Recent Developments in Differentiation Therapy of Acute Myeloid Leukemia



Simple Summary
Abstract
1. Introduction
2. IDH Inhibitors
2.1. Biology and Mechanisms of IDH Mutations
2.2. Small Molecule Inhibitors of IDH1: Ivosidenib
2.3. Clinical Studies with Ivosidenib
2.4. Small Molecule Inhibitors of IDH1: Olutasidenib
2.5. Clinical Studies with Olutasidenib
2.6. Small Molecule Inhibitor of IDH2: Enasidenib
2.7. Clinical Studies with Enasidenib
2.8. Differentiation Syndrome in Patients Treated with IDH Inhibitors
3. Inhibitors of Lysine-Specific Demethylase 1 (LSD1 or KMD1A)
3.1. Role of LSD1 Inhibitors in HSC Differentiation and in Myeloid Leukemia
3.2. Small Molecule Inhibitors of LSD1
3.3. Clinical Trials with LSD1 Inhibitors
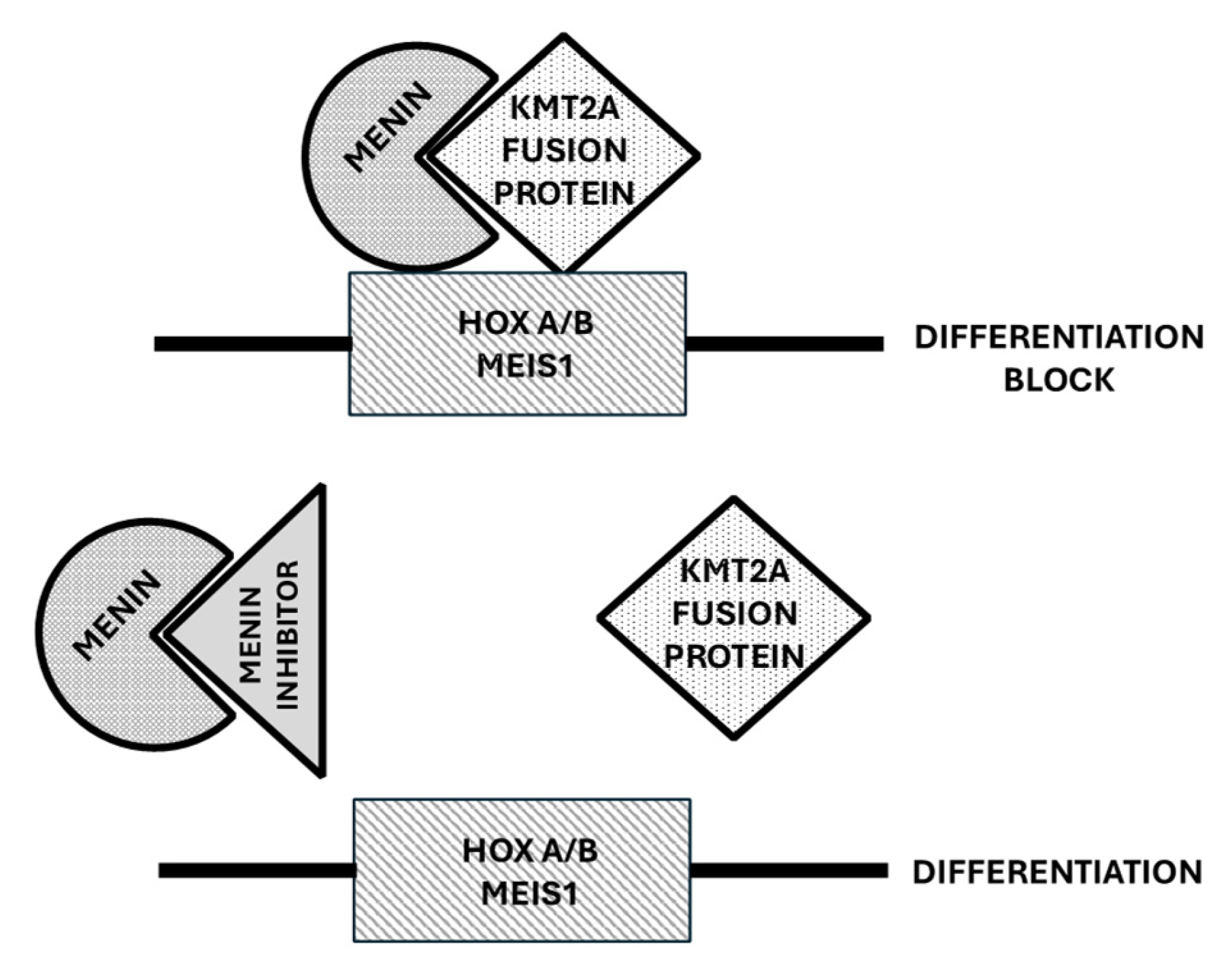
4. Menin Inhibitors
4.1. Role of Menin in the Control of Normal and Leukemic Hematopoiesis
4.2. Menin Inhibitors
4.3. Clinical Studies with Menin Inhibitors
4.3.1. Revumenib
4.3.2. Bleximenib
4.3.3. DSPP-5336 (Enzomenib)
4.3.4. KO-539 (Ziftomenib)
4.3.5. BMF-219 (Icovamenib)
4.3.6. Menin Inhibitors in AML Patients with NUP98 Rearrangements
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Breitman, T.R.; Collins, S.J.; Keene, B.R. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood 1981, 57, 100–1004. [Google Scholar] [CrossRef]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhou, L.; Gu, L.J.; Zhen, Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Degos, L.; Chomienne, C.; Daniel, M.T.; Berger, R.; Dombret, H.; Fenaux, P.; Castaigne, S. Treatment of first relapse in acute promyelocytic leukemia with all-trans retinoic acid. Lancet 1990, 336, 1440–1441. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Chomienne, C.; Daniel, M.T.; Ballerini, P.; Berger, R.; Fenaux, P.; Degos, L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia, I: Clinical results. Blood 1990, 76, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejan, A. The t(15;17) translocation of acute promyelocytic leukemia fuses the retinoic acid receptor alpha geneto a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Grignani, F.; Ferrucci, P.F.; Testa, U.; Talamo, G.; Fagioli, M.; Alacalay, M.; Mencarelli, A.; Grignani, F.; Peschle, C.; Nicoletti, I.; et al. The acute promyelocytic leukemia-specific PML-RARα fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993, 74, 423–431. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazzi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Isocitrate dehydrogenase mutations in myelodysplastic syndromes and in acute myeloid leukemia. Cancers 2020, 12, 2427. [Google Scholar] [CrossRef]
- Hoff, F.W.; Huang, Y.; Welkie, R.L.; Swords, R.T.; Traer, E.; Stein, E.M.; Lin, T.L.; Patel, P.A.; Collins, R.H.; Baer, M.R.; et al. IDH2 mutation is associated with favorable outcome among older adults with newly diagnosed acute myeloid leukemia treated with lower-intensity therapy. Blood 2024, 144 (Suppl. S1), 4325. [Google Scholar] [CrossRef]
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, T. IDH1-R132 changes vary according to NPM1 and other mutations status in AML. Leukemia 2019, 33, 1043–1047. [Google Scholar] [CrossRef]
- Zamegar-Lumley, S.; Alonzo, T.A.; Gerbing, R.B.; Othus, M.; Sun, Z.; Reis, R.E.; Wang, J.; Leonti, A.; Kutny, M.A.; Ostronoff, F.; et al. Characteristics and prognostic impact of IDH mutations in AML: A COG, SWOG and ECOG analysis. Blood Adv. 2023, 7, 5941–5953. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Heigth, J.; et al. IDH1(R132H) mutation increase murine hematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Buegess, K.; Bhagarva, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant Idh2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar]
- Wang, D.; Zheng, L.; Cheng, B.Y.L.; Sin, C.F.; Li, R.; Tsui, S.P.; Yi, X.; Ma, A.C.H.; He, B.L.; Leung, A.Y.H.; et al. Transgenic IDH2R172K and IDH2R140Q zebrafish model recapitulated features of human acute myeloid leukemia. Oncogene 2023, 42, 1272–1281. [Google Scholar] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagvat, N.; Vasannthakumar, A.; Fernandez, H.F.; et al. Leukemic Idh1 and Idh2 mutations result in a hypermethylation phenotype, disrupt Tet2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohe, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block of cell differentiation. Nature 2012, 483, 474–478. [Google Scholar]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, P.; Schneider, R.K.; McMahon, R.K.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G. (8R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar]
- Wang, F.; Trafins, J.; DeLa Barre, B.; Penard-Lacronique, V.; Schalm, V.; Hansen, E.; Starley, K.; Kewrnytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant Idh2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar]
- Pierangeli, S.; Donnini, S.; Ciaurro, V.; Milano, F.; Cardinali, V.; Sciabolacci, S.; Cimino, G.; Gianfriddo, I.; Ranieri, R.; Cipriani, S.; et al. The leukemic isocitrate dehydrogenase (IDH) 1/2 mutations impair myeloid and erythroid cell differentiation of primary human hematopoietic stem and progenitor cells (HSPCs). Cancers 2024, 16, 2675. [Google Scholar] [CrossRef]
- Landberg, N.; Koehnke, T.; Nakauchi, Y.; Fan, A.; Linde, M.H.; Karigane, D.; Thomas, D.; Majeti, R. Targeting Idh1-mutated pre-leukemic hematopoietic stem cells in myeloid disease, including CCUS and AML. Blood 2022, 140 (Suppl. S1), 2234–2235. [Google Scholar] [CrossRef]
- Landberg, N.; Köhnke, T.; Feng, Y.; Nakauchi, Y.; Fan, A.C.; Linde, M.H.; Karigane, D.; Lim, K.; Sinha, R.; Malcovati, L.; et al. Idh1-mutant preleukemic hematopoietic stem cells can be eliminated by inhibition of oxidative phosphorylation. Blood Cancer Discov. 2024, 5, 114–131. [Google Scholar]
- Wilson, E.R.; Helton, E.M.; Heath, S.E.; Fulton, S.R.; Payton, J.E.; Welch, J.S.; Walter, M.J.; Westervelt, P.; DiPersio, J.F.; Link, D.C.; et al. Focal disruption of DNA methylation dynamics at enhancers in IDH-Mutant AML cells. Leukemia 2022, 36, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, J.K.; Altman, A.S.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with ivodisenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [PubMed]
- Di Nardo, C.D.; Stein, E.M.; Pigneux, A.; Altman, J.K.; Collins, R.; Erba, E.P.; Watts, J.M.; Uy, G.L.; Winkler, T.; Wang, H.; et al. Outcomes of patients with IDH1-mutant relapsed or refractory acute myeloid leukemia receiving ivosidenib who proceeded to hematopoietic stem cell transplant. Leukemia 2021, 35, 3278–3281. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycha, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.P.; et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar]
- Woods, A.; Nosworthy, K.J.; Wang, X.; Vallejo, J.; Chow, E.C.Y.; Li, R.J.; Sun, J.; Charlab, R.; Jiang, X.; Pazdur, R.; et al. FDA approval summary: Ivosidenib in combination with azacitidine for treatment of patients with newly diagnosed acute myeloid leukemia with an IDH1 mutation. Clin. Cancer Res. 2024, 30, 1226–1231. [Google Scholar]
- Lachowiez, C.A.; Loghavi, S.; Zeng, Z.; Tanaka, T.; Kim, Y.J.; Uryu, H.; Turkalj, S.; Jakobsen, N.A.; Luskin, M.R.; Duose, D.Y.; et al. A phase Ib/II study of ivosidenib with venetoclax ± azacitidine in IDH1-mutated myeloid maliugnancies. Blood Cancer J. 2023, 4, 276–293. [Google Scholar] [CrossRef]
- Marvin-Peek, J.; Garcia, J.S.; Borthakur, G.; Garcia-Manero, G.; Short, N.J.; Kadia, T.M.; Loghavi, S.; Masarova, L.; Daver, N.A.; Mati, A. A phase Ib/II study of Ivosidenib with Venetoclax ± Azacitidine in IDH1-mutated hematologic malignancies: A 2024 update. Blood 2024, 144 (Suppl. S1), 219–221. [Google Scholar] [CrossRef]
- Smith, B.D.; Lachowiez, C.A.; Ambinder, A.J.; Binder, G.; Angiolillo, A.; Vestin, A.; Paglia, R.; Potluri, R.; Papademnetriou, E.; LeBlanc, T.W. A comparison of acute myeloid leukemia (AML) regimens: Hypomethylating agents combined with ivosidenib or venetoclax in newly diagnosed patients with IDH1 mutations: A real-world evidence study. Blood 2023, 142 (Suppl. S1), 971. [Google Scholar]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase I study. Blood 2021, 137, 1792–1803. [Google Scholar]
- Mason, E.F.; Pozdynakova, O.; Roshal, M.; Fathi, A.T.; Stein, E.M.; Ferrell, P.B.; Shaver, A.C.; Frattini, M.; Wang, H.; Hua, L.; et al. A novel differentiation response with combination IDH inhibitor and intensive induction therapy for AML. Blood Adv. 2021, 5, 2279–2283. [Google Scholar] [PubMed]
- Fathi, A.T.; Kim, H.T.; Soiffer, R.J.; Levis, M.J.; Li, S.; Kim, A.S.; DeFilipp, Z.; El-Jewari, A.; MCAfee, S.; Brunner, A.M.; et al. Multicenter phase I trial of ivosidenib as maintenance treatment following as maintenance treatment following allogeneic hematopoietic cell transplantation for IDH1-mutated acute myeloid leukemia. Clin. Cancer Res. 2023, 29, 2034–2042. [Google Scholar] [PubMed]
- Choe, S.; Wang, H.; Di Nardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following Ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar]
- Turkalj, S.; Stoilova, B.; Groom, A.J.; Radtke, F.E.; Mecklenbrauck, R.; Jakobsen, N.A.; Lachowiez, C.A.; Metzner, M.; Usukhbayar, B.; Salazar, M.A.; et al. Clonal basis of resistance and response to ivosidenib combination therapies is established early during treatment in IDH1-mutated myeloid malignancies. Blood 2024, 144, 642–644. [Google Scholar]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafsson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gofur, D.; et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar]
- Lin, J.; Lu, W.; Caravella, J.A.; Campbell, A.M.; Diebold, F.B.; Ericsson, A.; Fritzen, E.; Gustafson, G.R.; Lancia, D.R.; Shelekhin, T.; et al. Discovery and optimization of quinolone derivatives as potent, selective, and orally bioavailable mutant ioscitreate dehydrogenase 1 (mIDH1) inhibitors. J. Med. Chem. 2019, 62, 6575–6592. [Google Scholar]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.N.; Pigneux, A.; Montesinos, P.; Wang, A.S.; et al. Olutasedinib alone or with azicitidine in IDH1-mutated acute myeloid leukemia and myelodysplastic syndrome: Phase 1 results fo a phase 1-2 trial. Lancet Haematol. 2023, 10, e46–e58. [Google Scholar]
- De Botton, S.; Fenaux, P.; Yee, K.; Récher, C.; Wei, A.H.; Montesinos, P.; Taussig, D.C.; Pigneux, A.; Braun, T.; Curti, A.; et al. Olutasedinib (FT-2102) induces durable complete remissions in patients with relapsed or refractory IDH1-muatted AML. Blood Adv. 2023, 7, 3117–3127. [Google Scholar]
- Cortes, J.E.; Jonas, B.A.; Watts, J.M.; Chao, M.M.; De Botton, S. Olutasidenib for mutated IDH1 acute myeloid leukemia: Final five-year results from the phase 2 pivotal cohort. J. Clin. Oncol. 2024, 42 (Suppl. S16), 6528. [Google Scholar]
- De Botton, S.; Jonas, B.A.; Ferrell, B.; Choa, M.M.; Mims, A.S. Safety and efficacy of olutasidenib treatment in elderly patients with relapsed/refractory mIDH1 acute myeloid leukemia. J. Clin. Oncol. 2024, 42 (Suppl. S16), 6527. [Google Scholar]
- Cortes, J.E.; Esteve, J.; Bajel, A.; Yee, K.; Braun, T.; De Botton, S.; Peterlin, P.; Recher, C.; Thomas, X.; Watts, J.; et al. Olutasidenib (FT-2102) in combination with azacitidine induces durable complete remissions in patients with mIDH1 acute myeloid leukemia. Blood 2021, 138, 698–701. [Google Scholar]
- Cortes, J.E.; Roboz, G.J.; Watts, J.; Baer, M.R.; Jonas, B.A.; Schiller, G.J.; Yee, K.; Ferrell, B.; Yang, J.; Wang, E.S.; et al. Combination of olutasidenib and azacitidine induces durable complete remissions in mIDH1 acute myeloid leukemia: A multicohort open-label phase 1-2 trial. Blood 2024, 144 (Suppl. S1), 2886. [Google Scholar]
- Cortes, J.E.; Roboz, G.J.; Watts, J.; Baer, M.R.; Jonas, B.A.; Schiller, G.J.; Yee, K.; Ferrell, B.; Yang, J.; Wang, E.S.; et al. Olutasidenib in combination with azacitidine induces durable complete remissions in patients with relapsed or refractory mIDH1 acute myeloid leukemia: A multicohort open-label phase 1-2 trial. J. Hematol. Oncol. 2025, 18, 7. [Google Scholar]
- DiNardo, C.D.; Chien, K.S.; Mullin, J.; Hammond, D.; Ramdial, J.; Kadia, T.M.; Haddad, F.G.; Yilmaz, M.; Sasaki, K.; Issa, G.C.; et al. Phase ½ study of decitabine and venetoclax in combination with the targeted mutant IDH1 inhibitor olutasidenib for patients with relapsed/refractory AML, high risk MDS, or newly diagnosed AML not eligible for chemotherapy with an IDH1 mutation. Blood 2024, 144 (Suppl. S1), 617. [Google Scholar]
- Cortes, J.; Jonas, B.A.; Schiller, G.; Mims, A.; Roboz, G.J.; Wei, A.H.; Montesinos, P.; Ferrell, P.B.; Yee, K.; Fenaux, P.; et al. Olutasidenib in post-venetoclax patients with mutant isocitrate dehydrogenase 1 (IDH1) acute myeloid leukemia (AML). Leuk. Lymphoma 2024, 65, 1145–1152. [Google Scholar]
- Lai, C.E.; Leahy, T.P.; Turner, A.; Thomassen, A.; Wang, L.; Sheppard, A.; Cortes, J.E. Effectiveness of olutasidenib versus ivosidenib in patients with mutated isocitrate dehydrogenase 1 acute myeloid leukemia who are relapsed or refractory to venetoclax: The 2102-HEM-101 trial versus a US electronic health record-based external control arm. Blood 2024, 144, 1525. [Google Scholar]
- Watts, J.M.; Shaw, S.J.; Jona, B.A. Looking beyond the surface: Olutasidenib and ivosidenib for treatment of mIDH1 acute myeloid leukemia. Curr. Treat. Options Oncol. 2024, 25, 1345–1353. [Google Scholar]
- Yen, K.; Travens, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLa Barre, B.; Tobin, E.; et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar]
- Stein, E.M.; Di Nardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; De Angelo, D.J.; Levine, R.L.; Finn, J.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Tallman, M.S.; De Botton, S.; Komborjian, A.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed]
- De Botton, S.; Montesinos, P.; Schuh, A.C.; Papayannidis, C.; Vyes, P.; Wei, A.H.; Ommen, H.; Semochkin, S.; Kim, H.J.; Larsom, R.A.; et al. Enasidenib versus conventional care in older patients with late-stage mutant IDH2-relapsed/refractory AML: A randomized phase 3 trial. Blood 2023, 141, 156–164. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.H.; de Botton, S.; Ziedan, A.M.; Fathi, A.T.; Kantarjian, H.M.; Bennett, J.M.; et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukemia (AG221-AML-005): A single-arm, phase 1b and randomized, phase 2 trial. Lancet Oncol. 2021, 22, 1597–1608. [Google Scholar] [CrossRef]
- Cai, S.F.; Huang, Y.; Lance, J.R.; Mao, H.C.; Dunbar, A.J.; McNulty, S.N.; Druley, T.; Li, Y.; Baer, M.R.; Stock, W.; et al. A study to assess the efficacy of enasidenib and risk-adapted addition of azacitidine in newly diagnosed IDH2-mutant AML. Blood Adv. 2024, 8, 429–440. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Gupta, G.; Cameron, C.; Chatelin, S.; Bankar, A.; Davidson, M.B.; Gupta, V.; Maze, D.C.; Minden, M.D.; Murphy, T.; et al. Final results of the phase Ib/II study evaluating enasidenib in combination with venetoclax in patients with IDH2-mutated relapsed/refractory myeloid malignancies. Blood 2023, 142 (Suppl. S1), 159–161. [Google Scholar] [CrossRef]
- Salhotra, A.; Bejanyan, N.; Yang, D. Multicenter pilot clinical trial of enasidenib as maintenance therapy after allogeneic hematopoietic cell transplantation (allo-HCT) in patients with acute myeloid leukemia (AML) carrying IDH2 mutations. In Proceedings of the Transplantation and Cellular Therapies Meeting, Sant Antonio, TX, USA, 21–24 February 2024. abst 10. [Google Scholar]
- Ball, B.J.; Zhang, J.; Afkhami, M.; Robbins, M.; Chang, L.; Humpal, S.; Porras, V.; Liu, Y.; Synold, T.; Farzinkhou, S.; et al. Study of IDH inhibition with Enasidenib and MEK inhibition with Cobimetinib in patients with relapsed or refractory acute myeloid leukemia who have co-occurring IDH2 and RAS signling gene mutations. Blood 2024, 144 (Suppl. S1), 6043–6044. [Google Scholar] [CrossRef]
- Sebert, M.; Chevrer, S.; Dimicoli-Salazar, S.; Cluzeau, T.; Rauzy, O.; Bastard, A.S.; Lribi, K.; Fosard, G.; Thépot, S.; Gloaguen, S.; et al. Enasidenib (ENA) monotherapy in patients with IDH2 muatted myelodysplastic syndrome (MDS), the ideal phase 2 study by the GFM and Emsco groups. Blood 2024, 144 (Suppl. S1), 1839–1840. [Google Scholar] [CrossRef]
- Montesinos, P.; Bergua, J.M.; Vellenga, E.; Rayon, C.; Parody, R.; de la Serna, J.; Leon, A.; Esteve, J.; Milone, G.; Debén, G.; et al. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: Characteristics, outcome, and prognostic factors. Blood 2009, 113, 775–783. [Google Scholar] [CrossRef]
- Luesink, M.; Pennings, J.; Wissink, W.; Linssen, P.; Muus, P.; mPfundt, R.; de Witte, T.; van der Reijden, B.; Jansen, J.H. Chemokine induction by all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia: Triggering the differentiation syndrome. Blood 2009, 114, 5512–5521. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Montesinos, P. How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood 2014, 123, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; AG221-C-001 Study Investigators. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: Analysis of a phase 1–2 study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef]
- Nosworthy, K.J.; Mulkey, F.; Scott, E.C.; Ward, A.F.; Przepiorka, D.; Charlab, R.; Dorff, S.E.; Deisseroth, A.; Kazandjian, D.; Sridhara, R.; et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: A U.S. food and drug administration systematic analysis. Clin. Cancer Res. 2020, 26, 4280–4288. [Google Scholar] [CrossRef]
- Montesinos, P.; Fathi, A.T.; de Botton, S.; Stein, E.M.; Zeidan, A.M.; Zhu, Y.; Prtebet, T.; Vigil, C.E.; Bluemmert, I.; Yu, X.; et al. Differentiation syndrome associated with treatment with IDH2 inhibitor enasidenib: Pooled analysis from clinical trials. Blood Adv. 2024, 6, 2509–2520. [Google Scholar] [CrossRef]
- Sacilotto, N.; Dessanti, P.; Lufino, M.M.P.; Ortega, A.; Rodriguiez-Gimeno, A.; Salas, J.; Maes, T.; Bues, C.; Mascarò, C.; Soliva, R. Comprehensive in vitro characterization of the LSD1 small molecule inhibitor class in oncology. ACS Pharmacol. Transl. Sci. 2021, 4, 1818–1834. [Google Scholar] [CrossRef] [PubMed]
- Baby, S.; Shinde, S.D.; Kulkarni, N.; Sahu, B. Lysine-specific demethylase 1 (LSD1) inhibitors: Peptides as emerging class of therapeutics. ACS Chem. Biol. 2023, 18, 2144–2155. [Google Scholar] [CrossRef]
- Sprussel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Handschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; Konig, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef]
- Cai, W.; Xia, C.; Fan, T.; Deng, Z.; Wang, D.; Liu, Y.; Li, C.; He, J. Targeting LSD1 in cancer: Molecular elucidation and recent advances. Cancer Lett. 2024, 598, 217093. [Google Scholar] [CrossRef]
- Lynch, J.T.; Spence, G.J.; Harris, W.J.; Maiques-Diaz, A.; Ciceri, F.; Huang, X.; Somervaille, T. Pharmacological inhibitors of LSD1 promote differentiation of myeloid leukemia cells through a mechanism independent of histone demethylation. Blood Adv. 2014, 124 (Suppl. S1), 267. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef] [PubMed]
- Barth, J.; Abou-El-Ardat, K.; Dalic, D.; Kurrle, N.; Maier, A.M.; Mohr, S.; Schutte, J.; Vassen, L.; Greve, G.; Schulz-Fincke, J.; et al. Lsd1 inhibition by tranylcypromine derivatives interferes with GFI1.mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia 2019, 33, 1411–1426. [Google Scholar]
- Cusan, M.; Cai, S.F.; Mohammad, H.P.; Krivstov, A.; Chramiec, A.; Loizou, E. LSD1 inhibition exerts in antileukemic effect by recommissioning PU.1 and C/EBPα-dependent enhancers in AML. Blood 2018, 131, 1730–1742. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Nicosia, L.; Bosma, N.J.; Romero-Camarero, I.; Camera, F.; Spencer, G.J.; Amad, F.; Sineom, F.; Wingelhafer, B.; Williamson, A.; et al. HMG20B stabilizes association of LSD1 with GFI1 on chromatin to confer transcription repression and leukemia differentiation block. Oncogene 2022, 44, 4841–4854. [Google Scholar]
- Cai, S.F.; Chu, S.H.; Goldberg, A.D.; Parvin, S.; Koche, R.P.; Glass, J.L.; Tallman, M.S.; Sen, F.; Famulare, C.A.; Cusen, M.; et al. Leukemic cell of origin influences apoptotic priming and sensitivity to LSD1 inhibition. Cancer Discov. 2020, 10, 1500–1513. [Google Scholar]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by GFI1 and GFI1B is mediated by the cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [PubMed]
- Moroy, T.; Vassen, L.; Wilkes, B.; Khandanpour, C. From cytopenia to leukemia. The role of GFI1 and GFI1B in blood formation. Blood 2015, 126, 2561–2569. [Google Scholar]
- Waterbury, A.L.; Kwok, H.S.; Lee, C.; Narducci, D.N.; Freedy, A.M.; Su, C.; Raval, S.; Reiter, A.H.; Hawkins, W.; Lee, K. An autoinhibitory switch of the LSD1 disordered region controls enhancer silencing. Mol. Cell 2024, 84, 2238–2254. [Google Scholar] [PubMed]
- Maes, T.; Mascarò, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdone, A.; Lufino, M.M.; Somervaille, T.; et al. ORY-1001, a potent and selective covalent KMD1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef]
- Salamero, O.; Montesinos, P.; Willekens, C.; Perez-Simon, J.A.; Pigneux, A.; Rècher, C.; Popat, R.; Carpo, C.; Molinero, C.; Mascaro, C.; et al. First-in-human phase I study of Iadademstat (ORY-1001): A first-in-class lysine-specific histone demethylase 1A inhibitor, in relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar]
- Salamero, O.; Molero, A.; Perez-Simon, J.A.; Arnan, M.; Cokli, R.; Garcia-Avila, S.; Acuna-Criz, E.; Cano, I.; Somervaille, T.; Gutierrez, S.; et al. Iadademstat in combination with azacitidine in patients with newly diagnosed acute myeloid leukemia (ALICE): An open-label, phase 2a dose-finding study. Lancet Hematol. 2024, 11, e487–e498. [Google Scholar]
- Fathi, A.; Braun, T.P.; Abinder, A.J.; Borthakur, G.; Redner, R.L.; Arevalo, M.; Gutierrez, S.; Limon, A.; Faller, D.V. Iadademstat and gilteritinib for the treatment of FLT3-mutated relapsed/refractory acute myeloid leukemia: The Frida study. Blood 2023, 142 (Suppl. S1), 5974–5975. [Google Scholar]
- Fathi, A.; Braun, T.P.; Abinder, A.J.; Palmisano, N.; Khurana, S.; Strickland, S.; Murthy, G.; Venugopal, S.; Feld, J.; Sanchez, E.; et al. Preliminary Results of the FRIDA Study: Iadademstat and Gilteritinib in FLT3-Mutated R/R AML; EHA: Hague, The Netherlands, 2024. [Google Scholar]
- Brunett, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabradan, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512. [Google Scholar]
- Uckelmann, H.J.; Haaer, E.L.; Takeda, R.; Wong, E.M.; Hatton, C.; Marinaccio, C.; Perner, F.; Rajput, M.; Antonissen, N.J.C.; Wen, Y.; et al. Mutant NPM1 directly regulates oncogenic transcription in acute myeloid leukemia. Cancer Discov. 2023, 13, 746–765. [Google Scholar] [PubMed]
- Wang, X.Q.D.; Fan, D.; Han, Q.; Liu, Y.; Miao, H.; Wang, X.; Li, Q.; Chen, D.; Gore, H.; Himadewi, P.; et al. Mutant NPM1 hijacks transcriptional hubs to maintain pathogenic gene programs in acute myeloid leukemia. Cancer Discov. 2023, 13, 724–745. [Google Scholar]
- Khan, I.; Amin, M.A.; Eklund, E.A.; Gartel, A.L. Regulation of HOX gene expression in AML. Blood Cancer J. 2024, 14, 42. [Google Scholar]
- Kubota, Y.; Reynold, M.; Williams, N.D.; Kawashima, N.; Bravo-Perez, C.; Guarnera, L.; Haddad, C.; Mandala, A.; Guarnari, C.; Durmaz, A.; et al. Genomic analyses unveil the pahtogenesis and inform on therapeutic targeting in KMT2A-PTD AML. Blood 2023, 142 (Suppl. S1), 5696. [Google Scholar] [CrossRef]
- Yokoyama, A.; SKhan Somervaille, T.C.P.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Wallace, L.K.; Peplinski, J.H.; Ries, R.E.; Kirkey, D.C.; Meshinchi, S. The AML HOX-ome: The landscape of developmental transcription factors across pediatric acute myeloid leukemia. Blood 2024, 144 (Suppl. S1), 618. [Google Scholar] [CrossRef]
- Dhillon, V.; Aguilar, J.; Kim, P.; Padmanabhan, D.; Dhiman, S.; Dyson, G.; Maciejewski, J.; Balasubramanian, S.K. Clinical significance and Meis gene expression profiling in acute myeloid leukemia. Blood 2024, 144 (Suppl. S1), 6162–6163. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Muari, M.J.; Belcher, A.M.; Hartley, T.; et al. Meinin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [PubMed]
- Krivstov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged lkeukemia. Cancer Cell 2019, 36, 660–673. [Google Scholar]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. InvestIG. 2020, 130, 981–997. [Google Scholar]
- Soto-Feliciano, Y.; Sanchez-Rovera, F.; Perner, F.; Barrows, D.; Kastenhuber, E.; Ho, Y.J.; Carool, T.; Xiong, Y.; Aand, D.; Soshnev, A.A.; et al. A molecular switch between mammalian MLL complexes dictates response to menin-MLL inhibition. Cancer Discov. 2023, 13, 146–169. [Google Scholar]
- Ciaurro, V.; Skwarska, A.; Daver, N.; Konopleva, M. Menin inhibtor DS-1594b drives differentiation and induces synergistic lethality in combination with venetoclax in AML cells with MLL-rearranged and NPM1 mutation. Blood 2022, 140 (Suppl. S1), 3082–3083. [Google Scholar]
- Issa, C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukemia. Nature 2023, 615, 920–926. [Google Scholar] [PubMed]
- Muftuoglu, M.; Basyal, M.; Ayoub, E.; Lv, J.; Bedoy, A.; Patsilevas, T.; Bidikian, A.; Issa, G.S.; Andreef, M. Single-cell proteome analysis reveal menin inhibition-induced proteomic alterations in AML patients treated with Revumenib. Blood 2023, 142 (Suppl. S1), 2933. [Google Scholar]
- Issa, G.C.; Aldoss, I.; Thirman, M.J.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. Menin inhibition with Revumenib for KMT2A-rearranged relapsed or refractory acue leukemia (AUGMANY-101). J. Clin. Oncol. 2024, 43, 75–84. [Google Scholar]
- Aldoss, I.; Issa, G.C.; Blachly, J.; Thirman, M.J.; Mannis, G.N.; Arellano, M.; DiPersio, J.; Traer, E.; Zwaan, M.; Shukla, N.; et al. Updated results and longer follow-up from AUGMENT-101 phase 2 study of remuvenib in all patients with relapsed or refractory (R/R) KMT2A-Ar acute leukemia. Blood 2024, 144 (Suppl. S1), 211. [Google Scholar]
- Syndax Pharmaceuticals Announced Pivotal Topline Results from Relapsed or Refractory AML Cohort in AUGMAENT-101 Trial of Revumenib; New release; Syndax Pharmaceuticals: Waltham, MA, USA, 2024.
- Loo, S.; Iland, H.; Tiong, I.S.; Westerman, D.; Othman, J.; Masrlton, P.; Chua, C.C.; Putill, D.; Rose, H.; Fleming, S.; et al. Revumenib as pre-emptive therapy for measurable residual disease in NPM1 mutated or KMT2A-rearranged acute myeloid leukemia: A domain of the multi-arm ALLG AMLM26 intercept platform trial. Blood 2024, 144, 223–225. [Google Scholar] [CrossRef]
- Issa, G.C.; Cuglievan, B.; Daver, N.; DiNardo, C.; Farhat, A.; Short, N.J.; McCall, D.; Pike, A.; Tan, S.; Kamerer, B.; et al. Phase I/II stud of the all-oral combination of remivenib (SNDX-5613) with decitabine/cedazuridine (ASTX727) and venetoclax (SAVE) in R/R AML. Blood 2024, 144 (Suppl. S1), 216. [Google Scholar] [CrossRef]
- Scheidegger, N.; Alexa, G.; Khalid, D.; Ries, R.E.; Wang, J.; Alonzo, T.A.; Perr, J.; Armstrong, S.A.; Meshinchi, S.; Pikman, Y.; et al. Combining menin and MEK inhibition to target poor prognostic KMT2A-rearranged RAS pathway-mutant acute leukemia. Blood 2023, 142 (Suppl. S1), 166–167. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef]
- Perner, F.; Cai, S.F.; Wenge, D.V.; Kim, J.; Cutler, J.; Nowak, R.; Cassel, J.; Singh, S.; Bijpuria, S.; Miller, W.H.; et al. Characterization of acquired resistance mutations to Menin inhibitors. Cancer Res. 2023, 83 (Suppl. S7), 3457. [Google Scholar]
- Ray, J.; Clegg, B.; Grembecka, J.; Cierpicki, T. Drug-resistant menin variants retain high binding affinity and interactions with MLL1. J. Biol. Chem. 2024, 300, 107777. [Google Scholar]
- Kwon, M.C.; Thuring, J.W.; Querolle, O.; Dai, X.; Verhulst, T.; Pande, V.; Marien, A.; Goffin, D.; Wenge, D.V.; Yue, H.; et al. Preclinical efficacy of the potent, selective menin-KMT2A inhibitor JNJ-75276617 (bleximenib) in KMT2A- and NPM1-altered leukemias. Blood 2024, 144, 1206–1214. [Google Scholar] [CrossRef]
- Jabbour, E.; Searle, E.; Abdul-Hay, M.; Abedin, S.; Aldoss, I.; Pierola, A.A.; Alonso-Dominguez, J.M.; Chevallier, P.; Cost, C.; Diskalakis, N.; et al. A first-in-human phase 1 stud of the menin-KMT2A (MLL-1) inhibitor JNJ-75276617 in adult patients relapsed/refractory acute leukemia harboring KMT2A or NPM1 alterations. Blood 2023, 142 (Suppl. S1), 57–60. [Google Scholar] [CrossRef]
- Searle, E.; Recher, C.; Abdul-Hay, M.; Abedin, S.; Aldoss, I.; Pierole, A.; Aolnso-Dominguiez, J.; Chevallier, P.; Cost, C.; Daskalakis, N.; et al. Bleximenib dose optimization and determination of RP2D from a phase 1 stud in relapsed/refractor acute leukemia patients with KMt2A and NPM1 alterations. Blood 2024, 144 (Suppl. S1), 212. [Google Scholar] [CrossRef]
- Recher, C.; O’Nions, J.; Aldoss, I.; Pierola, A.A.; Allred, A.; Alonso-Dominguez, J.M.; Barreyro, L.; Bories, P.; Curtis, M.; Daskalakis, N.; et al. Phase Ib study of menin-KMT2A inhibitor Blixemenib in combination with intensive chemotherapy in newly diagnosed acute myeloid leukemia with KMT2A or NPM1 alterations. Blood 2024, 144 (Suppl. S1), 215. [Google Scholar] [CrossRef]
- Wei, A.H.; Searle, E.; Aldoss, I.; Alfonso-Pierole, A.; Alonso-Dominguez, J.M.; Curtis, M.; Dsakalakis, N.; Della Porta, M.; Dohner, H.; D’Souza, A.; et al. A Phase 1B Study of the Menin-KMT2A Inhibitor JNJ-75276617 in Combination with Venetoclax and Azacytidine in Relapsed/Refractory Acute Myeloid Leukemic with Alterations in KMT2A or NPM1; abst S133; EHA: Hague, The Netherlands, 2024. [Google Scholar]
- Hogeling, S.M.; Le, D.M.; La Rose, N.; Kwon, M.C.; Wierenga, A.; van den Heuvei, F.; van den Boom, V.; Kucknio, A.; Philippar, U.; Huls, G.; et al. Bleximinab, the novel menin-KMT2A inhibitor JNJ-75276617, impairs long-term proliferation and immune evasion in acute myeloid leukemia. Haematologica 2025, in press. [Google Scholar]
- Collins, C.; Wang, J.; Miao, H.; Bronstein, J.; Nawer, H.; Xu, T.; Figueroa, M.; Muntean, A.; Hess, J.L. C/ERBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 9899–9904. [Google Scholar]
- Schmidt, L.; Heyes, E.; Scheiblecker, L.; Eder, T.; Volpe, G.; Frampton, J.; Nerlov, C.; Valent, P.; Germbecka, J.; Grebien, F. CEBPA-mutated leukemia is sensitive to genetic and pharmacological targeting of the MLL1 complex. Leukemia 2019, 33, 1608–1619. [Google Scholar]
- Eguchi, K.; Shimizu, T.; Kato, D.; Furuta, Y.; Kamioka, S.; Ban, H.; Ymamoto, S.; Yokoyama, A.; Kitabaayshi, I. Preclinical evaluation of a novel orally bioavailable menin-MLL interaction inhibitor, DSP-5336, for the treatment of acute leukemia patients with MLL-rearrangement of NPM1 mutation. Blood 2021, 138 (Suppl. S1), 3339. [Google Scholar]
- Daver, N.; Zeidner, J.F.; Yuda, J.; Watts, J.M.; Levis, M.J.; Fukushima, K.; Ikezoe, T.; Ogawa, Y.; Brandwein, J.; Wang, E.S.; et al. Phase 1-2 first-in-human study of the menin-MLL inhibitor DSP-5336 in patients with relapsed or refractory acue leukemia. Blood 2023, 142 (Suppl. S1), 2911. [Google Scholar]
- Zeidner, J.F.; Yuda, J.; Watts, J.M.; Levis, M.J.; Erba, H.P.; Fukushima, K.; Shaima, T.; Palmisano, N.D.; Wang, E.S.; Borate, U.; et al. Phase 1 results: First-in-human phase 1-2 study of the menin-MLL inhibitor enzomenib (DSP-5336) in patients with relapsed or refractor acute leukemia. Blood 2024, 144 (Suppl. S1), 213. [Google Scholar]
- Fisku, W.; Daver, N.; Boettcher, S.; Mill, C.P.; Sasaki, K.; Bindwell, C.E.; Davis, J.A.; Das, K.; Takashi, K.; Kadia, T.M.; et al. Activity of menin inhibitor zitfomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia 2022, 36, 2729–2733. [Google Scholar]
- Rauch, J.; Dzama, M.M.; Dolgikh, N.; Stiller, H.; Bohl, S.; Lahrmann, C.; Kunz, K.; Kessler, L.; Echannoni, H.; Chei, C.W.; et al. Menin inhibitor ziftomeninb (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax. Haematologica 2023, 108, 2837–2847. [Google Scholar]
- Wang, E.S.; Issa, G.C.; Erba, H.P.; Altman, J.K.; Montesinos, P.; DeBotton, S.; Walter, R.B.; Pettit, K.; Savona, M.R.; Shah, M.V.; et al. Ziftomenib in relapsed or refractory acute myeloid leukemia (KOPMET-001): A multicenter, open-label, multi-cohort, phase I trial. Lancet Oncol. 2024, 25, 1310–1324. [Google Scholar]
- Zeidan, A.M.; Wang, E.S.; Issa, G.C.; Altman, J.; Balasubramianan, S.K.; Strickland, S.A.; Roboz, G.J.; Schiller, G.J.; McMahon, C.M.; Palmisano, N.D.; et al. Ziftomenib combined with intensive induction (7+3) in newly diagnosed NPM1-m or KMT2A-r acute myeloid leukemia: Interim phase 1a results from KOMET-007. Blood 2024, 144 (Suppl. S1), 214. [Google Scholar]
- Golobberg, A.D.; Corun, D.; Ahsan, J.; Nie, K.; Koziek, T.; Leoni, M.; Dale, S. Kamet-008: A phase 1 study to determine the safety and tolerability of Ziftomernib combinations for the treatment of KMT2A-rearranged or NPM1-mutant relapsed/refractory acute myeloid leukemia. Blood 2023, 142 (Suppl. S1), 1553. [Google Scholar] [CrossRef]
- Lancet, J.; Ravandi, F.; Montesinos, P.; Barrientos, J.C.; Badar, T.; Alegre, A.; Bashey, A.; Bergua Bugues, J.M.; Brunetti, L.; Curran, E.K.; et al. Covalent menin inhibitor Bmf-219 in patients with relapsed or refractory (R/R) leukemia (AL): Preliminary phase 1 data from the Covalent-101 study. Blood 2023, 142 (Suppl. S1), 2916–2918. [Google Scholar] [CrossRef]
- Rasouli, M.; Troester, S.; Grebien, F.; Goemans, B.F.; Zwaan, G.M.; Heidenreich, O. NUP98 oncofusions in myeloid malignancies: An update on molecular mechanisms and therapeutic opportunities. HemaSphere 2024, 8, 70013. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.P.; Hu, W.; Chen, C.W.; Chu, S.H.; Brien, G.L.; Park, C.Y.; et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Heinroich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Golalakrishnapillai, A.; Xu, H.; Uckelmann, H.J.; et al. The menin-MLL1 interaction in a molecular dependency in NUP98-rearranghed AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef]
- Rasouli, M.; Blair, H.; Troester, S.; Szoltysek, K.; Cameron, R.; Ashtiani, M.; Krippmner-Heidenreich, A.; Grebien, F.; McGeehan, G.; McGeehanm, G.; et al. The MLL-menin interaction is a therapeutic vulnerability in NUP98-rearranged AML. HemaSphere 2023, 7, e935. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, E.D.; Martucci, C.; Henrich, J.A.; Neel, D.S.; Mahendra-Rajah, S.; Rice, H.; Wenge, D.V.; Perner, F.; Wen, Y.; Hatton, C.; et al. NUP98 fusion proteins and KMT2A-Menin antagonize PRC1.1 to drive gene expression in AML. Cell Rep. 2024, 43, 114901. [Google Scholar] [CrossRef]
- Carraway, H.E.; Nakitandwe, J.; Cacovean, A.; Ma, Y.; Mumeke, B.; Waghmare, G.; Mandap, C.; Ahmed, U.; Kawalkezyk, N.; Butler, T.; et al. Complete remission of NUP98 fusion-positive acute myeloid leukemia with the covalent menin inhibitor BMF-219, icovamenib. Haematologica 2025, in press. [Google Scholar] [CrossRef]
- Miao, H.; Cheri, D.; Ropa, J.; Purohit, T.; Kim, E.; Sulis, M.L.; Ferrando, L.; Cierpicki, T.; Grembecka, J. Combination of menin and kinase inhibitors as an effective treatment for leukemia with NUP98 translocations. Leukemia 2024, 34, 1674–1686. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Molecular Structure | Mechanism of Action | Major Effects on Tumor Target | Registration Status |
|---|---|---|---|---|---|
| Ivosidenib (AG-120) | IDH1 It inhibits mutant IDH1 at much lower doses than WT IDH1 | Small molecule inhibitor (MW 583) | Inhibition of binding pocket of IDH1 dimers | Reduction in 2-HG levels, inhibition of cell proliferation, and induction of cell differentiation | Approved for R/R IDH1-mut AML patients |
| Olutasidenib (FT-2102) | IDH1 Selective inhibitor of mutant IDH1; no inhibition of WT IDH1 | Small molecule inhibitor (MW 355) | Inhibition of binding pocket of IDH1 dimers | Reduction in 2-HG levels, inhibition of cell proliferation, and induction of cell differentiation | Approved for R/R IDH1-mut AML patients |
| Enasidenib (AG-221) | IDH2 It inhibits mutant IDH2 (R172K, R172S, R140Q) at much lower doses than WT IDH2 | Small molecule inhibitor (MW 569) | Inhibition of binding pocket of IDH2 dimers | Reduction in 2-HG levels, inhibition of cell proliferation, and induction of cell differentiation | Approved for R/R IDH2-mut AML patients |
| Iadademstat (ORY-1001) | LSD1 Highly selective covalent LSD1 inhibitor | Small molecule inhibitor (MW 230) | Inhibition of both demethylating activity of LSD1 and scaffolding function | Reduced proliferation of blast leukemic cells and induction of their differentiation | Orphan Drug-Designated |
| Ravumenib | Menin | Small molecule inhibitor (MW 841) | Inhibition of Menin-MLL binding | It impairs proliferation and induces cell differentiation in MLL-r and NPM1-mut AML | Approved for R/R AML |
| Bleximenib (JNJ-75276617) | Menin | Small molecule inhibitor (MW 600) | Inhibition of Menin-MLL binding | It impairs proliferation and induces cell differentiation in MLL-r and NPM1-mut AML | Under investigation |
| Enzomenib (DSP-5336) | Menin | Small molecule inhibitor (MW 590) | Inhibition of Menin-MLL binding | It impairs proliferation and induces cell differentiation in MLL-r and NPM1-mut AML | Under investigation |
| Ziftomenib | Menin | Small molecule inhibitor (MW 718) | Inhibition of Menin-MLL binding | It impairs proliferation and induces cell differentiation in MLL-r and NPM1-mut AML | Under investigation |
| NCT Identifier Phase | Patient Number and Disease Status | Therapeutic Regimen | Efficacy | Toxicity |
|---|---|---|---|---|
| NCT02074839 Phase I | 179 R/R IDH1-mut AML | Ivosidenib 500 mg QD (single arm) | ORR 41.6% CR 21.6% CR + CRi 30.4% MDR 6.5 mo Among responders: 7% IDH1-mut negative | QT interval prolongation 7.8% Differentiation syndrome 3.9% Anemia 2.2% Thrombocytopenia 3.4 Leukocytosis 1.7% |
| NCT02074839 Phase I | 34 ND IDH1-mut AML not eligible for standard therapy | Ivosidenib 500 mg QD (single arm) | CR 30.3% CR + CRi 42.4% Median OS 12.6 mo 77.8% of responding patients in remission at 1 year | Differentiation syndrome 9% Anemia 12% Thrombocytopenia 15% |
| NCT03173248 Phase III | 146 ND IDH1-mut AML | Azacitine (75 mg/m2) Ivosidenib (500 mg/QD) vs. Azacitidine + placebo | ORR 62% vs. 19% CR + CRi 58% vs. 19% EFS at 12 mo 38% vs. 11% mEFS 22.9 vs. 4.1 mo mOS 24 vs. 7.9 mo | Differentiation syndrome 4% vs. 4% Febrile neutropenia 20% vs. 34% Thrombocytopenia 20% vs. 15% Infection 21% vs. 30% |
| NCT02719574 Phase I/II | 126 (expansion cohort) R/R IDH1-mut AML | Olutasideb 150 mg BID (single arm) | 0RR 48% CR + CRi 35% mOS 11.6 mo In responding patients mOS Not reached MDR 11.6 mo MDR in responding patients 25.9 mo | Differentiation syndrome 9% Febrile neutropenia 20% Thrombocytopenia 16% Anemia 16% |
| NCT02719524 Phase I/II | 67 R/R IDH1-mut AML | Olutasideb 150 mg BID + Azacitidine (75 mg/m2) (sigle arm) | ORR 51% CR + CRi 31% mOS 12.5 mo mOS in patients achieving CR + CRi 36 mo | Anemia 25% Thrombocytopenia 37% Febrile neutropenia 19% Leukocytosis 6% |
| NCT Identifier Phase | Patient Number and Disease Status | Therapeutic Regimen | Efficacy | Toxicity |
|---|---|---|---|---|
| NCT01915498 Phase I/II | 153 (expansion phase) R/R IDH2-mut AML | Enasidenib 100 mg QD (single arm) | ORR 38.5% CR + CRi 26.6% MDR 5.6 mo MDR in CR 8.8 mo mOS 9.3 mo mOS in CR 19.3 mo | Differentiation syndrome 7% Hyperbilirubinemia 8% |
| NCT02577406 Phase I | 319 R/R IDH2-mut AML | Enasidenib 100 mg QD vs. Conventional therapy | ORR 40.5% vs. 9.9% CR 23.4% vs. 3.7% CR + CRi 29.7% vs. 6.2% OS at 12 mo 38% vs. 26% mEFS 4.9 mo vs. 2.6 mo | Differentiation syndrome 5% vs. 0.0% Hyperbilirubinemia 10.8% vs. 0.0% |
| NCT03013998 Phase Ib/II | 60 ND IDH2-mut AML not suitable for standard therapy | Total of 60 ND IDH2-mut AML patients treated with 5 cycles of Enasidenib: patients with CR + CRi continued Enasidenib, patients not responding were treated with Ena + AZA Enasidenib 100 mg QD (phase II) Enasidenib + Azacitidine (75 mg/m2) (phase Ib) | Phase II vs. phase Ib CR + CRi 48% vs. 40% MDR 11.1 mo vs. 14.6 mOS 17.1 mo vs. 12.5 mo at 24 months, mOS 41% vs. 47% | Differentiation syndrome 20% vs. 11.8% Thrombocytopenia 5% vs. 29% Anemia 5% vs. 17% Leukopenia 3% vs. 33% |
| NCT04092179 Phase II | 27 R/R IDH2-mut AML or MDS | Enasidenib 100 mg QD BID (single arm) Venetoclax 400 mg QD | 0RR 70% CR 57% Responses were higher in IDH2R140 than in IDH2R172-mutant patients | Febrile neutropenia 41% Thrombocytopenia 26% Hyperbilirubinemia 48% Leukocytosis 8% |
| Compound | NCT Identifier | Patient Number | Patient Typology | Toxicity | Efficacy |
|---|---|---|---|---|---|
| Iadademstat (ORY-1001) | EDRA CT 2013-002447-29 | 27 dose-escalation (5–220 μg/m2/day) 14 dose-expansion (140 ug/m2/day) | R/R AML | Myelosuppression and non- hematological adverse events (infections, asthenia, mucositis diarrhea) Differentiation syndrome two patients. One grade 3 CS and One patient was fatal | 2CRi Blast reduction and cell differentiation in 2/4 MLL-rearranged and 2/4 erythroleukemia |
| Iadademstat (ORY-1001) Azacitidine | Phase II | 60 or 90 mg/m2/day | Unfit AML patients | Myelosuppression with anemia, Thrombocytopenia, and granulocytpenia One grade 3 DS and one fatal grade 5 intrachranial hemorrhagia | ORR of 81% in responding patients and 64% CR and 36% PR Responding AML subtypes: Flt3-mutated (3/3), TP53-mutated (75%), FABM 4/M5 86% |
| Iadademstat (ORY-1001) Gilteritinib | PhaseI/II | Dose-escalation from 75 to 150 μg/m2/day 13 patients at 75 or 100 μg/m2/day | FLT3-mutant R/R AML | Not reported | At 100 μg/m2/day, 5/6 patients cleared BML blasts; at 75 μg, 2/7 had no response, 1/7 CR, and 1/7 CRi; 1 patient cleared BM blast; and 2 patients not yet assessed |
| Compound | Revumenib | Bleximenib | Enzomenib | Ziftomenib |
|---|---|---|---|---|
| Trial | AUGMENT-101 Phase I/II | CAMELOT-1 Phase I/II | DSP-5336-101 Phase I/II | KOMET-001 Phase I/II |
| Number of patients | 161 | 21 | 40 | 58 |
| ORR | KMT2Ar 64% NPM1m 47% | KMT2Ar 30% NPM1m 50% | KMT2Ar 59% NPM1m 54% | KMT2Ar 17% NPM1m 42% |
| CR + CRi | KMT2Ar 23% NPM1m 23% | KMT2Ar 33% NPM1m 33% | KMT2Ar 30% NPM1m 47% | KMT2Ar 17% NPM1m 35% |
| MRD negativity (in CR + CRi) | KMT2Ar 58% NPM1m 64% | NR | NR | KMT2Ar 100% NPM1m 63% |
| HSCT among responders | KMT2Ar 36% NPM1m 17% | NR | NR | KMT2Ar 33% NPM1m 33% |
| Differentiation Syndrome (%) | 22% | 19% | 11% | 11% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. Recent Developments in Differentiation Therapy of Acute Myeloid Leukemia. Cancers 2025, 17, 1141. https://doi.org/10.3390/cancers17071141
Testa U, Castelli G, Pelosi E. Recent Developments in Differentiation Therapy of Acute Myeloid Leukemia. Cancers. 2025; 17(7):1141. https://doi.org/10.3390/cancers17071141
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2025. "Recent Developments in Differentiation Therapy of Acute Myeloid Leukemia" Cancers 17, no. 7: 1141. https://doi.org/10.3390/cancers17071141
APA StyleTesta, U., Castelli, G., & Pelosi, E. (2025). Recent Developments in Differentiation Therapy of Acute Myeloid Leukemia. Cancers, 17(7), 1141. https://doi.org/10.3390/cancers17071141