Fan, J.; Lu, L.; Fan, C.; Wang, Y.; Chen, T.; Zhang, S.; Yu, Y.; Xia, C.; Lu, J.; et al. Long Term Outcome of Prevention of Liver Cancer by Hepatitis B Vaccine: Results from an RCT with 37 Years. Cancer Lett. 2022, 536, 215652. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Thorne, S.H.; Liang, W.; Sampath, P.; Schmidt, T.; Sikorski, R.; Beilhack, A.; Contag, C.H. Targeting Localized Immune Suppression Within the Tumor Through Repeat Cycles of Immune Cell-Oncolytic Virus Combination Therapy. Mol. Ther. 2010, 18, 1698–1705. [Google Scholar] [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of Virus-Mediated Immunogenic Cancer Cell Death and the Consequences for Oncolytic Virus-Based Immunotherapy of Cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way Is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef]
- Yan, Z.; Zhang, Z.; Chen, Y.; Xu, J.; Wang, J.; Wang, Z. Enhancing Cancer Therapy: The Integration of Oncolytic Virus Therapy with Diverse Treatments. Cancer Cell Int. 2024, 24, 242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; McGray, A.J.R.; Jiang, W.; Lu, B.; Kalinski, P.; Guo, Z.S. Improving Cancer Immunotherapy by Rationally Combining Oncolytic Virus with Modulators Targeting Key Signaling Pathways. Mol. Cancer 2022, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic Virus Immunotherapy: Future Prospects for Oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Peter, M.; Kühnel, F. Oncolytic Adenovirus in Cancer Immunotherapy. Cancers 2020, 12, 3354. [Google Scholar] [CrossRef]
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic Viruses for Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic Cancer Vaccines: Advancements, Challenges and Prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol. Ther.-Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef] [PubMed]
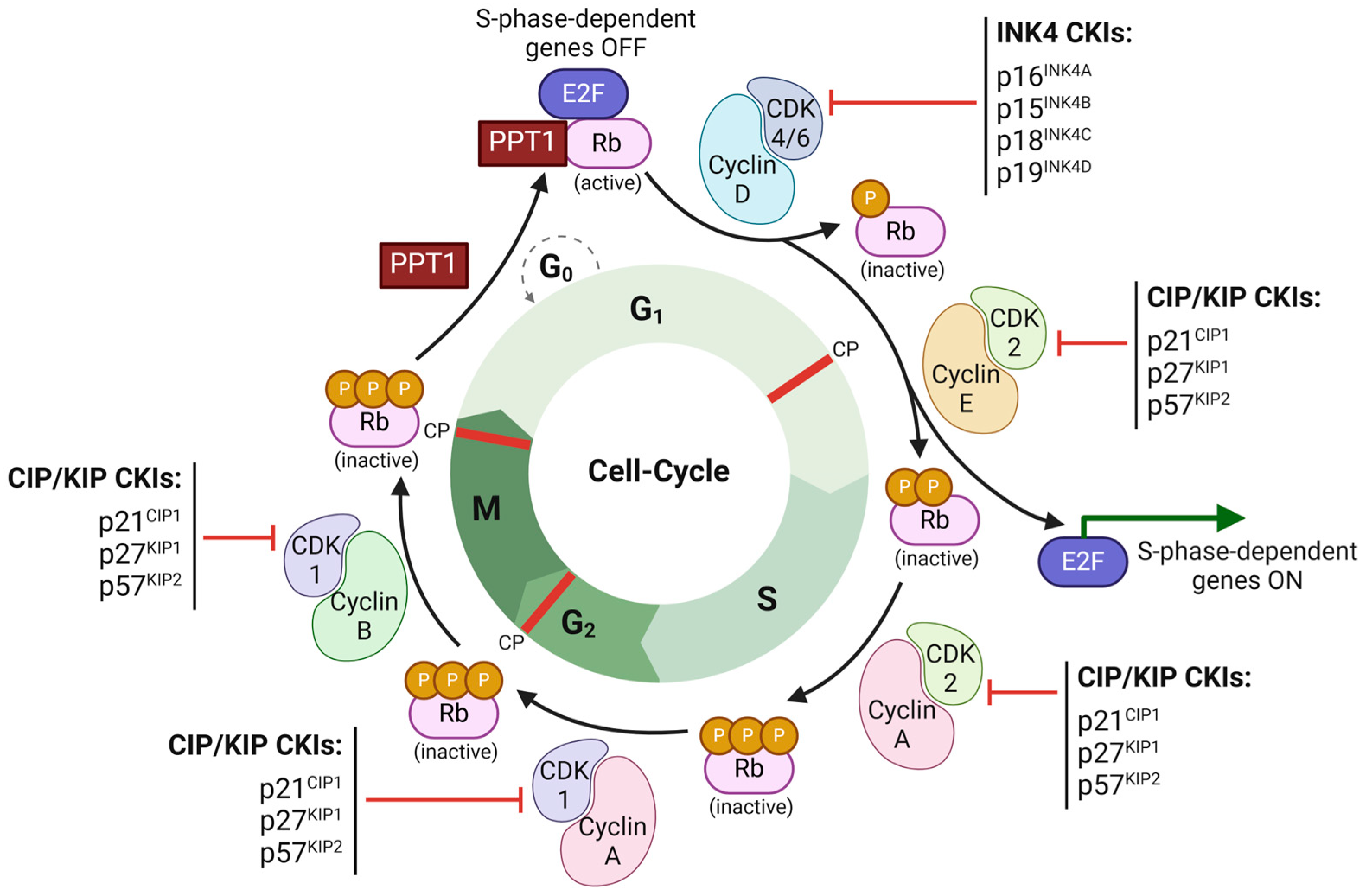
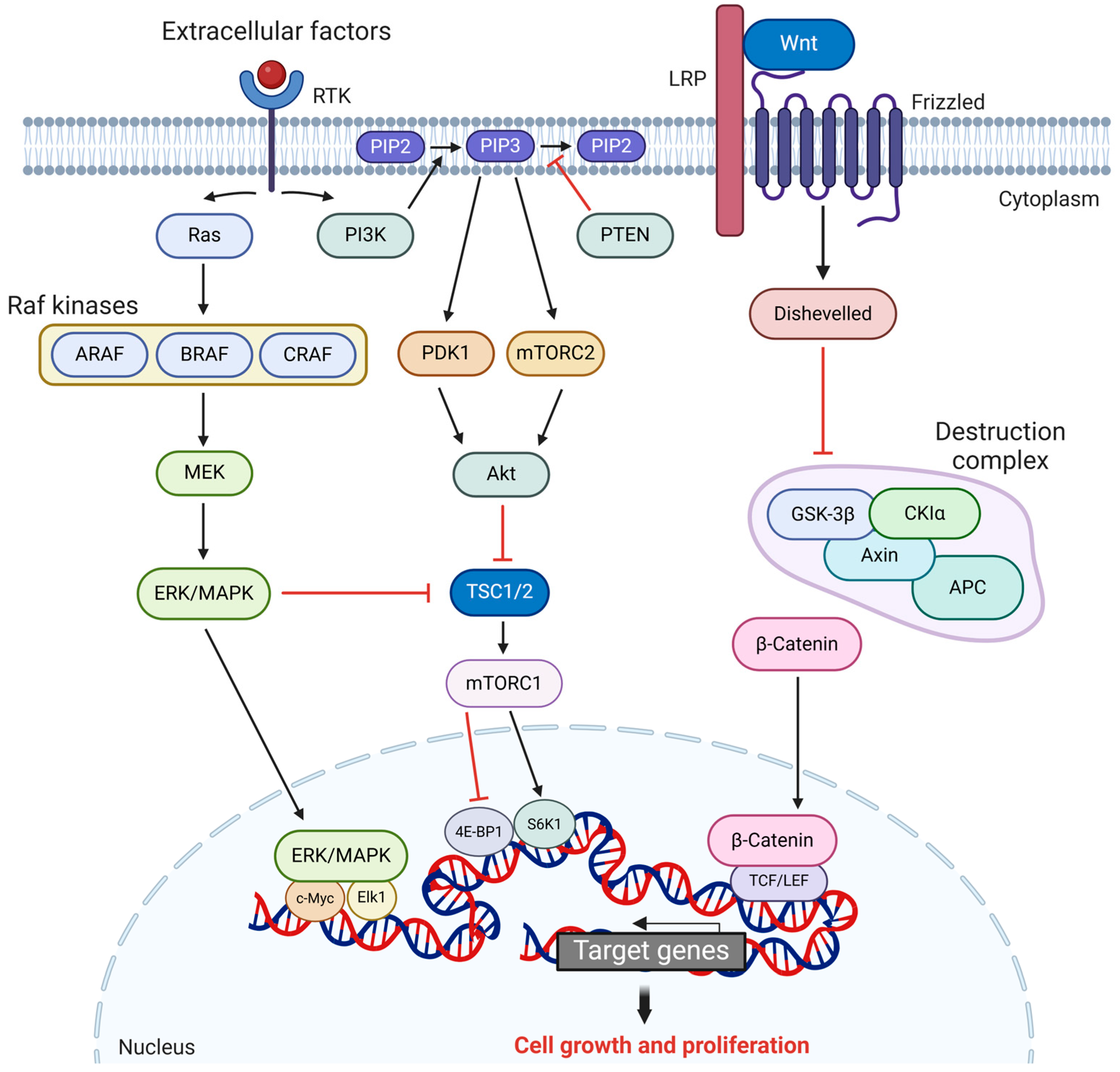
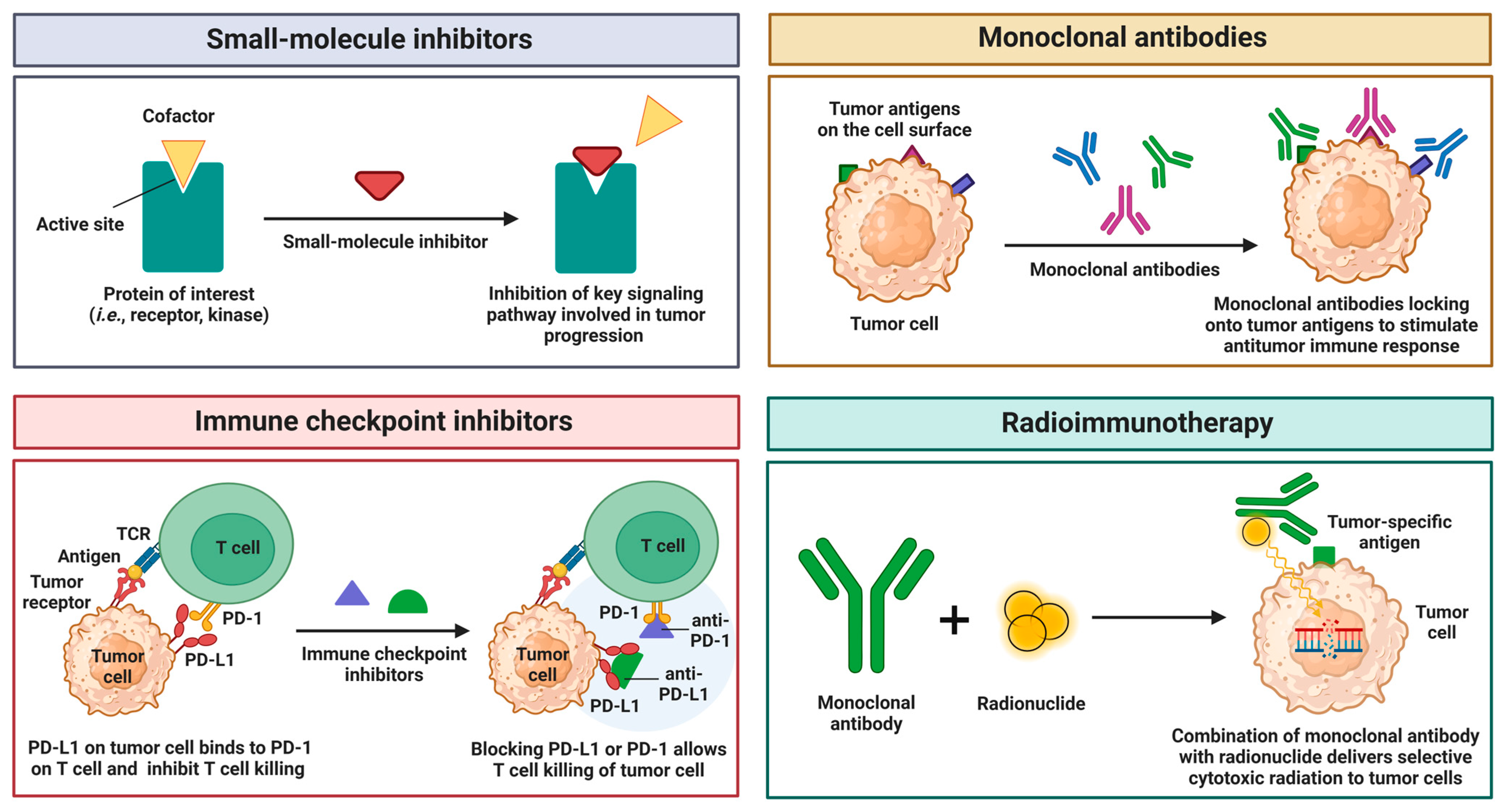

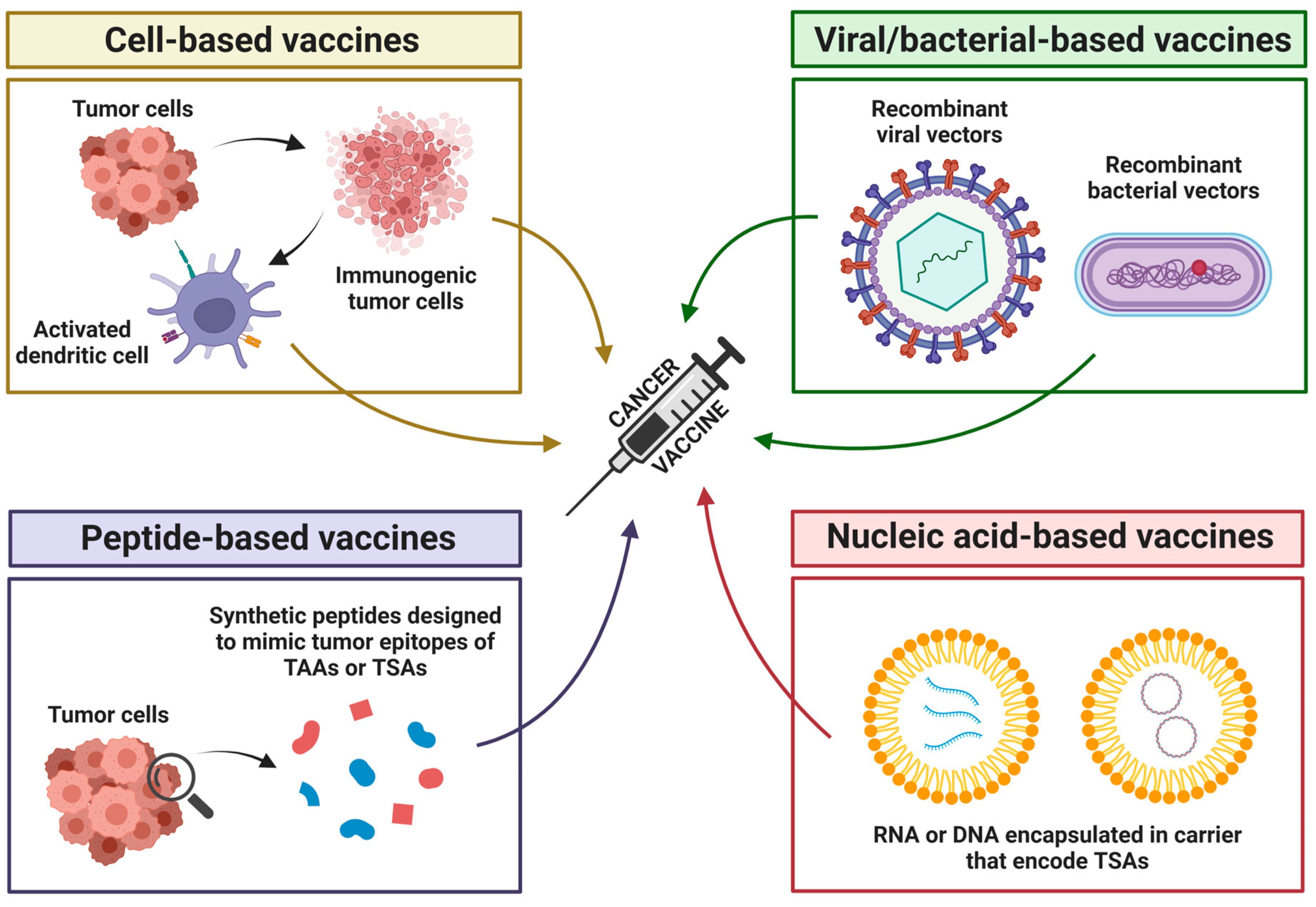

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primary Role | Dual Role | Condition | Biological Mechanism |
|---|---|---|---|---|
| TP53 | Tumor Suppressor | Oncogene | GOF mutations (e.g., R175, R248) | Altered transcriptional activity; activating pro-oncogenic genes (e.g., growth factors) |
| MYC | Oncogene | Tumor Suppressor | Overexpression | MDM2-p53 apoptotic stress response; upregulating pro-apoptotic targets (e.g., BAX) |
| TGFβ1 | Tumor Suppressor | Oncogene | Pathway defects (e.g., SMAD loss) | Signaling cascade activation; enhancing angiogenesis via VEGF and immune evasion |
| Inhibitor | Target | Mechanism of Action | FDA Approval |
|---|---|---|---|
| Erlotinib | EGFR (RTK) | Competitively inhibits ATP binding to EGFR; Blocks downstream signaling | 2004 (NSCLC), 2005 (Pancreatic cancer) |
| Gefitinib | EGFR (RTK) | Inhibits EGFR tyrosine kinase activity; reduces cell proliferation | 2003 (NSCLC) |
| Lapatinib | HER2, EGFR (RTKs) | Dual inhibitor; prevents phosphorylation and signaling | 2007 (HER+ Breast cancer) |
| Sunitinib | VEGFR, PDGFR, KIT (RTKs) | Inhibits multiple RTKs; leads to antiangiogenic and antiproliferative effects | 2006 (pNET, RCC, GIST) |
| Sorafenib | VEGFR, PDGFR, KIT (RTKs) | Inhibits RTKs and RAF kinase; blocks angiogenesis and tumorigenesis | 2005 (HCC, RCC, Thyroid cancer) |
| Vemurafenib | BRAF V600E (MAPK Pathway) | Selectively inhibits mutant BRAF; prevents aberrant MAPK activation | 2011 (Melanoma, Erdheim–Chester disease) |
| Dabrafenib | BRAF V600E (MAPK Pathway) | Inhibits mutant BRAF kinase; reduces MAPK-driven cell proliferation | 2013 (Melanoma, NSCLC, Anaplastic thyroid cancer) |
| Encorafenib | BRAF V600E (MAPK Pathway) | BRAF kinase inhibitor; blocks mutant BRAF kinase; inhibits MAPK pathway signaling. | 2018 (Melanoma), 2020 (Colorectal cancer), 2023 (NSCLC) |
| Binimetinib | MEK1/2 (MAPK Pathway) | Blocks MEK1/2 activity; inhibits downstream MAPK pathway signaling. | 2018 (Melanoma), 2023 (NSCLC) |
| Trametinib | MEK1/2 (MAPK Pathway) | Inhibits MEK1/2; blocks MAPK activation downstream of BRAF | 2013 (Melanoma, NSCLC, Anaplastic thyroid cancer) |
| Cobimetinib | MEK1/2 (MAPK Pathway) | Selectively inhibits MEK; suppresses MAPK signaling | 2015 (Melanoma) |
| ADC Generic Name | Target Antigen | Cytotoxic Payload | FDA Approval |
|---|---|---|---|
| Loncastuximab tesirine | CD19 | SG3199, alkylating agent (DNA targeting) | 2021 (Diffuse Large B-Cell Lymphoma—DLBCL) |
| Inotuzumab ozogamicin | CD22 | Calicheamicin (cytotoxic antibiotic) | 2017 (B-cell Acute Lymphoblastic Leukemia—ALL) |
| Brentuximab vedotin | CD30 | Monomethyl auristatin E (microtubule targeting) | 2011, 2015, 2018 (Hodgkin lymphoma—HL; 2011, 2017, 2018 (Anaplastic Large Cell Lymphoma—ALCL); 2018 (Peripheral T-Cell Lymphoma—PTCL) |
| Gemtuzumab ozogamicin | CD33 | Calicheamicin (cytotoxic antibiotic) | 2017 (Acute Myeloid Leukemia—AML) |
| Polatuzumab vedotin | CD79b | Monomethyl auristatin E (microtubule targeting) | 2019, 2023 (DLBCL) |
| Trastuzumab emtansine | HER2 | DM1 (microtubule targeting) | 2013, 2019 (HER2+ Breast Cancer) |
| Trastuzumab deruxtecan | HER2 | Topoisomerase I inhibitor (DNA targeting) | 2019, 2022 (HER2+ Breast Cancer); 2021 (Gastric Adenocarcinoma—GAC or Gastroesophageal Junction—GEJ Adenocarcinoma); 2022 (NSCLC) |
| Tisotumab vedotin | Tissue Factor | Monomethyl auristatin E (microtubule targeting) | 2021 (Cervical Cancer) |
| Mirvetuximab soravtansine–gynx | Folate Receptor Alpha | DM4 (microtubule targeting) | 2022 (Ovarian Cancer, Fallopian Tube Cancer, and Peritoneal Cancer) |
| Enfortumab vedotin | Nectin-4 | Monomethyl auristatin E (microtubule targeting) | 2019, 2023 (Urothelial Cancer) |
| Sacituzumab govitecan | Trop-2 | SN-38 topoisomerase-1 inhibitor (DNA targeting) | 2020 (Triple-Negative Breast Cancer—TNBC); 2021 (Urothelial Cancer); 2023 (HER2- Breast Cancer, HR+ Breast Cancer) |
| CAR T-Cell Product Generic Name | Target Antigen | FDA Approval |
|---|---|---|
| Tisagenlecleucel | CD19 | 2017 (ALL); 2018 (DLBCL); 2022 (Follicular lymphoma—FL) |
| Axicabtagene ciloleucel | CD19 | 2017, 2022 (DLBCL, PMBCL); 2021 (FL) |
| Brexucabtagene autoleucel | CD19 | 2020 (Mantle Cell Lymphoma—MCL); 2021 (ALL) |
| Lisocabtagene maraleucel | CD19 | 2021, 2022, 2024 (DLBCL, PMBCL) |
| Idecabtagene vicleucel | BCMA | 2021, 2024 (Multiple Myeloma—MM) |
| Ciltacabtagene autoleucel | BCMA | 2022, 2023 (MM) |
| Omics Level | Description | Analytical Techniques |
|---|---|---|
| Genome | Study of the complete genome of organisms | NGS, WGS, Sanger Sequencing, SNP Sequencing |
| Transcriptome | Study of the messenger RNA transcripts and gene expression | Northern Blotting, Serial Analysis of Gene Expression, RNA Sequencing, DNA Microarrays |
| Proteome | Study of protein levels present in the organism | Mass Spectrometry, SDS-PAGE, Multidimensional Protein Identification Technology |
| Metabolome | Study of small molecules (i.e., amino acids, sugars, and fatty acids) that are metabolized | Mass Spectrometry, Nuclear Magnetic Resonance |
| Interactome | Study of protein–protein interactions in signal transduction, transcriptional regulation, and metabolic pathways | Mass Spectrometry, Tandem Affinity Purification, Two-Hybrid System |
| Vaccine Name | Type | Key Details | Prophylactic/ Therapeutics | FDA Approval |
|---|---|---|---|---|
| Sipuleucel-T (Provenge) | Cell based | Autologous dendritic cells activated with PAP-GM-CSF fusion protein | Therapeutic | 2010 (Metastatic castration- resistant prostate cancer mCRPC) |
| Bacillus Calmette-Guerin (BCG) | Bacterial based | Live attenuated bacterium; stimulates immune response against bladder tumors | Therapeutic | 1990 (Non-muscle-invasive bladder cancer NMIBC) |
| Talimogene Laherparepvec (T-VEC, Imlygic) | Viral based (Oncolytic) | Modified herpes virus; lyses tumors and enhances antitumor immunity | Therapeutic | 2015 (Melanoma) |
| Hepatitis B (HBV) Vaccine (Recombivax HB, Energix-B) | Viral based (Recombinant protein) | Prevents HBV infection, indirectly reducing HCC | Prophylactic | 1986 Recombivax HB 1989 Energix-B, (Hepatitis B virus—prevents HCC) |
| HPV Vaccines (Cervarix, Gardasil 9) | Viral based (Virus replicon particle) | Targets HPV strains (i.e., 16/18) directly linked to HPV-related cancer and prevents infection | Prophylactic | 2009 Cervarix, 2014 Gardasil 9 (HPV-related cancers— prevents cervical, anal, and other types of cancers) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojchevski, R.; Sutanto, E.A.; Sutanto, R.; Hadzi-Petrushev, N.; Mladenov, M.; Singh, S.R.; Sinha, J.K.; Ghosh, S.; Yarlagadda, B.; Singh, K.K.; et al. Translational Advances in Oncogene and Tumor-Suppressor Gene Research. Cancers 2025, 17, 1008. https://doi.org/10.3390/cancers17061008
Stojchevski R, Sutanto EA, Sutanto R, Hadzi-Petrushev N, Mladenov M, Singh SR, Sinha JK, Ghosh S, Yarlagadda B, Singh KK, et al. Translational Advances in Oncogene and Tumor-Suppressor Gene Research. Cancers. 2025; 17(6):1008. https://doi.org/10.3390/cancers17061008
Chicago/Turabian StyleStojchevski, Radoslav, Edward Agus Sutanto, Rinni Sutanto, Nikola Hadzi-Petrushev, Mitko Mladenov, Sajal Raj Singh, Jitendra Kumar Sinha, Shampa Ghosh, Bhuvaneshwar Yarlagadda, Krishna Kumar Singh, and et al. 2025. "Translational Advances in Oncogene and Tumor-Suppressor Gene Research" Cancers 17, no. 6: 1008. https://doi.org/10.3390/cancers17061008
APA StyleStojchevski, R., Sutanto, E. A., Sutanto, R., Hadzi-Petrushev, N., Mladenov, M., Singh, S. R., Sinha, J. K., Ghosh, S., Yarlagadda, B., Singh, K. K., Verma, P., Sengupta, S., Bhaskar, R., & Avtanski, D. (2025). Translational Advances in Oncogene and Tumor-Suppressor Gene Research. Cancers, 17(6), 1008. https://doi.org/10.3390/cancers17061008