


Effects of Induced Pluripotent Stem Cell-Derived Astrocytes on Cisplatin Sensitivity in Pediatric Brain Cancer Cells

Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human iPSC, Normal Human Astrocyte, and Cancer Cell Culture
2.2. Differentiation of Human iPSCs into Astrocytes
2.3. Tumor Cell Coculture with iPSC-Astrocytes and Drug Treatment
2.4. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
2.5. Immunocytochemistry (ICC)
2.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.7. Quantitative Analysis of Marker Expression Using Flow Cytometry
2.8. Statistical Analysis
3. Results
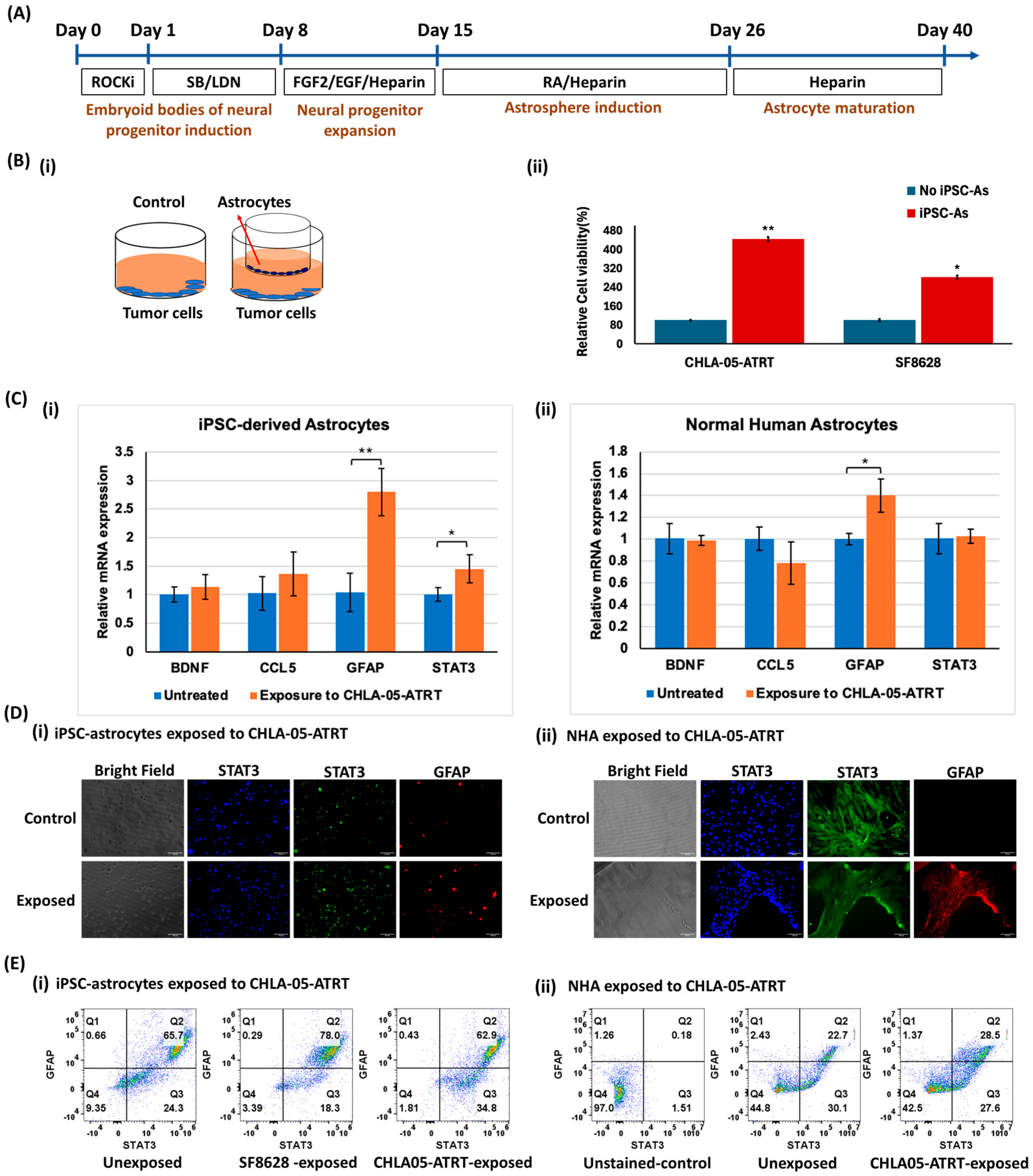
3.1. Effects of Crosstalk Between iPSC-Astrocytes and Brain Tumor Cells
3.1.1. Characterization of iPSC-Astrocytes and Comparison of Their Phenotype with NHA
3.1.2. Tumor Cell and iPSC-Astrocyte Paracrine Interaction Resulted in Astrocyte Activation and Tumor Cell Proliferation
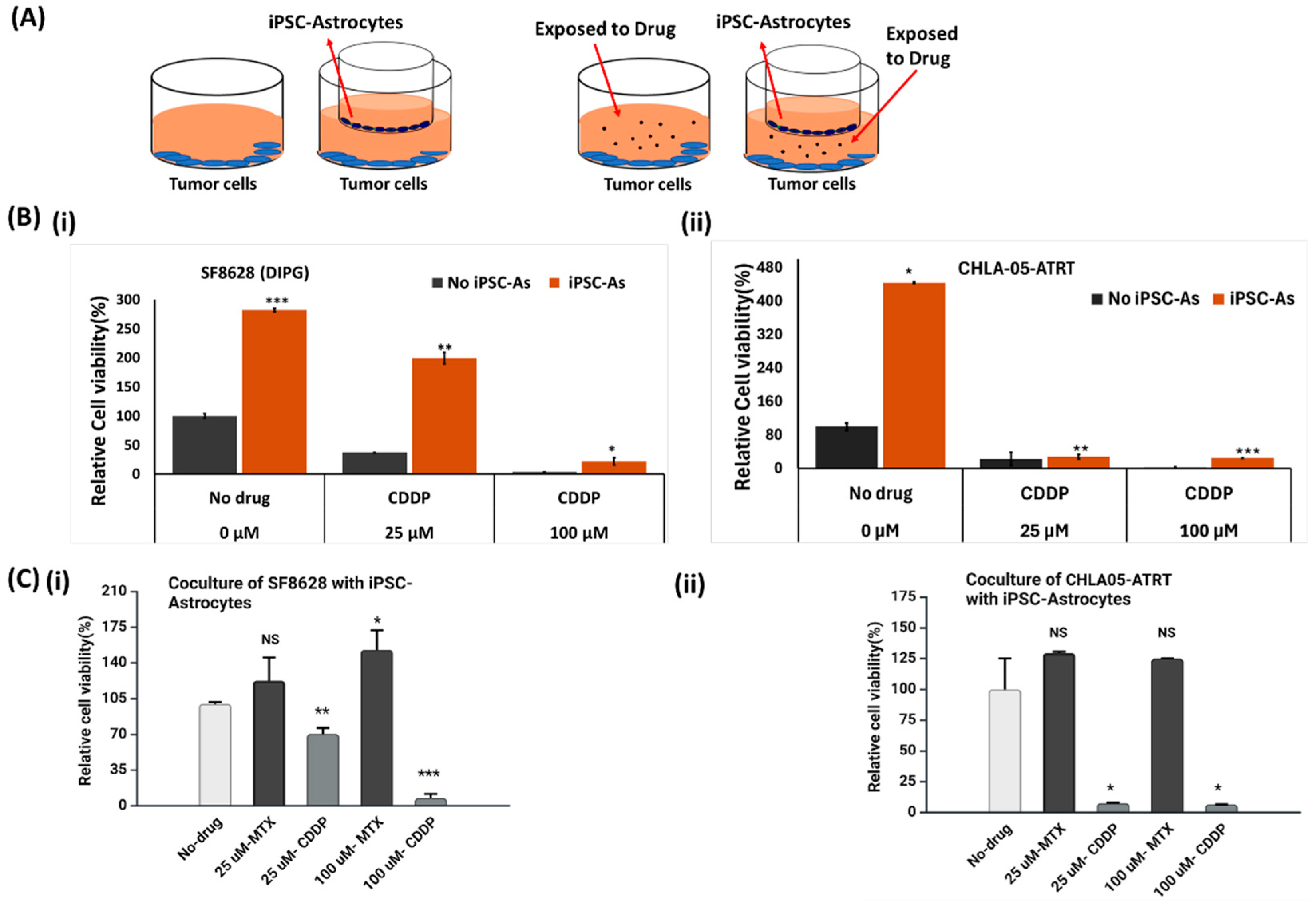
3.2. Cytotoxic Effect of Anti-Cancer Drugs on Malignant Pediatric Brain Tumor Cells
3.2.1. Cisplatin or Cis-Diamminedichloroplatinum (II)
3.2.2. Methotrexate
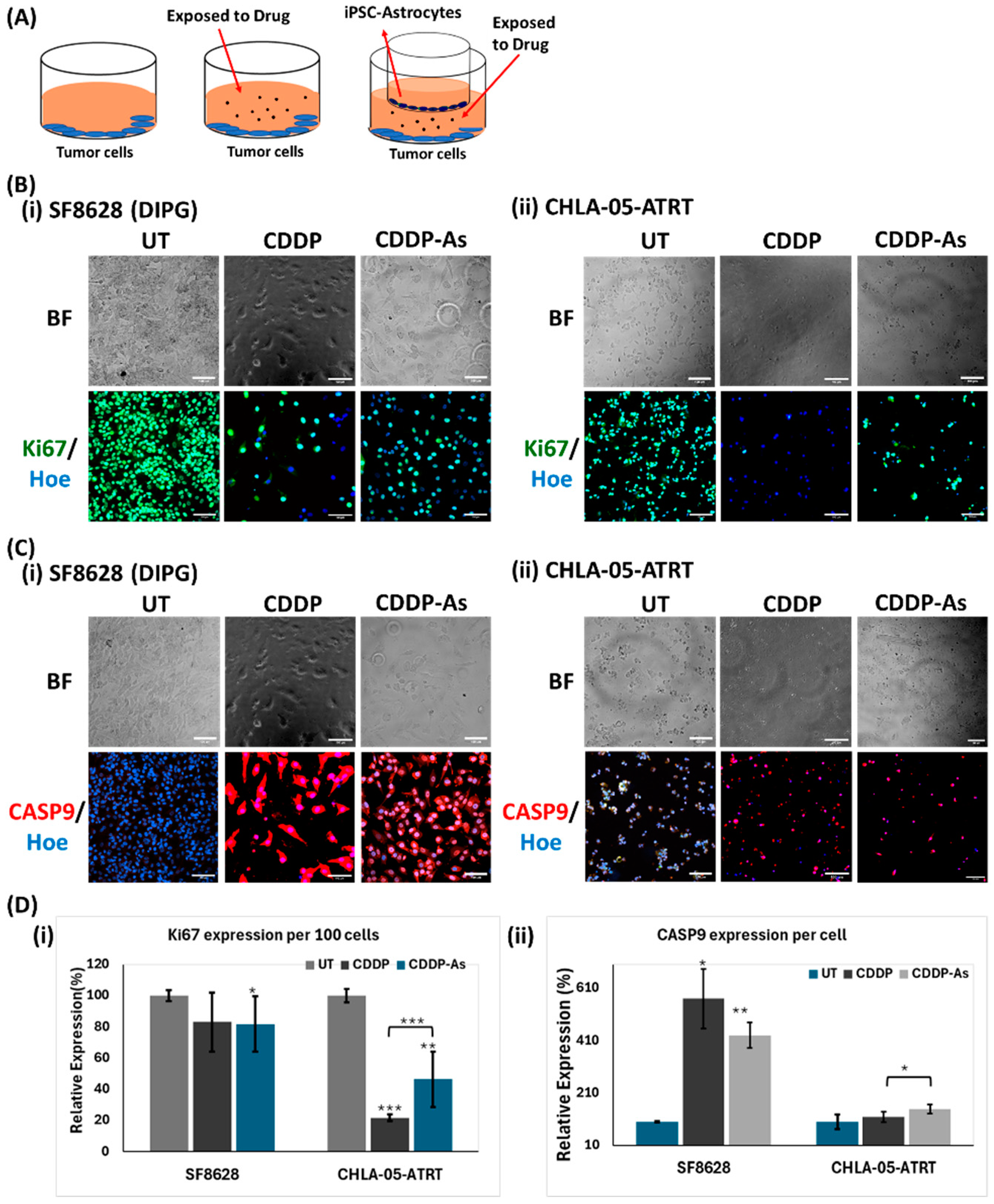
3.3. Activity of Anti-Tumor Drugs in ATRT and DIPG Cells Cocultured with iPSC-Astrocytes
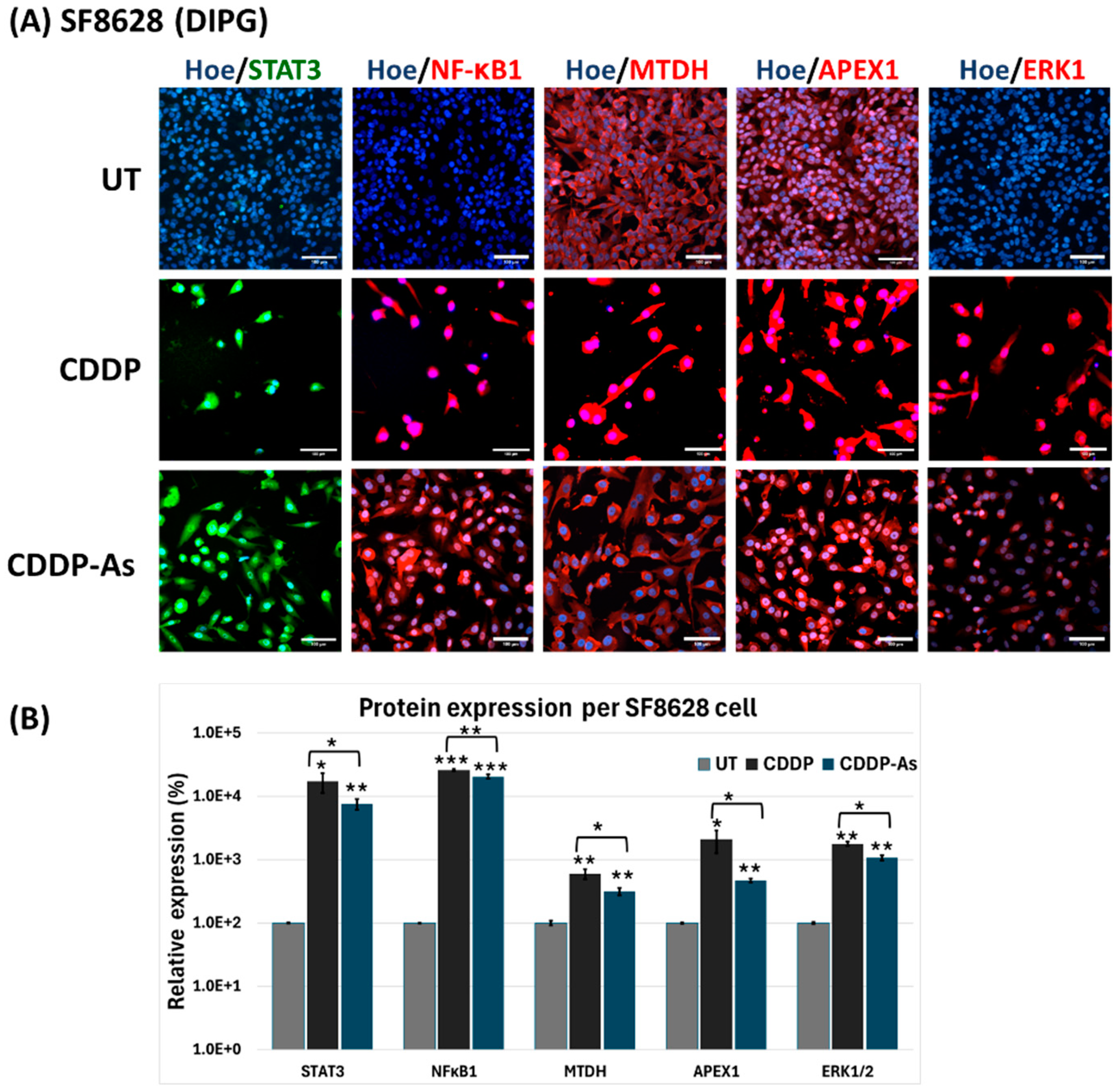
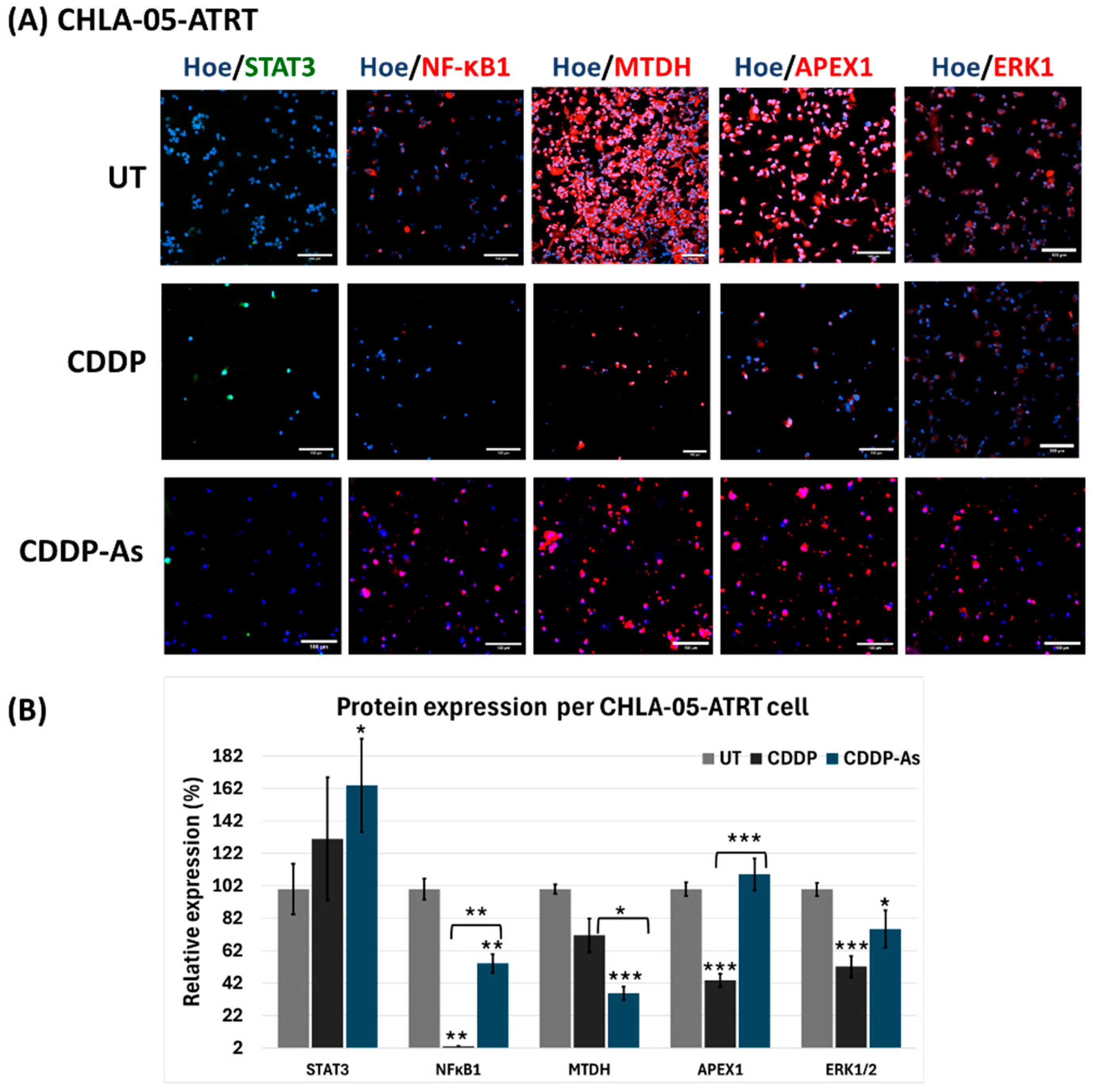
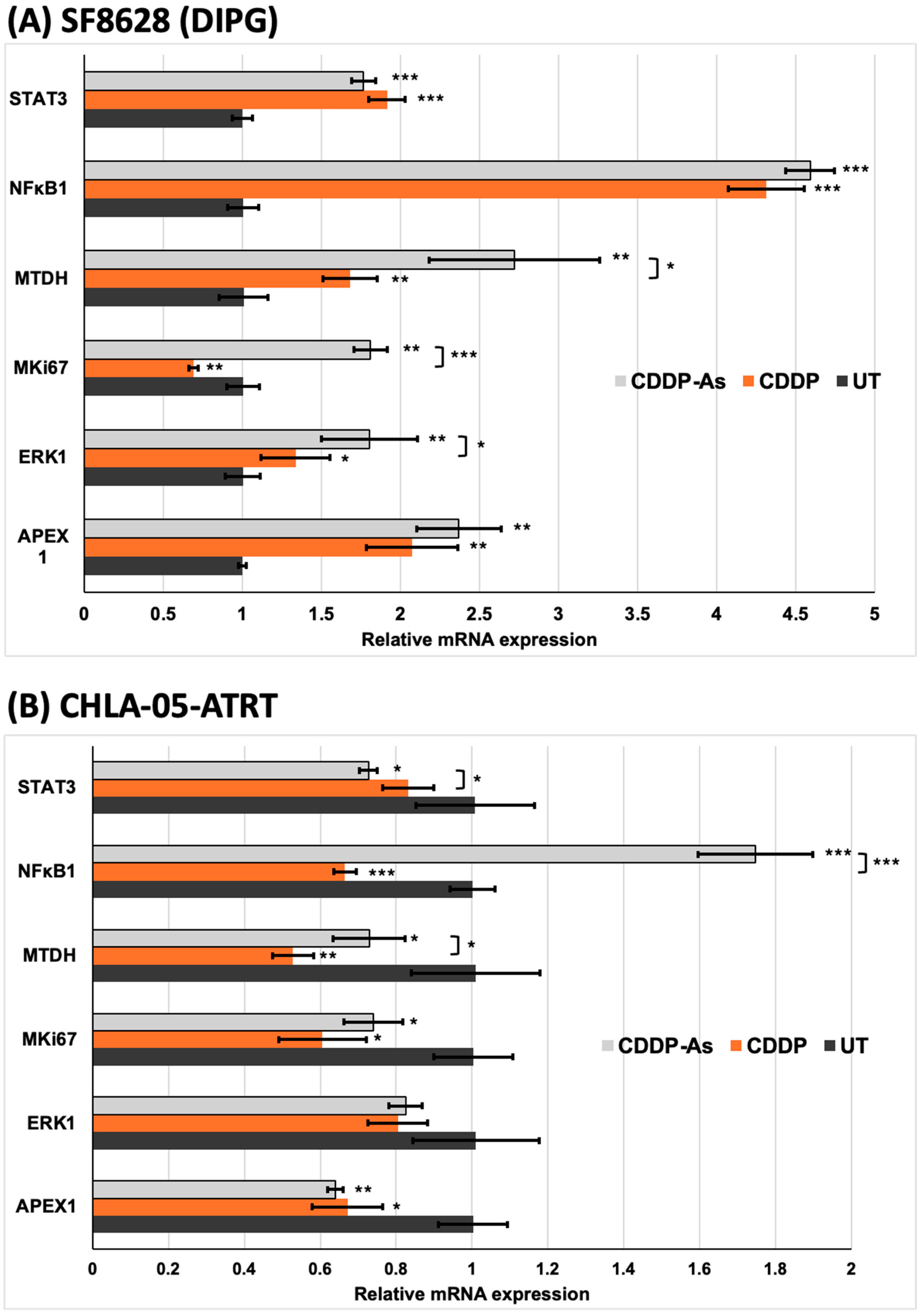
3.4. iPSC-Astrocytes Modulated Cisplatin Resistance and Proliferation Pathways in ATRT and DIPG Cells
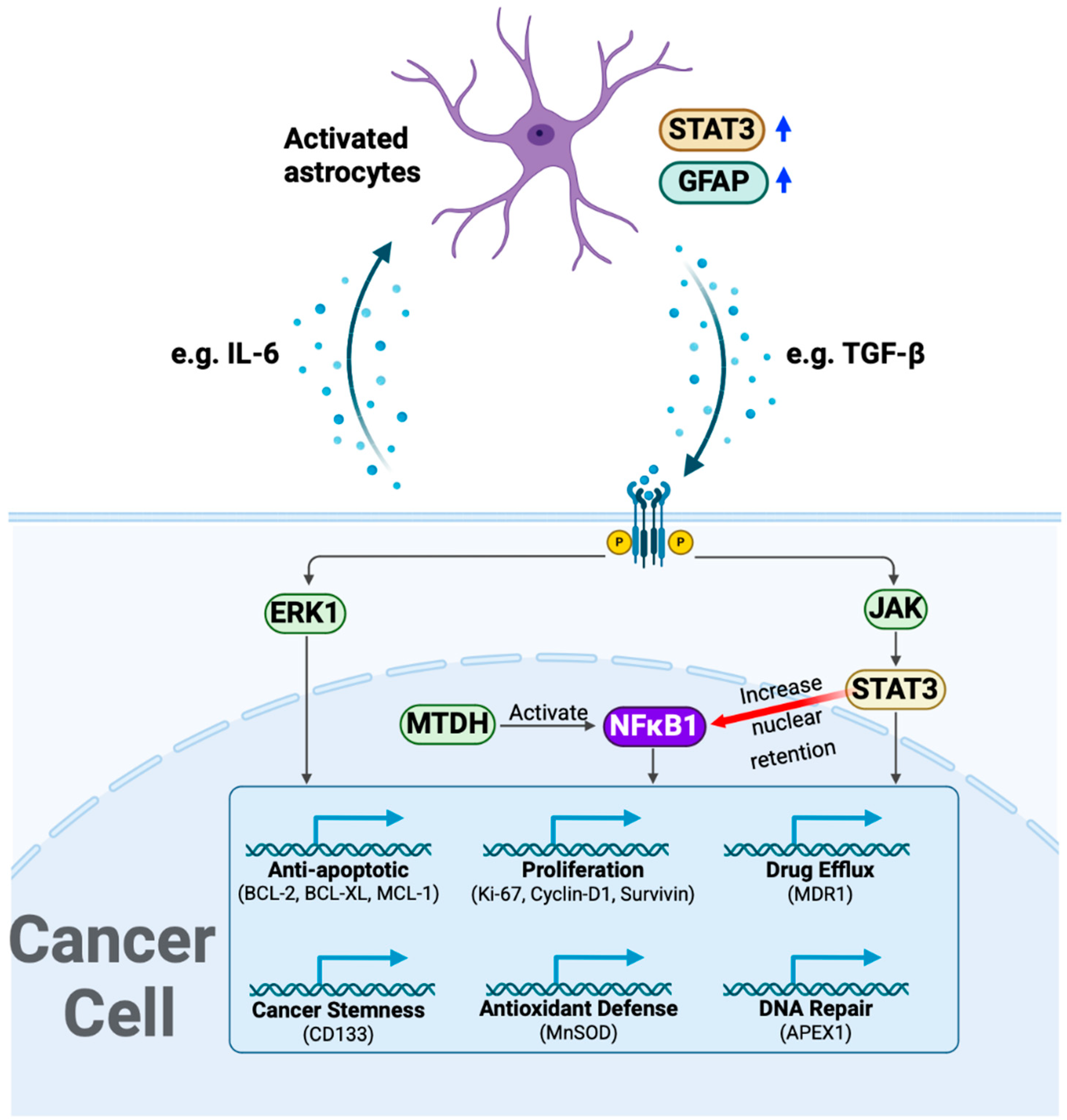
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roehrig, A.; Indelicato, D.J.; Paulino, A.C.; Ermoian, R.; Hartsell, W.; Perentesis, J.; Hill-Kayser, C.; Lee, J.Y.; Laack, N.N.; Mangona, V.; et al. Radiotherapy for Atypical Teratoid/Rhabdoid Tumor (ATRT) on the Pediatric Proton/Photon Consortium Registry (PPCR). J. Neurooncol. 2023, 162, 353–362. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e7. [Google Scholar] [CrossRef] [PubMed]
- Gastberger, K.; Fincke, V.E.; Mucha, M.; Siebert, R.; Hasselblatt, M.; Fruhwald, M.C. Current Molecular and Clinical Landscape of ATRT—The Link to Future Therapies. Cancer Manag. Res. 2023, 15, 1369–1393. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.J.; Hukin, J.; Rassekh, R.; Fryer, C.; Kim, J.; Stemmer-Rachamimov, A.; Birks, D.K.; Huang, A.; Yip, S.; Dunham, C. Atypical Teratoid Rhabdoid Tumors (ATRTs): The British Columbia’s Children’s Hospital’s Experience, 1986–2006. Brain Pathol. 2012, 22, 625–635. [Google Scholar] [CrossRef]
- Lobon-Iglesias, M.J.; Andrianteranagna, M.; Han, Z.Y.; Chauvin, C.; Masliah-Planchon, J.; Manriquez, V.; Tauziede-Espariat, A.; Turczynski, S.; Bouarich-Bourimi, R.; Frah, M.; et al. Imaging and Multi-Omics Datasets Converge to Define Different Neural Progenitor Origins for ATRT-SHH Subgroups. Nat. Commun. 2023, 14, 6669. [Google Scholar] [CrossRef] [PubMed]
- Keane, L.; Cheray, M.; Saidi, D.; Kirby, C.; Friess, L.; Gonzalez-Rodriguez, P.; Gerdes, M.E.; Grabert, K.; McColl, B.W.; Joseph, B. Inhibition of Microglial EZH2 Leads to Anti-Tumoral Effects in Pediatric Diffuse Midline Gliomas. Neurooncol. Adv. 2021, 3, vdab096. [Google Scholar] [CrossRef]
- Katagi, H.; Takata, N.; Aoi, Y.; Zhang, Y.; Rendleman, E.J.; Blyth, G.T.; Eckerdt, F.D.; Tomita, Y.; Sasaki, T.; Saratsis, A.M.; et al. Therapeutic Targeting of Transcriptional Elongation in Diffuse Intrinsic Pontine Glioma. Neuro-oncology 2021, 23, 1348–1359. [Google Scholar] [CrossRef]
- Vanan, M.I.; Eisenstat, D.D. DIPG in Children—What Can We Learn from the Past? Front. Oncol. 2015, 5, 237. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Monje, M. Diffuse Intrinsic Pontine Glioma: From Diagnosis to Next-Generation Clinical Trials. Curr. Treat. Options Neurol. 2019, 21, 37. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef]
- Egiz, A.; Kannan, S.; Asl, S.F. The Impact of Surgical Resection and Adjuvant Therapy on Survival in Pediatric Patients with Atypical Teratoid/Rhabdoid Tumor: Systematic Review and Pooled Survival Analysis. World Neurosurg. 2022, 164, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.G.; Ruggiero, A.; Centonze, A.; Carrieri, A.; Ferorelli, S.; Scilimati, A. Diffuse Intrinsic Pontine Glioma (DIPG): Breakthrough and Clinical Perspective. Curr. Med. Chem. 2021, 28, 3287–3317. [Google Scholar] [CrossRef]
- Verma, V.; Johnson, C.P.; Bennion, N.R.; Bhirud, A.R.; Li, S.; McComb, R.D.; Lin, C. Atypical Teratoid Rhabdoid Tumor: Long-Term Survival after Chemoradiotherapy. Child’s Nerv. Syst. 2015, 31, 37. [Google Scholar] [CrossRef] [PubMed]
- Malik, J.R.; Podany, A.T.; Khan, P.; Shaffer, C.L.; Siddiqui, J.A.; Baranowska-Kortylewicz, J.; Le, J.; Fletcher, C.V.; Ether, S.A.; Avedissian, S.N. Chemotherapy in Pediatric Brain Tumor and the Challenge of the Blood–Brain Barrier. Cancer Med. 2023, 12, 21075–21096. [Google Scholar] [CrossRef]
- Alva, E.; Rubens, J.; Chi, S.; Rosenberg, T.; Reddy, A.; Raabe, E.H.; Margol, A. Recent Progress and Novel Approaches to Treating Atypical Teratoid Rhabdoid Tumor. Neoplasia 2023, 37, 100880. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The Rediscovery of Platinum-Based Cancer Therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Liu, Q.H.; Shi, M.L.; Sun, C.; Bai, J.; Zheng, J.N. Role of the ERK1/2 Pathway in Tumor Chemoresistance and Tumor Therapy. Bioorg. Med. Chem. Lett. 2015, 25, 192–197. [Google Scholar] [CrossRef]
- Meng, X.; Thiel, K.W.; Leslie, K.K. Chapter Five—Drug Resistance Mediated by AEG-1/MTDH/LYRIC. In Advances in Cancer Research; Sarkar, D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 120, pp. 135–157. ISBN 0065-230X. [Google Scholar]
- Sriramulu, S.; Sun, X.F.; Malayaperumal, S.; Ganesan, H.; Zhang, H.; Ramachandran, M.; Banerjee, A.; Pathak, S. Emerging Role and Clinicopathological Significance of AEG-1 in Different Cancer Types: A Concise Review. Cells 2021, 10, 1497. [Google Scholar] [CrossRef]
- Osama, M.; Mostafa, M.N.; Alvi, M.A. Astrocyte Elevated Gene-1 as a Novel Therapeutic Target in Malignant Gliomas and Its Interactions with Oncogenes and Tumor Suppressor Genes. Brain Res. 2020, 1747, 147034. [Google Scholar] [CrossRef] [PubMed]
- Grundy, G.J.; Parsons, J.L. Base Excision Repair and Its Implications to Cancer Therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Placone, A.L.; Quinones-Hinojosa, A.; Searson, P.C. The Role of Astrocytes in the Progression of Brain Cancer: Complicating the Picture of the Tumor Microenvironment. Tumour Biol. 2016, 37, 61–69. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the Rising Stars of the Glioblastoma Microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Lin, Q.; Balasubramanian, K.; Fan, D.; Kim, S.J.; Guo, L.; Wang, H.; Bar-Eli, M.; Aldape, K.D.; Fidler, I.J. Reactive Astrocytes Protect Melanoma Cells from Chemotherapy by Sequestering Intracellular Calcium through Gap Junction Communication Channels. Neoplasia 2010, 12, 748–754. [Google Scholar] [CrossRef]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-Associated Reactive Astrocytes Aid the Evolution of Immunosuppressive Environment in Glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef]
- O’Brien, E.; Howarth, C.; Sibson, N.R. The Role of Astrocytes in CNS Tumors: Pre-Clinical Models and Novel Imaging Approaches. Front. Cell. Neurosci. 2013, 7, 40. [Google Scholar] [CrossRef]
- Porter, H.D.; McCrorie, P.; Diksin, M.; Moloney, C.; Grundy, R.; Rahman, R. ATRT-01. MODELLING ATYPICAL TERATOID/ RHABDOID TUMOUR AND HEALTHY BRAIN CROSSTALK. Neuro-oncology 2024, 26, 0. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, J.S.; Park, E.S.; Lee, J.S.; Lin, Q.; Langley, R.R.; Maya, M.; He, J.; Kim, S.W.; Weihua, Z.; et al. Astrocytes Upregulate Survival Genes in Tumor Cells and Induce Protection from Chemotherapy. Neoplasia 2011, 13, 286. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; Ransohoff, R.M.; McMahon, J.T.; Jacobs, B.S.; Barna, B.P. Characterization of Adult Human Astrocytes Derived from Explant Culture. J. Neurosci. Res. 1990, 27, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Loss of Wild-Type P53 Bestows a Growth Advantage on Primary Cortical Astrocytes and Facilitates Their in Vitro Transformation—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/7796398/ (accessed on 20 January 2025).
- Lattke, M.; Guillemot, F. Understanding Astrocyte Differentiation: Clinical Relevance, Technical Challenges, and New Opportunities in the Omics Era. WIREs Mech. Dis. 2022, 14, e1557. [Google Scholar] [CrossRef]
- Khamis, Z.I.; Sarker, D.B.; Xue, Y.; Al-Akkary, N.; James, V.D.; Zeng, C.; Li, Y.; Sang, Q.X.A. Modeling Human Brain Tumors and the Microenvironment Using Induced Pluripotent Stem Cells. Cancers 2023, 15, 1253. [Google Scholar] [CrossRef]
- Jessa, S.; Blanchet-Cohen, A.; Krug, B.; Vladoiu, M.; Coutelier, M.; Faury, D.; Poreau, B.; De Jay, N.; Hébert, S.; Monlong, J.; et al. Stalled Developmental Programs at the Root of Pediatric Brain Tumors. Nat. Genet. 2019, 51, 1702. [Google Scholar] [CrossRef] [PubMed]
- Malone, K.; Dugas, M.; Earl, N.; Alain, T.; LaCasse, E.C.; Beug, S.T. Astrocytes and the Tumor Microenvironment Inflammatory State Dictate the Killing of Glioblastoma Cells by Smac Mimetic Compounds. Cell Death Dis. 2024, 15, 592. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.; Cui, B.; Liu, Z.; Shen, H. Novel Insights into Astrocyte-Mediated Signaling of Proliferation, Invasion and Tumor Immune Microenvironment in Glioblastoma. Biomed. Pharmacother. 2020, 126, 110086. [Google Scholar] [CrossRef]
- Lazic, A.; Balint, V.; Ninkovic, D.S.; Peric, M.; Stevanovic, M. Reactive and Senescent Astroglial Phenotypes as Hallmarks of Brain Pathologies. Int. J. Mol. Sci. 2022, 23, 4995. [Google Scholar] [CrossRef]
- Kiran, S.; Xue, Y.; Sarker, D.B.; Li, Y.; Sang, Q.A. Feeder-Free Differentiation of Human IPSCs into Natural Killer Cells with Cytotoxic Potential against Malignant Brain Rhabdoid Tumor Cells. Bioact. Mater. 2024, 36, 301–316. [Google Scholar] [CrossRef]
- Hua, T.; Xue, Y.; Sarker, D.B.; Kiran, S.; Li, Y.; Sang, Q.A. Modeling Human Brain Rhabdoid Tumor by Inactivating Tumor Suppressor Genes in Induced Pluripotent Stem Cells. Bioact. Mater. 2024, 31, 136–150. [Google Scholar] [CrossRef]
- Griffin, K.; Bejoy, J.; Song, L.; Hua, T.; Marzano, M.; Jeske, R.; Sang, Q.X.A.; Li, Y. Human Stem Cell-Derived Aggregates of Forebrain Astroglia Respond to Amyloid Beta Oligomers. Tissue Eng. Part A 2020, 26, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Blagosklonny, M. V Analysis of FDA Approved Anticancer Drugs Reveals the Future of Cancer Therapy. Cell Cycle 2004, 3, 1033–1040. [Google Scholar] [CrossRef]
- Wu, S.; Shah, D.K. Determination of ADC Cytotoxicity in Immortalized Human Cell Lines. In Antibody-Drug Conjugates; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2078. [Google Scholar]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, Mechanisms and Clinical Application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef] [PubMed]
- Uxa, S.; Castillo-Binder, P.; Kohler, R.; Stangner, K.; Müller, G.A.; Engeland, K. Ki-67 Gene Expression. Cell Death Differ. 2021, 28, 3357–3370. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common Markers of Proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef]
- Bentayebi, K.; El Aked, R.; Ezzahidi, O.; Alami, A.B.; Louati, S.; Ouadghiri, M.; Aanniz, T.; Amzazi, S.; Belyamani, L.; Ibrahimi, A.; et al. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in DIPG: A Review of Current and Future Strategies. Brain Disord. 2024, 14, 100132. [Google Scholar] [CrossRef]
- Hu, Y.; Li, Z.; Zhang, Y.; Wu, Y.; Liu, Z.; Zeng, J.; Hao, Z.; Li, J.; Ren, J.; Yao, M. The Evolution of Tumor Microenvironment in Gliomas and Its Implication for Target Therapy. Int. J. Biol. Sci. 2023, 19, 4311–4326. [Google Scholar] [CrossRef]
- Messiaen, J.; Jacobs, S.A.; De Smet, F. The Tumor Micro-Environment in Pediatric Glioma: Friend or Foe? Front. Immunol. 2023, 14, 1227126. [Google Scholar] [CrossRef]
- Gonzalez Castro, L.N.; Liu, I.; Filbin, M. Characterizing the Biology of Primary Brain Tumors and Their Microenvironment via Single-Cell Profiling Methods. Neuro-oncology 2023, 25, 234–247. [Google Scholar] [CrossRef]
- Sait, S.F.; Giantini-Larsen, A.M.; Tringale, K.R.; Souweidane, M.M.; Karajannis, M.A. Treatment of Pediatric Low-Grade Gliomas. Curr. Neurol. Neurosci. Rep. 2023, 23, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Dalianis, T.; Lukoseviciute, M.; Holzhauser, S.; Kostopoulou, O.N. New Approaches Towards Targeted Therapy for Childhood Neuroblastoma. Anticancer Res. 2023, 43, 3829–3839. [Google Scholar] [CrossRef]
- Cui, Y.; Lee, P.; Reardon, J.J.; Wang, A.; Lynch, S.; Otero, J.J.; Sizemore, G.; Winter, J.O. Evaluating Glioblastoma Tumour Sphere Growth and Migration in Interaction with Astrocytes Using 3D Collagen-Hyaluronic Acid Hydrogels. J. Mater. Chem. B 2023, 11, 5442–5459. [Google Scholar] [CrossRef]
- Almad, A.; Maragakis, N.J. A Stocked Toolbox for Understanding the Role of Astrocytes in Disease. Nat. Rev. Neurol. 2018, 14, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Rota, C.; Segal, R.A. Astrocytes Promote Pediatric Glioma Progression and Enhance Ex Vivo Tumor Modeling. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 2022. [Google Scholar]
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of Gp130-Related Cytokines and Activation of JAK2/STAT3 Pathway in Astrocytes Precedes Up-Regulation of Glial Fibrillary Acidic Protein in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Neurodegeneration: KEY SIGNALING PATHWAY FOR ASTROGLIOSIS IN VIVO? J. Biol. Chem. 2004, 279, 19936–19947. [Google Scholar] [CrossRef]
- Lu, Y.; He, M.; Zhang, Y.; Xu, S.; Zhang, L.; He, Y.; Chen, C.; Liu, C.; Pi, H.; Yu, Z.; et al. Differential Pro-Inflammatory Responses of Astrocytes and Microglia Involve STAT3 Activation in Response to 1800 MHz Radiofrequency Fields. PLoS ONE 2014, 9, e108318. [Google Scholar] [CrossRef]
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of Tumor Cells and Astrocytes Promotes Breast Cancer Brain Metastases through TGF-Β2/ANGPTL4 Axes. NPJ Precis. Oncol. 2019, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Ávila, M.; Gonzalez, J.; El-Bachá, R.S.; Báez, E.; García-Segura, L.M.; Coronel, J.C.J.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic Modulation of Blood Brain Barrier: Perspectives on Parkinson’s Disease. Front. Cell. Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef]
- Chi, G.; Lu, J.; He, T.; Wang, Y.; Zhou, X.; Zhang, Y.; Qiu, L. High Mobility Group Box-1 Protein Promotes Astrocytic CCL5 Production through the MAPK/NF-ΚB Pathway Following Spinal Cord Injury. Sci. Rep. 2024, 14, 22344. [Google Scholar] [CrossRef]
- Liu, W.-H.; Chen, M.-T.; Wang, M.-L.; Lee, Y.-Y.; Chiou, G.-Y.; Chien, C.-S.; Huang, P.-I.; Chen, Y.-W.; Huang, M.-C.; Chiou, S.-H.; et al. Cisplatin-Selected Resistance Is Associated with Increased Motility and Stem-like Properties via Activation of STAT3/Snail Axis in Atypical Teratoid/Rhabdoid Tumor Cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [CrossRef]
- Yang, K.; Luan, L.; Li, X.; Sun, X.; Yin, J. ERK Inhibition in Glioblastoma Is Associated with Autophagy Activation and Tumorigenesis Suppression. J. Neurooncol. 2022, 156, 123–137. [Google Scholar] [CrossRef]
- Ullah, A.; Leong, S.W.; Wang, J.; Wu, Q.; Ghauri, M.A.; Sarwar, A.; Su, Q.; Zhang, Y. Cephalomannine Inhibits Hypoxia-Induced Cellular Function via the Suppression of APEX1/HIF-1α Interaction in Lung Cancer. Cell Death Dis. 2021, 12, 490. [Google Scholar] [CrossRef] [PubMed]
- Woessmann, W.; Chen, X.; Borkhardt, A. Ras-Mediated Activation of ERK by Cisplatin Induces Cell Death Independently of P53 in Osteosarcoma and Neuroblastoma Cell Lines. Cancer Chemother. Pharmacol. 2002, 50, 397–404. [Google Scholar] [CrossRef]
- Concetti, J.; Wilson, C.L. NFKB1 and Cancer: Friend or Foe? Cells 2018, 7, 133. [Google Scholar] [CrossRef]
- Gaptulbarova, K.A.; Tsyganov, M.M.; Pevzner, A.M.; Ibragimova, M.K.; Litviakov, N. V NF-KB as a Potential Prognostic Marker and a Candidate for Targeted Therapy of Cancer. Exp. Oncol. 2020, 42, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; Lucks, P.; Borghouts, C. The Function of Stat3 in Tumor Cells and Their Microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug Resistance via Feedback Activation of Stat3 in Oncogene-Addicted Cancer Cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a Target for Inducing Apoptosis in Solid and Hematological Tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent Activation of Stat3 Signaling Induces Survivin Gene Expression and Confers Resistance to Apoptosis in Human Breast Cancer Cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, W.; Bai, X.; Liu, Y. Targeting the Transcriptional Activity of STAT3 by a Novel Fusion Protein. BMC Cancer 2022, 22, 751. [Google Scholar] [CrossRef]
- Lin, W.; Sun, J.; Sadahira, T.; Xu, N.; Wada, K.; Liu, C.; Araki, M.; Xu, A.; Watanabe, M.; Nasu, Y.; et al. Discovery and Validation of Nitroxoline as a Novel STAT3 Inhibitor in Drug-Resistant Urothelial Bladder Cancer. Int. J. Biol. Sci. 2021, 17, 3255–3267. [Google Scholar] [CrossRef]
- Abyaneh, H.S.; Gupta, N.; Radziwon-Balicka, A.; Jurasz, P.; Seubert, J.; Lai, R.; Lavasanifar, A. STAT3 but Not HIF-1α Is Important in Mediating Hypoxia-Induced Chemoresistance in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancers 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, J.; Tang, M.; Zhang, K.; Gao, X.; Fang, L.; Liu, W. STAT3 and P63 in the Regulation of Cancer Stemness. Front. Genet. 2022, 13, 909251. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Fuchs, B.C.; Tanabe, K.K. STAT3 Is a Key Transcriptional Regulator of Cancer Stem Cell Marker CD133 in HCC. Hepatobiliary Surg. Nutr. 2016, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Tschopp, J. Signals from within: The DNA-Damage-Induced NF-ΚB Response. Cell Death Differ. 2006, 13, 773–784. [Google Scholar] [CrossRef]
- Ros, J.E.; Schuetz, J.D.; Geuken, M.; Streetz, K.; Moshage, H.; Kuipers, F.; Manns, M.P.; Jansen, P.L.M.; Trautwein, C.; Müller, M. Induction of Mdr1b Expression by Tumor Necrosis Factor-α in Rat Liver Cells Is Independent of P53 but Requires NF-ΚB Signaling. Hepatology 2001, 33, 1425–1431. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Barbu, V.; Fillet, M.; Chariot, A.; Relic, B.; Jacobs, N.; Gielen, J.; Merville, M.P.; Bours, V. NF-ΚB Transcription Factor Induces Drug Resistance through MDR1 Expression in Cancer Cells. Oncogene 2003, 22, 90–97. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2010, 21, 103–115. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently Activated Stat3 Maintains Constitutive NF-ΚB Activity in Tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-ΚB and STAT3 Signaling Pathways Collaboratively Link Inflammation to Cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, C.; Zhang, C.; Li, Z.; Zhu, T.; Chen, J.; Ren, Y.; Wang, X.; Zhang, L.; Zhou, X. TGF-β-Induced STAT3 Overexpression Promotes Human Head and Neck Squamous Cell Carcinoma Invasion and Metastasis through Malat1/MiR-30a Interactions. Cancer Lett. 2018, 436, 52–62. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain Tumour Cells Interconnect to a Functional and Resistant Network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jin, X.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; et al. Tumoral RANKL Activates Astrocytes That Promote Glioma Cell Invasion through Cytokine Signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; López-Picazo, J.M.; Toledo, G.; Lozano, M.D.; Torre, W.; Garcia-Corchón, C.; Quero, C.; Soria, J.C.; Martín-Algarra, S.; Manzano, R.G.; et al. ERK 1/2 Is Activated in Non-Small-Cell Lung Cancer and Associated with Advanced Tumours. Br. J. Cancer 2004, 90, 1047–1052. [Google Scholar] [CrossRef]
- Balmanno, K.; Cook, S.J. Tumour Cell Survival Signalling by the ERK1/2 Pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. ROLES OF THE RAF/MEK/ERK PATHWAY IN CELL GROWTH, MALIGNANT TRANSFORMATION AND DRUG RESISTANCE. Biochim. Biophys. Acta 2006, 1773, 1263. [Google Scholar] [CrossRef] [PubMed]
- Ki67 Is a Promising Molecular Target in the Diagnosis of Cancer (Review). Available online: https://www.spandidos-publications.com/10.3892/mmr.2014.2914 (accessed on 20 November 2024).
- Wu, S.Y.; Liao, P.; Yan, L.Y.; Zhao, Q.Y.; Xie, Z.Y.; Dong, J.; Sun, H.T. Correlation of MKI67 with Prognosis, Immune Infiltration, and T Cell Exhaustion in Hepatocellular Carcinoma. BMC Gastroenterol. 2021, 21, 416. [Google Scholar] [CrossRef]
- Liu, Z.M.; Bao, Y.; Li, T.K.; Di, Y.B.; Song, W.J. MKI67 an Potential Oncogene of Oral Squamous Cell Carcinoma via the High Throughput Technology. Medicine 2022, 101, e32595. [Google Scholar] [CrossRef]
- He, H.; Song, F.; Gao, Q.; Lu, Z.; Yuan, Y.; Li, X.; Chen, L.; Jia, C.; Yang, R.; Yang, J.; et al. The APEX1/MiRNA-27a-5p Axis Plays Key Roles in Progression, Metastasis and Targeted Chemotherapy of Gastric Cancer. Int. J. Pharm. 2021, 599, 120446. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Song, L.; Xiong, H.; Wang, L.; Yan, X.; Yuan, J.; Wu, J.; Li, M. Astrocyte Elevated Gene-1 Is a Proliferation Promoter in Breast Cancer via Suppressing Transcriptional Factor FOXO1. Oncogene 2009, 28, 3188–3196. [Google Scholar] [CrossRef]
- Manna, D.; Sarkar, D. Multifunctional Role of Astrocyte Elevated Gene-1 (Aeg-1) in Cancer: Focus on Drug Resistance. Cancers 2021, 13, 1792. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, R.; Zhang, Y. MiR-296-3p Targets APEX1 to Suppress Cell Migration and Invasion of Non-Small-Cell Lung Cancer. Oncol. Lett. 2019, 18, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 Biology into Effective Treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef]
- Kim, B.G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel Therapies Emerging in Oncology to Target the TGF-β Pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef] [PubMed]
- Parsels, L.A.; Wahl, D.R.; Koschmann, C.; Morgan, M.A.; Zhang, Q. Developing H3K27M Mutant Selective Radiosensitization Strategies in Diffuse Intrinsic Pontine Glioma. Neoplasia 2023, 37, 100881. [Google Scholar] [CrossRef]
- Entz-werlé, N.; Poidevin, L.; Nazarov, P.V.; Poch, O.; Lhermitte, B.; Chenard, M.P.; Burckel, H.; Guérin, E.; Fuchs, Q.; Castel, D.; et al. A DNA Repair and Cell Cycle Gene Expression Signature in Pediatric High-grade Gliomas: Prognostic and Therapeutic Value. Cancers 2021, 13, 2252. [Google Scholar] [CrossRef]
- Hol, E.M.; Pekny, M. Glial Fibrillary Acidic Protein (GFAP) and the Astrocyte Intermediate Filament System in Diseases of the Central Nervous System. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the Glial Scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Sherman, L.S.; Back, S.A. A “GAG” Reflex Prevents Repair of the Damaged CNS. Trends Neurosci. 2008, 31, 44–52. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte Intermediate Filaments in CNS Pathologies and Regeneration. J. Pathol. 2004, 204, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2012, 13, 24–57. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiran, S.; Xue, Y.; Sarker, D.B.; Sang, Q.-X.A. Effects of Induced Pluripotent Stem Cell-Derived Astrocytes on Cisplatin Sensitivity in Pediatric Brain Cancer Cells. Cancers 2025, 17, 997. https://doi.org/10.3390/cancers17060997
Kiran S, Xue Y, Sarker DB, Sang Q-XA. Effects of Induced Pluripotent Stem Cell-Derived Astrocytes on Cisplatin Sensitivity in Pediatric Brain Cancer Cells. Cancers. 2025; 17(6):997. https://doi.org/10.3390/cancers17060997
Chicago/Turabian StyleKiran, Sonia, Yu Xue, Drishty B. Sarker, and Qing-Xiang Amy Sang. 2025. "Effects of Induced Pluripotent Stem Cell-Derived Astrocytes on Cisplatin Sensitivity in Pediatric Brain Cancer Cells" Cancers 17, no. 6: 997. https://doi.org/10.3390/cancers17060997
APA StyleKiran, S., Xue, Y., Sarker, D. B., & Sang, Q.-X. A. (2025). Effects of Induced Pluripotent Stem Cell-Derived Astrocytes on Cisplatin Sensitivity in Pediatric Brain Cancer Cells. Cancers, 17(6), 997. https://doi.org/10.3390/cancers17060997